

Abstract
Here, we report the solution NMR structure of the isolated thumb subdomain of HIV-1 reverse transcriptase (RT). A detailed comparison of the current structure with dozens of the highest resolution crystal structures of this domain in the context of the full-length enzyme reveals that the overall structures are very similar, with only two regions exhibiting local conformational differences. The C-terminal capping pattern of the αH helix is subtly different, and the loop connecting the αI and αJ helices in the p51 chain of the full-length p51/p66 heterodimeric RT differs from our NMR structure due to unique packing interactions in mature RT. Overall, our data show that the thumb subdomain folds independently and essentially the same in isolation as in its natural structural context.
Keywords: HIV-1 thumb subdomain, Ensemblator, nuclear magnetic resonance
INTRODUCTION
HIV-1 reverse transcriptase (HIV-1 RT) is an essential protein in the viral life cycle and a major drug target (Das et al. 2005; Sarafianos et al. 2009; De Béthune 2010). Previous crystallographic studies have shown that mature HIV-1 RT is an asymmetric heterodimer, composed of two subunits, p66 and p51. The p66 subunit contains two enzymatically active units: the polymerase and RNase H (RNH) domains. The polymerase domain is divided into subdomains named fingers, palm, thumb, and connection (Kohlstaedt et al. 1992). The names of the first three subdomains were inspired by the shape of the p66 subunit of this and related polymerases, in which the spatial arrangement of the thumb, fingers and palm subdomains resembles a right hand ready to clasp a piece of DNA. The p51 subunit shares the same sequence as the p66 subunit but lacks the RNH domain. The finger, palm, thumb and connection subdomains are also present in p51, although they exhibit different spatial arrangements from those in the p66 subunit (Fig. 1) (Hsiou et al. 1996; Huang 1998; Sarafianos et al. 2002; Lansdon et al. 2010).
Fig. 1.
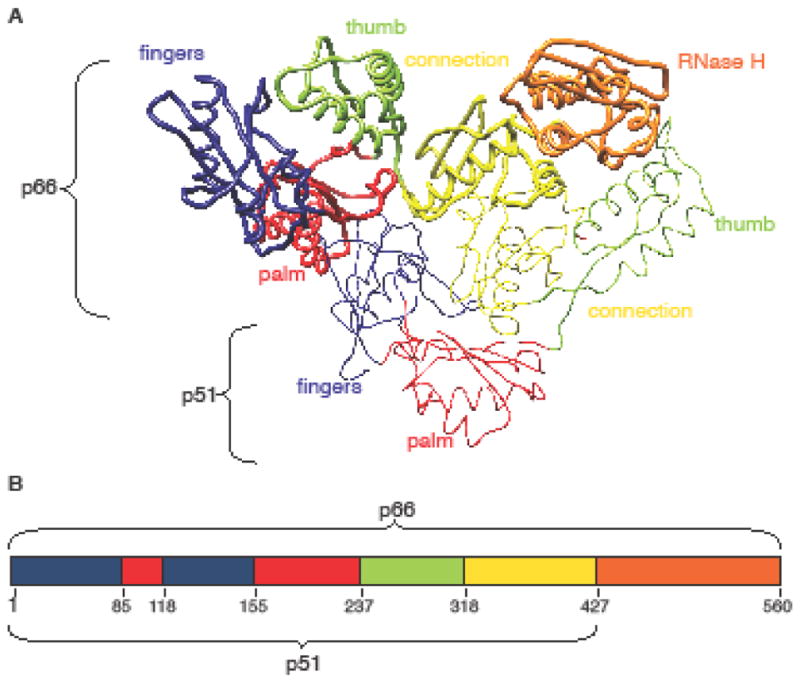
Overall HIV-1 RT structure. a Tube representation of apo-RT (PDB ID: 1DLO), with the p66 and p51 subunits shown in thick and thin tube representation, respectively. b Schematic diagram of (sub)domain organization of HIV-1 RT. The palm, fingers, connection, thumb and RNase H (sub)domains in a and b are colored in red, blue, yellow, green and orange, respectively.
