

Abstract
We use single-molecule fluorescence lifetimes to probe dynamics of photoinduced reversible electron transfer occurring between triphenylamine (donor) and perylenediimide (acceptor) in single molecules of a polyphenylenic rigid dendrimer embedded in polystyrene. Here, reversible electron transfer in individual donor-acceptor molecules results in delayed fluorescence that is emitted with a high photon count rate. By monitoring fluorescence decay times on a photon-by-photon basis, we find fluctuations in both forward and reverse electron transfer spanning a broad time range, from milliseconds to seconds. Fluctuations are induced by conformational changes in the dendrimer structure as well by polystyrene chain reorientation. The conformational changes are related to changes in the dihedral angle of adjacent phenyl rings located in the dendritic branch near the donor transferring the charge, a torsional motion that results in millisecond fluctuations in the “through-bond” donor-acceptor electronic coupling. Polymer chain reorientation leads to changes in the local polarity experienced by the donors and to changes in the solvation of the charge-separated state. As a result, switching between different donor moieties within the same single molecule becomes possible and induces fluctuations in decay time on a time scale of seconds.
Single-molecule detection (SMD) has been extensively used to answer questions of both biophysical chemistry and chemical physics relevance (1-5). An elegant way to probe conformational changes is to take advantage of distance-dependent processes involving pairs of fluorophores. Förster resonant energy transfer (FRET) between two fluorophores has gained popularity as a sensitive tool to probe distance changes in the nanometer range (5-8). With an efficiency scaling with the inverse of the 6th power of the interchromophore distance, FRET can be used to track distance changes in the 2- to 8-nm range in various biological structures (5). The distance scale can be downsized to the ångstrom scale if one uses photoinduced electron transfer (ET). ET usually involves a pair of electron donor (D) and acceptor (A) chromophores spatially separated by an edge-to-edge distance of ≈1 nm or less (9). Because the efficiency scales exponentially with the D-A distance, ET can probe distance changes with subångstrom resolution. However, whereas highly efficient FRET results in fluorescence emitted mainly from the acceptor fluorophore, highly efficient ET usually leads to a strong quenching of the fluorescence of the emitting chromophore. Thus, reports on single-molecule photoinduced ET are rather limited (10-14), in contrast to reports on single-molecule FRET. A particular situation of ET refers to the case in which the locally excited state (LES) and the charge-separated state (CSS) are relatively close in energy (Fig. 1A, with CSS in position 2). In this case, upon optical excitation, the LES deactivates mainly by forward ET to the CSS and, if the radiationless deactivation of the CSS to the ground state (GS) is inefficient (see below), the CSS is forced to decay through the LES by reverse ET (14, 15). The fluorescence is delayed but can retain a high quantum yield. If present, reversible ET can be probed by SMD if the excited chromophore shows bright fluorescence and high photostability. In some cases, competition between the deactivation of the CSS to the LES or to the GS may be influenced by molecular oxygen (15).
Fig. 1.

Electron D-A compounds and energetic considerations. (A) Energetic scheme for photoinduced ET in PN8 or PN16: level 1, apolar solvent/polymer leads to no ET; level 2, solvent/polymer of intermediate polarity leads to reversible ET; level 3, polar solvent/polymer leads to forward-only ET. GS, ground state. (B) Chemical structures of PN8 (R = R1) and PN16 (R = R2), in which PN indicates perylenediimide acceptor (P) and triphenylamine donor (N).
Here we use single-molecule fluorescence decay times to probe the dynamics of reversible ET between an excited perylenediimide (acceptor, P) and triphenylamine (donor, N) as parts in a dendritic structure. Triphenylamines are attached to the core perylenediimide by means of a polyphenylene skeleton that imposes some rigidity on the whole structure. The D-A distance is thus modified, in a controlled way, from the first (PN8, Fig. 1B with R = R1) to the second (PN16, Fig. 1B with R = R2) generation; hence, we expected that the parameters of the ET change as well.
Materials and Methods
Ensemble stationary and time-resolved fluorescence spectroscopy was performed on 10-6 M solutions of PN8 and PN16 (16) in solvents of different polarity: methylcyclohexane, toluene, and ethyl acetate, all of spectral-grade purity. Before measurements, solutions were degassed by freeze-pump-thaw cycling. Fluorescence decays were recorded at 543-nm excitation by the time-correlated single photon counting technique (TCSPC). Details are given in Supporting Text, which is published as supporting information on the PNAS web site.
For single-molecule detection, individual PN8 or PN16 molecules were immobilized in different polymer matrices by spin-casting solutions of compounds (10-10 M) and polymer [5 mg/ml Zeonex E48R (ZE; Zeon, Dusseldorf, Germany) or polystyrene (PS; polystyrene standard, Mw 106, Aldrich)] in chloroform on a cover glass. Single-molecule lifetime measurements at 543-nm pulsed excitation were performed by the TCSPC technique using a confocal microscope. Single-molecule fluorescence trajectories were collected in FIFO (first-in, first-out) mode, allowing the registering, for each detected photon, of both the microtime (time lag with respect to the excitation pulse) and macrotime (time lag between consecutive photons) information. Details are given in Supporting Text.