At present, the precise mechanism of HIV-1 RT maturation has not been elucidated. Based on model system data, a dimeric p66 immature precursor is formed first, which is subsequently cleaved at the p51-RNH processing site on one of the p66 subunits. This removes the RNH domain and generates the p51 subunit (Tomasselli et al. 1993; Wang et al. 1994; Sluis-Cremer et al. 2004; Wapling et al. 2005). No atomic structures of the p66:p66 immature precursor are available, although several maturation models for HIV-1 RT have been proposed (Jacobo-Molina and Arnold 1991; Davies et al. 1991; Tomasselli et al. 1993; Zheng et al. 2014). In addition, numerous biochemical/biophysical data characterizing the properties of the immature precursor exist (Beard and Wilson 1993; Divita et al. 1995; Cabodevilla et al. 2001; Braz et al. 2010; Marko et al. 2013; Sharaf et al. 2014; Zheng et al. 2015). The p66 immature precursor and mature HIV-1 RT exhibit similar polymerase and RNH activities, but differ in their inter-subunit affinity (Fletcher et al. 1996). The mature HIV-1 RT (i.e a p66:p51 heterodimer) is a tighter dimer with a dissociation constant (Kd) of 0.23 μM, while the Kd for the p66:p66 immature precursor is 4.4 μM (Venezia et al. 2006).
To structurally characterize RT maturation, we previously investigated the conformation of the p66 homodimer by solution NMR (Sharaf et al. 2014). Given the protein’s large size (132 kDa), NMR studies of the p66 immature precursor are challenging. However, using TROSY-type, 1H-15N HSQC spectroscopy, it was possible to assess the structures of the (sub)domains within the p66 immature precursor and the isolated (sub)domains of HIV-1 RT. 1H-15N HSQC resonances of the isolated HIV-1 thumb subdomain superimposed well with the equivalent resonances in the 1H-15N HSQC spectrum of the homodimer, suggesting that the thumb domains in the p66 immature precursor exhibit very similar conformations as the isolated thumb domain (Sharaf et al. 2014). Here, we extend this work by reporting the solution structure of the isolated HIV-1 RT thumb subdomain, and showing that it too is very similar to the crystal structures of the thumb domains in the mature RT.
EXPERIMENTAL METHODS
Protein expression and purification
Gene construction, protein expression, and purification of the thumb domain of HIV-1 reverse transcriptase (Kohlstaedt et al. 1992) were performed as previously described (Sharaf et al. 2014). Briefly, the thumb domain coding sequence, comprising residues 237-318 of RT, was inserted between the NdeI and XhoI sites in the pET21 plasmid (Novagen). Expression from this plasmid results in a protein that possesses an N-terminal methionine and a C-terminal hexa-histidine tag. Uniformly 15N- or 13C,15N-labeled thumb domain protein was produced in E. coli BL21(DE3) gold cells (Agilent Technologies, Santa Clara, CA), in modified minimal medium at 27°C, using 15NH4Cl and 13C6-glucose as the sole nitrogen and carbon sources, respectively. Proteins were purified over a 5 mL HisTrap column (GE Healthcare), followed by 5 mL HiTrap SP column (GE Healthcare), and then a HiLoad 26/60 Superdex 200 gel filtration column (GE Healthcare). Protein fractions were pooled and concentrated in an Amicon Ultra concentrator (EMD Millipore, Billerica, MA) to ~10 μM. Glycerol (50% v/v glycerol) was added to the purified protein and samples were stored at −20°C for future use.
NMR spectroscopy
Uniformly 15N- and 13C,15N-labeled proteins were buffer exchanged into 25 mM sodium phosphate, 100 mM NaCl, 10% v/v D2O, pH 6.8 in an Amicon Ultra concentrator (EMD Millipore) to a final volume of 350 μL and final protein concentration of 1.0 mM. All NMR spectra were acquired at 30 °C on Bruker AVANCE600 and AVANCE700 spectrometers, equipped with 5 mm triple resonance, Z-axis gradient cryoprobes (Bruker Biospin, Billerica, MA). Backbone and side chain resonance assignments were carried out using two-dimensional (2D) 1H-15N HSQC, three-dimensional (3D) HN(CO)CACB, HNCACB, H(CCCO)NH, C(CCO)NH, HCCH-TOCSY spectra (Clore and Gronenborn 1998). Distance restraints were derived from 3D simultaneous 13C- and 15N-edited NOESY spectra (Sattler et al. 1995), using a mixing time of 0.15 s. All NMR data were processed with TOPSPIN 2.1 or 3.1 (Bruker) and NMRPipe (Delaglio et al. 1995), and analyzed with Collaborative Computing Project for NMR (CCPN) (Vranken et al. 2005)
NMR structure calculation
Structure calculations were performed using the anneal.py protocol in XPLOR-NIH (Schwieters et al. 2006). An iterative approach with extensive manual cross-checking of all distance restraints against the NOESY data and intermediate structures was employed using CCPN. The final number of the NMR-derived restraints was 2,782, with 2,620 NOE distances, 46 H-bond distances identified from NOE patterns for α-helices and β-sheets, and 116 ϕ and ψ backbone torsion angles from TALOS calculations (Cornilescu et al. 1999). Two hundred and fifty six structures were generated and the 30 lowest energy structures were selected and analyzed using PROCHECK-NMR (Laskowski et al. 1996) and MolProbity (Davis et al. 2007). Atomic coordinates of the structures have been deposited in the Protein Data Bank with accession code 5T82, and chemical shift assignments have been deposited in the Biological Magnetic Resonance Data Bank accession number 30171. Structures were visualized with MOLMOL (Koradi et al. 1996) and VMD (Humphrey et al. 1996). Structural figures were generated using UCSF Chimera (Pettersen et al. 2004) and the PyMOL Molecular Graphics System, Version 1.8 (Schrödinger, LLC 2015).
Ensemblator comparisons of NMR and crystal structure ensembles
A representative set of high-resolution RT crystal structures, refined at a resolution of 2.4 Å or better, was selected from all deposited structures. This yielded the following set of 28 PDB entries: 1RTJ, 1S1T, 2OPS, 2RKI, 2YKN, 2YNF, 2YNG, 2ZD1, 3BGR, 3DLG, 3DLK, 3LAK, 3LP1, 3MEC, 3MEE, 3QIP, 3T1A, 4DG1, 4I2P, 4ID5, 4IDK, 4IFV, 4IFY, 4IG3, 4KFB, 4KO0, 4KV8, and 5D3G. Analyses were carried out using Ensemblator version 3 (https://github.com/atomoton/ensemblator; Brereton and Karplus, unpublished), a greatly enhanced version of the recently described general ensemble-ensemble comparison program (Clark et al. 2015). The Ensemblator “Prepare” stage was used to combine the 30-member NMR ensemble with all the crystal structures to generate a file that contained all of the backbone and side-chain atoms that were in common between the NMR models and the 28 individual p51 and p66 chains from the X-ray structures. For all Ensemblator “Analysis” stage runs, a distance cutoff of 2 A was used to define “core” atoms; with this cutoff, 26.9 % of the atoms in the ensemble qualified for the common-core, which was used to guide the global best-fit. Based on these best-fit models, two automated clustering methods inherent to the program were used, as well as a “manual” approach, in which the NMR models, the p51 chains and the p66 chains were separated into three groups.
For each comparison, the standard Ensemblator output (Clark et al. 2015) included residue-level plots of the global and local comparisons for each pair of groups. In order to identify the most significant/consistent regions of similarity and any differences between a given pair of grouped structures, a residue-level “silhouette index” was calculated with Ensemblator v3. This index combines both global and local comparison information. For each of the two groups in a given M,N pair of grouped structures, every atom’s global silhouette score is calculated as the mean pairwise distance between the groups minus the mean pairwise distance within the group, divided by the larger of the two values: (<dinter>−<dintra>)/max(<dintra>,<dinter>). The silhouette scores for each atom are averaged across the two groups, and a residue-based value is obtained by averaging the values for the N, CA, C′, and O atoms of each residue. A second silhouette score for comparing local backbone conformations is similarly calculated for each residue based on the “locally-overlaid dipeptide residual” (LODR) distances (Clark et al. 2015). The final “silhouette index” for a residue is the average of the global and local silhouette scores. The level of detectable difference between the groups increases with the index as it goes from near 0 to 1. For indices 0.4 – 0.6, we have considered the groups to be neither notably similar nor different; within this range, more fine clustering may permit the identification of subgroups with some differences.