Results and Discussion
Ensemble Spectroscopy. Shown in Fig. 1B are the chemical structures of PN8 and PN16. Molecular modeling calculations (Fig. 6, which is published as supporting information on the PNAS web site) indicate an increase in the D-A distance from 9.6 to 10.8 Å in PN8 up to 14.9 to 19.3 Å in PN16.
In methylcyclohexane, both PN16 and PN8 fluoresce with a quantum yield approaching unity. Fluorescence decays are best fitted with a single-exponential lifetime of 6.5 ns (Fig. 7 B and C, which is published as supporting information on the PNAS web site). Thus, fluorescence is the main deactivation channel of the LES (Fig. 1 A, with CSS at level 1).
In ethyl acetate, a much more polar solvent, fluorescence from both PN8 and PN16 is strongly quenched. For PN16, the fluorescence quantum yield and decay time drop to 0.04 and 0.6 ns, respectively (Fig. 7B). For PN8, we cannot detect any fluorescence. This fact suggests that quenching is due to photoinduced ET from one of the triphenylamines to the excited perylenediimide (Fig. 1 A, with CSS at level 3) followed by ET to the GS (14, 15).
In toluene, a solvent of intermediate polarity, the fluorescence quantum yield is 0.65 for PN16 and 0.33 for PN8. Both compounds show multiexponential decays with decay times spanning a broad range from tens of picoseconds to tens of nanoseconds. The decays of PN16 and PN8 (Fig. 7 B and C, respectively) can be best fitted with four exponentials: 0.14 (15% contribution), 1.6 (14%), 6.5 (65%), and 12 ns (6%) for PN16 (14) and 0.07 (62% contribution), 1.2 (10%), 6.5 (9%), and 22.2 ns (19%) for PN8. In addition to a decay time with a value similar to that seen in methylcyclohexane (6.5 ns), we recover faster and slower decay times. The fast components reflect quenching of the LES by forward ET to the CSS. Components longer than the lifetime of the unquenched perylenediimide (6.5 ns) are attributed to delayed fluorescence (Fig. 1 A, with CSS at level 2). The long decay times decrease substantially, both in value and contribution in aerated toluene (data not shown), suggesting that oxygen influences the competition between the deactivation of the CSS to the GS and LES (15). From PN8 to PN16, the efficiency of ET decreases considerably as seen from the overall contribution of the fast and slow decay times we recover in toluene.
Ensemble data show that by changing the polarity of the solvent we can tune the respective position of the energy levels of the LES and CSS to get into one of the following situations: (i) no ET is present (Fig. 1 A, case 1); (ii) ET takes place only in forward direction, i.e., from LES to CSS (Fig. 1 A, case 3); and (iii) reversible ET is present, i.e., forward ET from LES to CSS and reverse ET from CSS to LES (Fig. 1 A, case 2). However, ensemble data are averaged over a large number of molecules and hide differentiated behavior at the molecular level. Dynamic information is smeared out and eventual heterogeneity is difficult or even impossible to detect. Information hidden in ensemble data can be extracted by single-molecule spectroscopy. Here, we use fluorescence decay times to probe dynamics of single-molecule ET. Decay times are less sensitive to changes in experimental conditions, mainly excitation intensity and/or polarization fluctuations, and can access dynamic changes over a broad time range, i.e., from nanoseconds to seconds (see below) (10-14, 17).
Single-Molecule Detection. Because long-time observation requires immobilization, single molecules are embedded in thin polymer films. By choosing appropriate polymers, we can mimic the polarity conditions from ensemble experiments and hence we can tune the energy levels of the LES and the CSS. ZE and PS are the best equivalents in terms of polarity for methylcyclohexane and toluene, respectively (18, 19).
In ZE, an apolar matrix, we do not expect ET to occur at all. Indeed, single molecules of PN16 and PN8 display a behavior reminiscent of that of single dye molecules (20-22). Fluorescence is emitted with one stable level of intensity at an average photon count rate of 100-175 counts per 10 ms (Fig. 2 A and B) and decays single-exponentially with lifetimes (calculated by averaging over all detected photons) distributed around 6 ns (Figs. 2 A and B and 3B). Hence, immobilization of single PN16 and PN8 molecules in ZE leads to a CSS energetically higher than LES. Consequently, single molecules of PN16 and PN8 in ZE behave “photophysically” as single perylenediimide dye molecules (20), an observation supported by the strong resemblance between the photon count rate vs. lifetime correlations from Fig. 2 A (PN16) and B (PN8).