RESULTS AND DISCUSSION
Solution structure of the HIV-1 RT thumb domain
The 1H-15N HSQC spectrum of the thumb subdomain exhibits well-dispersed resonances (Fig. 2a), indicative of a well-folded structure. Near complete (> 95 %, backbone and side chain) NMR assignments were obtained. A superposition of the backbone atoms (N, Cα, and C′) of the final 30-member ensemble is shown in Fig. 2b. A summary of structural statistics is provided in Table 1, demonstrating that the core domain structure is well defined with an average atomic root mean square deviation (RMSD) of 0.45 ± 0.05 Å and 0.97 ± 0.08 Å for the backbone and all heavy atoms (residues 246-314). Fig. 2c displays the lowest energy structure in ribbon representation, illustrating the architecture of the protein. As seen in RT crystal structures, the fold of the thumb domain consists of three α-helices, commonly named αH, αI, and, αJ (Kohlstaedt et al. 1992) that are linked together by loop regions that lack regular secondary structure. In the solution structure of the isolated thumb domain determined here, the core of the structure similarly comprises three α-helices, with αH (254-270), αI (278-285), and αJ (297-311). The greatest backbone variation in the ensemble occurs at the two termini and the 285-295 loop between αI and αJ.
Fig. 2.

Assignments and solution structure of the thumb subdomain. a 600 MHz 1H-15N HSQC spectrum of the thumb subdomain (1.0 mM protein in 25 mM sodium phosphate, 100 mM NaCl, pH 6.8). The two resonances (Q278 and N306) that are located outside the displayed spectral range are shown in insets. b Stereoview of the final 30 conformer ensemble (N, Cα, and C′, residues 240-315). Regions of helical structure are colored in green and the remainder of the structure in grey. c Ribbon representation of the lowest energy structure using the same color scheme as in b.
Table 1.
Statistics for the final 30 conformer ensemble of the thumb subdomain of HIV-1 RT
| Number of NOE distance restraints | |
| Intra-residue (i−j=0) | 1091 |
| Sequential (|i−j|=1) | 617 |
| Medium range (2≤|i−j|≤4) | 456 |
| Long range (|i−j|≥5) | 456 |
| Total | 2620 |
| Number of hydrogen bond restraints | 46 |
| Number of dihedral angle restraints | |
| ϕ | 59 |
| ψ | 57 |
| Total | 116 |
| Structural Quality | |
| Violationsa | |
| Distances restraints (Å) | 0.029 ± 0.001 |
| Dihedral angles restraints (°) | 0.431 ± 0.091 |
| Deviation from idealized covalent geometry | |
| Bond lengths (Å) | 0.003 ± 0.000 |
| Bond Angles (°) | 0.478± 0.010 |
| Improper torsions (°) | 0.302 ± 0.012 |
| Average RMSD of atomic coordinates (Å)b | |
| Backbone heavy atoms | 0.45 ± 0.05 |
| All heavy atoms | 0.97 ± 0.08 |
| Ramachandran plot analysis (%)c | |
| Favored regions | 77.5 ± 3.0 |
| Allowed regions | 95.8 ± 1.9 |
No individual member of the ensemble exhibited distance violations > 0.5 Å or dihedral angle violations >5°.
The average RMSD of atomic coordinates for residues 246-314 was calculated for individual structures with respect to the mean structure. The terminal regions (residues 237-245 and 315-326) were excluded from the statistics.
Statistics were calculated using MolProbity (Davis et al 2007) for residues 246-314; of the 4.2% of residues in disallowed’ regions, all are near the allowed/disallowed borders
Comparisons with crystal structures of the thumb domain in the context of HIV-1 RT
The structure of HIV-1 RT thumb domain in the context of the full-length protein was first determined using X-ray crystallography (Kohlstaedt et al. 1992) (Fig. 1a) and since then has been seen in over 100 crystal structures deposited in the PDB. In the p66 subunit, the thumb subdomain interacts mostly with the connection domain in the same subunit and is poised to make extensive interactions with DNA. In the p51 chain, the thumb subdomain similarly interacts with its own connection domain but also packs against the RNase H domain of the p66 subunit (Fig. 1). Although the thumb subdomains in p66 and p51 are found in different positions in the mature enzyme, their structures are quite similar, and both contribute to binding and positioning of nucleic acid substrates (Jacobo-Molina et al. 1993; Huang 1998; Lapkouski et al. 2013). In the crystal structure of HIV-1 RT bound to DNA, αH of p66 interacts with the sugar-phosphate backbone of the primer strand, while the antiparallel αI interacts with the template strand (Jacobo-Molina et al. 1993). In p51, in which the thumb subdomain is located next to the RNase H domain of p66, the thumb domain forms the “floor” of the nucleic acid binding cleft, contributing to RNA/DNA binding (Jacobo-Molina et al. 1993; Huang 1998; Lapkouski et al. 2013; Balzarini et al. 2015). Amino acid changes in the αH and αI helices were shown to affect DNA binding, DNA synthesis, and frameshift fidelity (Beard et al. 1994; Hermann et al. 1994; Bebenek et al. 1995; Powell et al. 1999; Betancor et al. 2010). αH and αI are part of the helix-turn-helix segment, termed the “helix clamp” motif, with similar motifs found in many eukaryotic, prokaryotic and viral nucleic acid polymerases (Hermann et al. 1994).
In terms of overall chain-fold, the isolated domain NMR solution structure presented here (Fig. 2b and 2c) is similar to the crystallographic subdomain structures in over 100 available crystal structures of the p51 and p66 chains of heterodimeric HIV-1 RT (Fig. 3a,b). Within the complete set of RT crystal structures, many structures are of moderate resolution (3 Å or lower), and, in such structures, the conformational details are less reliable; we, therefore, selected a high-resolution subset of 28 RT crystal structures, determined at 2.4 Å resolution or better. This set was subjected to analysis by the Ensemblator (Clark et al. 2015) to identify systematic and significant differences between the NMR thumb domain structure and the p51 and p66 X-ray thumb domain structures. Automatic clustering by the Ensemblator separated the full set of models into distinct groups, also containing exclusively the NMR structure or the p51 or p66 thumb domain crystal structures. Therefore, we carried out our final analyses by manually defining these as distinct groups. The key Ensemblator output for a given comparison of two groups consists of a pair of plots that reflect the global and local conformational differences, respectively. In each plot, the intra-group variations are compared with the inter-group variation and the closest approach between the groups (Fig. 4, upper and middle panels). The regions of greatest systematic difference are characterized by high silhouette index values (Fig. 4, lower panels), which contain information from both the global and local comparisons.
Fig. 3.

Comparison between the NMR structure of the isolated thumb domain and the thumb domain in the p51 and p66 chains in X-ray structures of the heterodimeric HIV-1 RT. a Backbone superposition of p51 thumb domain X-ray structures and the mean NMR structure (cyan), with atoms in the X-ray structures colored from blue to red with increasing RMSD values. b Equivalent superposition as in a, but for the p66 thumb domain X-ray structures. c Detailed view of residues 265-271, including the side chain of Ser268, for conformers in the NMR ensemble (orange carbons), p51 chains (blue carbons) and p66 chains (green carbons). d Hydrogen bonds between the H-atom of the donor (sphere) and the backbone oxygen acceptor in the NMR (orange), p51 (blue), and p66 (green) X-ray structures of the region shown in panel c. e Ribbon diagram of the p51 thumb domain (light blue) from a representative RT crystal structure (PDB Code 4IFY), with residues 285-287 depicted in blue. The remainder of p51 and p66 is shown in blue and red, respectively. The NMR ensemble (grey) differs from the X-ray structure around residue 287, where the p51 thumb subdomain contacts p66. f Ribbon diagram of the p66 thumb domain (pink) in a representative RT crystal structure (PDB Code 4IFY), highlighting the solvent exposed position of residues 285-287 (red coil). The remainder of the p66 chain is shown in red surface representation. In the NMR ensemble (grey), a similar local conformation is seen for residues 285-287.
Fig. 4.

Comparison between the NMR ensemble and the collection of 28 p51 and p66 thumb domain crystal structures. a NMR vs. p51. b NMR vs. p66. c p51vs. p66. For each pair of groups, global (upper panels), and local (middle panels) comparisons are shown, along with a Silhouette Index (lower panels; black trace) plot that identifies the regions with significant differences (above the grey strip) and similarities (below the grey strip). In the global and local comparison plots, the colors are as follows: NMR average backbone intra-group pairwise RMSD (orange), p51 average backbone intra-group pairwise RMSD (blue), and p66 average backbone intra-group pairwise RMSD (green) are shown along with average backbone inter-group pairwise RMSDs (pale purple) and closest approach distances (red). A secondary structure diagram (based on PDB entry 1RTJ) indicates α-helical (red), PII-helical (blue), and other (green) segments.