Fig. 2.

Averaged photon count rate vs. decay time correlation from single molecules of PN16 in ZE (A), PN8 in ZE (B), PN16 in PS (C), and PN8 in PS (D). Decay times are calculated by binning all photons from a single molecule (details in Supporting Text).
Fig. 3.

Time-resolved single-molecule data for PN16. (A) Multiexponential fluorescence decay (filled circles) of a single PN16 molecule in PS. Also shown are the single (black dashed line) and multiexponential (gray line) fits. a.u., Arbitrary units. (B) Single-exponential fluorescence decay (filled circles) of a single PN16 molecule in ZE. Fit is shown in gray. (C) Forward (filled circles) and reverse (open circles) ET-related decay time trajectories of a single PN16 molecule in PS (10,000 photons per bin). (Inset) Graph of data from C featuring mean value and width of 0.6 and 0.4 ns for forward ET and of 8.6 and 0.9 ns for reverse ET. (D) Reverse ET decay time trajectory (black, 500 photons per bin) of the single PN16 molecule in PS accounting for A and lifetime trajectory (gray, 500 photons per bin) of the single PN16 molecule in ZE accounting for B.
In PS single PN16 and PN8 molecules emit in nitrogen atmosphere at an average photon count rate of 20-200 counts per 10 ms. Single molecules emitting at low photon count rate show multiexponential decay of fluorescence with long decay time components as large as 15 ns for PN16 and 44 ns for PN8 (Fig. 2 C and D). The photon count rate vs. the average decay time correlations for PS spread to longer decay times/lower photon count rates, for both PN16 and PN8. The detection of low photon count rates associated with long decay times (longer than the 6-ns lifetime of unquenched perylenediimide in ZE) from single PN16 and PN8 molecules in PS is a signature of the reversible ET. Assuming safely that single-molecule decay time components longer than 10 ns relate to delayed fluorescence and hence to reverse ET, within the probed population (100 individual molecules of each compound), we detected reversible ET in 10% of PN16 and 40% of PN8 (Fig. 2 C and D). We noticed a larger spread of the single-molecule decay times for PN8, i.e., up to 44 ns. At the ensemble level, we detect reverse ET in PN8 as a single long decay time component (22 ns). This component reflects actually an average value of a broad distribution of decay times detected at the single-molecule level (Fig. 2C). This broad distribution relates to a wide range of rate constants for the reverse ET process.
Figs. 3A and 5A show fluorescence decays of single molecules of PN16 and PN8, respectively, undergoing reversible ET in PS. The decays, histogrammed by using all of the detected photons from a single molecule, are multiexponential, similar to the ensemble-averaged decays in toluene, and carry decay time components as large as several tens of nanoseconds. Multiexponential single-molecule fluorescence decays have been reported for both biological and synthetic molecules and were related to fluctuating decay times during the measurement (10, 13, 14), contrary to the single-exponential decays of single dye molecules (refs. 20-22 and as seen here for single PN16 and PN8 molecules in ZE).
Fig. 5.

Time-resolved single-molecule data and results for PN8. (A) Multiexponential fluorescence decays (open, filled, and gray circles) and fits (black and gray lines) of two single PN8 molecules in PS. (B) Reverse ET-related decay time trajectories (500 photons per bin) accounting for the single molecules from A, according to color and symbol. (C and D) Autocorrelations of fluorescence decay time fluctuation (gray) calculated on a photon-by-photon basis for the single PN8 molecules in PS from A, according to symbol. Also shown are a biexponential fit (black line) to the autocorrelation from C and a single (black dashed line) and a stretched (black line) exponential fit to the autocorrelation from D. (E and F) Probability density of through-bond D-A electronic coupling coordinate θ calculated from B, according to color and symbol. (G and H) Potentials of mean force expressed in kBT (open circles) calculated from E and F, respectively. Also shown are fits according to a harmonic model (line).
Dynamics of Decay Time Fluctuations in Single PN16 Molecules. A conventional way of monitoring lifetime fluctuations from single molecules is to bin a certain number of photons in a decay, which is then analyzed by a maximum-likelihood estimator (MLE) (13, 17, 23). A trajectory of the lifetime of a single molecule can be obtained with a certain time resolution depending on the detected photon count rate and the bin size. Fig. 3D, gray, shows the lifetime trajectory of a single PN16 molecule in ZE when bins of 500 photons were used for which lifetimes were estimated by MLE fitting with a single-exponential model. The trace shows the behavior of a single dye molecule, i.e., no lifetime fluctuations during the measurement. In contrast, when a single PN16 molecule in PS undergoing reversible ET is probed, fluctuations are seen in both decay times (Fig. 3C). Here, single-molecule decay times were estimated from bins of 10,000 photons by MLE fitting with a biexponential model accounting for forward and reverse ET (see Supporting Text and Fig. 8A, which is published as supporting information on the PNAS web site). Fluctuations are even more clearly seen in the forward and backward ET-related decay time histograms (Inset in Fig. 3C), which are much broader than the expected standard deviation from MLE (23), as well as the lifetime distribution of single PN16 molecules in ZE (Fig. 4B).