In addition to some differences that occur near the N- and C-terminal residues extending from the core of the domain (<254 and >310), the silhouette indices reveal two areas of consistent differences (> ~0.6) between the NMR and crystal structures (Fig. 4a,b lower panels). One is near residue 270 and applies to the NMR group as compared to both the p51 and p66 thumb domain groups. The second is around residues 285-295 and applies to the NMR ensemble as compared to the p51 group, but not the p66 group (Fig. 4). In both of these regions, no individual member of the NMR ensemble is closer than 1 Å to any member in the crystal structure ensembles, either globally or locally (red traces in Fig. 4a,b upper and middle panels). In both cases, these emerged as real differences between the conformation of the isolated thumb subdomain and in the mature p66:p51 heterodimer.
The difference near residue 270 involves the last turn and C-terminal capping of the αH helix. In this segment, in all of the NMR conformers, the transition from an α- to a 310-helix occurs at residue 267, with hydrogen bonds between 269-N ⋯ 265-O, 270-N ⋯ 267-O and 271-N ⋯ 268-O. In contrast, in all of the crystal structures this transition occurs one residue earlier and the structures contain H-bonds between with 269-N ⋯ 266-O and 271-N ⋯ 267-O, along with a side chain-backbone H-bond from Ser268 Oγ to 265-O (Fig 3c,d). We carefully examined this difference and ascertained that indeed it is real; the observed NMR NOE pattern is incompatible with the 271-N ⋯ 267-O H-bond seen in the crystal structure, although the cause for this discrepancy is not clear. Inspection of this region in the p51 and p66 chains in the crystal structures shows diverse (i.e. subunit and crystal form-dependent) packing interactions with either distant parts of the chain or across crystal contacts. For example, the structural contexts of the p66 and p51 thumbs are distinct: the p66 helical cap packs against and makes H-bonds with the connection domain near residue 345, but this part of the p51 helix is more exposed while the sidechain of Ile270 packs deeply into a hydrophobic pocket that also includes Phe346 and Trp426. Because these interactions are not uniform and present in all structures, it is hard to ascertain whether and how they may influence the helix capping pattern. One set of interactions that is common between p66 and p51 involves side chain packing of Tyr271 against residues 310 through 314. These interactions are also similar in the isolated domain NMR structure, thus do not appear responsible for the difference between the X-ray and NMR structures.
The difference between the p51 and p66 thumb domains near residue 285 can easily be rationalized by the unique context of the p51 chain in the heterodimeric RT. In p51, the 285-295 loop between αI and αJ packs against the surface of p66 (Fig 3e), with a consistent hydrogen bond between the Lys287 backbone oxygen and Tyr441-OζH of the p66 chain. By contrast, in the p66 thumb domain, this loop is fully exposed to solvent in some crystal forms (e.g. Fig 3f) or involved in crystal contacts in others. This results in a number of diverse conformations that vary over ~3 Å among the different crystal structures. This spread is larger than the ~2.5 Å backbone intra-group RMSD observed in the NMR ensemble (Fig. 4b upper panel). Although the local conformation in this area is somewhat similar between the NMR ensemble and the p66 crystal structures, there appears to be a global shift of ~1 Å (Fig. 3f). Interestingly, there is a low silhouette index near the 285 region, when p51 and p66 crystal structures are compared (Fig. 4c), indicating that no large consistent difference is present; this occurs because among the broad conformations sampled in the p66 chains, some structures adopt a conformation similar to that seen in p51.
CONCLUSION
We previously suggested that the structure of the isolated HIV-1 thumb subdomain resembles that of the thumb domains in the p66 immature precursor, based on a qualitative comparison of their 1H-15N HSQC spectra (Sharaf et al. 2014). Here, we directly confirm that the NMR structure of the isolated HIV-1 thumb subdomain is very similar to the thumb domain in X-ray structures of mature RT, albeit with two regions of interesting local conformational difference. One pertains to the C-terminal capping pattern of the αH helix, with no apparent cause. The other involves the loop conformation between the αI and αJ helices in the p51 chains. This most likely originates from unique packing interactions of the p51 thumb domain in mature RT that are not present in the isolated domain. Taken together, we show that the thumb subdomains in the mature RT and the p66 immature precursor are independent units that can fold autonomously and exhibit very similar structures, whether in isolation or present in its two different natural structural contexts (p51 or p66). Our data also underscore the well-known fact that surface regions are malleable and are influenced by context.
Acknowledgments
This work was supported by National Institutes of Health grants P50GM082251 (to AMG) and R01GM083136 (to PAK). N.G.S was the recipient of a Graduate Research Fellowship, 1247842, from the National Science Foundation.
References
- Balzarini J, Das K, Bernatchez JA, et al. Alpha-carboxy nucleoside phosphonates as universal nucleoside triphosphate mimics. Proc Natl Acad Sci USA. 2015;112:3475–3480. doi: 10.1073/pnas.1420233112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard WA, Stahl SJ, Kim HR, Bebenek K. Structure/function studies of human immunodeficiency virus type 1 reverse transcriptase. Alanine scanning mutagenesis of an alpha-helix in the thumb subdomain. J Biol Chem. 1994;269:28091–28097. [PubMed] [Google Scholar]
- Beard WA, Wilson SH. Kinetic analysis of template:primer interactions with recombinant forms of HIV-1 reverse transcriptase. Biochemistry-Us. 1993;32:9745–9753. doi: 10.1021/bi00088a029. [DOI] [PubMed] [Google Scholar]
- Bebenek K, Beard WA, Casas-Finet JR, et al. Reduced frameshift fidelity and processivity of HIV-1 reverse transcriptase mutants containing alanine substitutions in helix H of the thumb subdomain. J Biol Chem. 1995;270:19516–19523. doi: 10.1074/jbc.270.33.19516. [DOI] [PubMed] [Google Scholar]
- Betancor G, Puertas MC, Nevot M, et al. Mechanisms involved in the selection of HIV-1 reverse transcriptase thumb subdomain polymorphisms associated with nucleoside analogue therapy failure. Antimicrob Agents Chemother. 2010;54:4799–4811. doi: 10.1128/AAC.00716-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz VA, Holladay LA, Barkley MD. Efavirenz binding to HIV-1 reverse transcriptase monomers and dimers. Biochemistry-Us. 2010;49:601–610. doi: 10.1021/bi901579y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabodevilla JF, Odriozola L, Santiago E, Martinez-Irujo JJ. Factors affecting the dimerization of the p66 form of HIV-1 reverse transcriptase. Eur J Biochem. 2001;268:1163–1172. doi: 10.1046/j.1432-1327.2001.01939.x. [DOI] [PubMed] [Google Scholar]
- Clark SA, Tronrud DE, Karplus PA. Residue-level global and local ensemble-ensemble comparisons of protein domains. Protein Science. 2015;24:1528–1542. doi: 10.1002/pro.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Gronenborn AM. Determining the structures of large proteins and protein complexes by NMR. Trends Biotechnol. 1998;16:22–34. doi: 10.1016/S0167-7799(97)01135-9. [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Das K, Lewi PJ, Hughes SH, Arnold EE. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog Biophys Mol Biol. 2005;88:209–231. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Davies JF, Hostomska Z, Hostomsky Z, et al. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science. 1991;252:88–95. doi: 10.1126/science.1707186. [DOI] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Béthune M-P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989–2009) Antiviral Res. 2010;85:75–90. doi: 10.1016/j.antiviral.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Divita G, Rittinger K, Geourjon C, et al. Dimerization kinetics of HIV-1 and HIV-2 reverse transcriptase: A two step process. J Mol Biol. 1995;245:508–521. doi: 10.1006/jmbi.1994.0042. [DOI] [PubMed] [Google Scholar]
- Fletcher RS, Holleschak G, Nagy E, et al. Single-step purification of recombinant wild-type and mutant HIV-1 reverse transcriptase. PREP. 1996;7:27–32. doi: 10.1006/prep.1996.0004. [DOI] [PubMed] [Google Scholar]
- Hermann T, Meier T, Götte M, Heumann H. The “helix clamp” in HIV-1 reverse transcriptase: a new nucleic acid binding motif common in nucleic acid polymerases. Nucleic Acids Res. 1994;22:4625–4633. doi: 10.1093/nar/22.22.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiou Y, Ding J, Das K, et al. Structure of unliganded HIV-1 reverse transcriptase at 2.7 Å resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure. 1996;4:853–860. doi: 10.1016/s0969-2126(96)00091-3. [DOI] [PubMed] [Google Scholar]
- Huang H. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Gr. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jacobo-Molina A, Arnold EE. Hiv Reverse-Transcriptase Structure-Function-Relationships. Biochemistry-Us. 1991;30:6351–6361. doi: 10.1021/bi00240a001. [DOI] [PubMed] [Google Scholar]
- Jacobo-Molina A, Ding J, Nanni RG, et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc Natl Acad Sci USA. 1993;90:6320–6324. doi: 10.1073/pnas.90.13.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlstaedt LA, Wang J, Friedman JM, Rice PA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Gr. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Lansdon EB, Brendza KM, Hung M, et al. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): implications for drug design. J Med Chem. 2010;53:4295–4299. doi: 10.1021/jm1002233. [DOI] [PubMed] [Google Scholar]
- Lapkouski M, Tian L, Miller JT, et al. Complexes of HIV-1 RT, NNRTI and RNA/DNA hybrid reveal a structure compatible with RNA degradation. Nat Struct Mol Biol. 2013;20:230–236. doi: 10.1038/nsmb.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Rullmann JAC, MacArthur MW, et al. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Marko RA, Liu H-W, Ablenas CJ, et al. Binding kinetics and affinities of heterodimeric versus homodimeric HIV-1 reverse transcriptase on DNA-DNA substrates at the single-molecule level. J Phys Chem B. 2013;117:4560–4567. doi: 10.1021/jp308674g. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Powell MD, Beard WA, Bebenek K, et al. Residues in the α H and α I helices of the HIV-1 reverse transcriptase thumb subdomain required for the specificity of RNase H-catalyzed removal of the polypurine tract primer. J Mol Biol. 1999;274:19885–19893. doi: 10.1074/jbc.274.28.19885. [DOI] [PubMed] [Google Scholar]
- Sarafianos SG, Clark AD, Das K, Tuske S. Structures of HIV-1 reverse transcriptase with pre-and post-translocation AZTMP-terminated DNA. EMBO J. 2002;21:6614–6624. doi: 10.1093/emboj/cdf637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafianos SG, Marchand B, Das K, et al. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol. 2009;385:693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Maurer M, Schleucher J. A simultaneous 15N,1H- and 13C,1H-HSQC with sensitivity enhancement and a heteronuclear gradient echo. J Biomol NMR. 1995;5:97–102. doi: 10.1007/BF00227475. [DOI] [PubMed] [Google Scholar]
- Schrödinger, LLC. The PyMOL molecular graphics system, version 1.8. 2015. [Google Scholar]
- Schwieters CD, Kuszewski JJ, Clore GM. Using Xplor–NIH for NMR molecular structure determination. Prog Nucl Magn Reson Spectrosc. 2006;48:47–62. [Google Scholar]
- Sharaf NG, Poliner E, Slack RL, et al. The p66 immature precursor of HIV-1 reverse transcriptase. Proteins. 2014;82:2343–2352. doi: 10.1002/prot.24594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluis-Cremer N, Temiz NA, Bahar I. Conformational changes in HIV-1 reverse transcriptase induced by nonnucleoside reverse transcriptase inhibitor binding. Curr HIV Res. 2004;2:323–332. doi: 10.2174/1570162043351093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasselli AG, Sarcich JL, Barrett LJ, et al. Human immunodeficiency virus type-1 reverse transcriptase and ribonuclease H as substrates of the viral protease. Protein Sci. 1993;2:2167–2176. doi: 10.1002/pro.5560021216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venezia CF, Howard KJ, Ignatov ME, Holladay LA. Effects of efavirenz binding on the subunit equilibria of HIV-1 reverse transcriptase. Biochemistry-Us. 2006;45:2779–2789. doi: 10.1021/bi051915z. [DOI] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Wang J, Smerdon SJ, Jäger J, et al. Structural basis of asymmetry in the human immunodeficiency virus type 1 reverse transcriptase heterodimer. Proc Natl Acad Sci USA. 1994;91:7242–7246. doi: 10.1073/pnas.91.15.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapling J, Moore KL, Sonza S, Mak J. Mutations That Abrogate Human Immunodeficiency Virus Type 1 Reverse Transcriptase Dimerization Affect Maturation of the Reverse Transcriptase Heterodimer. J Virol. 2005;79:10247–10257. doi: 10.1128/JVI.79.16.10247-10257.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Pedersen LC, Gabel SA, et al. Selective unfolding of one Ribonuclease H domain of HIV reverse transcriptase is linked to homodimer formation. Nucleic Acids Res. 2014;42:5361–5377. doi: 10.1093/nar/gku143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Perera L, Mueller GA, et al. Asymmetric conformational maturation of HIV-1 reverse transcriptase. Elife. 2015;4:11952. doi: 10.7554/eLife.06359. [DOI] [PMC free article] [PubMed] [Google Scholar]