Fig. 4.

Single-molecule results for PN16. (A) Correlation between forward and reverse ET rate constants calculated with data from Fig. 3C. (B) Distributions of fluorescence decay times from single PN16 molecules in PS (gray, data from Fig. 3D) and ZE (black, data from Fig. 3E) and Gauss fits featuring peaks and widths of 5.6 and 0.41 ns for ZE and 8.4 and 1.26 ns for PS. (C) Autocorrelation of fluorescence decay time fluctuation calculated on a photon-by-photon basis for a single PN16 molecule in PS (gray). Also shown are single (dashed black line) and stretched (solid black line) exponential fits. (D) Autocorrelation of fluorescence decay time fluctuation calculated on a photon-by-photon basis for a single PN16 molecule in ZE. (E) Distribution of through-bond D-A electronic coupling coordinate θ calculated from data shown in B (only data in PS). (F) Potential of mean force expressed in kBT calculated from data in E (open circles). Also shown is a fit according to a harmonic model (line).
We next focus on the origin of the decay time fluctuations we detect. The rate constant for nonadiabatic ET can be described as kET = (4π2/h)H2FCWD. Here h is the Planck constant, H is the electronic coupling between the LES and the CSS, and FCWD = (4πλkBT)-1/2exp[-(ΔG° + λ)2/(4λkBT)] is the Franck-Condon weighted density of states with λ the reorganization energy and ΔG° the free enthalpy. Hence, fluctuations in lifetime/rate constant can arise from changes in FCWD or in H, that is, from fluctuations in the free enthalpy or D-A distance/orientation coupling (see below). Assuming a three-state model, we compute from the related decay times and their contributions (Supporting Text) the forward and reverse ET rate constants, which show strong correlation, as seen in Fig. 4A. From these values we estimate a variation in the free energy of charge separation from -300 to 200 cal/mol. Thus, for this particular example, the LES and the CSS are energetically almost equal. If originating from changes in ΔG°, the fluctuations in decay time and hence rate constant for forward and reverse ET should anticorrelate. Instead, they are highly correlated (Fig. 4A), suggesting that the fluctuations originate from changes in the electronic coupling H.
According to molecular modeling calculations (Fig. 6C), the D-A distances are as short as 14 Å for PN16 and 10 Å for PN8. For such distances, ET is governed here by a through-bond mechanism (Fig. 9A, which is published as supporting information on the PNAS web site) and, hence, changes in mutual orientation and in through-space distance between donor and acceptor chromophores have little influence on the electronic coupling. More important are changes in the delocalization of the electronic density in the highest occupied molecular orbital (HOMO) of the donor (see below). The HOMO of the donor can delocalize over several phenyl rings of the polyphenylene branch connecting the donor and acceptor (see Supporting Text). For each dendritic branch, for both PN16 and PN8, delocalization is larger for the para- than the meta-substituted donor (Fig. 9 C and D). Next to the donor, adjacent phenyl rings from the dendritic branch have a dihedral angle of ≈75° (Fig. 9B Inset). Small changes in the dihedral angle, as small as few degrees, can lead to rather large changes in the delocalization of the donor HOMO over the polyphenylene branch (Fig. 9B). If the extent of the delocalization varies, the through-bond D-A electronic coupling will also vary. Consequently, fluctuations in decay times originate from small fluctuations of the dihedral angle of the adjacent phenyl rings next to the donor, a torsional motion known as libration (24). In this assumption, the electronic coupling H follows an angular dependence of the form H = H0cos(θ), with θ the torsion angle between adjacent phenyl rings next to the donor, and hence so does the rate constant for ET, kET(θ) = kET0cos2(θ) (24). Here, changes in the electronic coupling lead to correlated fluctuations of the rate constants for forward and reverse ET. This behavior allows us to extract information about the fluctuations in D-A coupling coordinate θ by following only the rate constant of reverse ET (see below). Within the photons detected from a single molecule undergoing reversible ET, the main contribution comes from the delayed photons. They can be easily separated from forward ET-related photons, which usually are detected within the first 3 ns after the excitation pulse. Using only delayed photons, we next construct decay histograms with bins of 500 photons that lack the contribution of forward ET and hence can be analyzed as a single exponential with MLE. Fig. 3D (black) shows, for the single PN16 molecule accounting for the data in Fig. 3C, the decay time trajectory constructed with delayed photons. Compared with the lifetime trajectory of a single PN16 molecule in ZE (Fig. 3D, gray), it shows a larger spread, which is indicative of the presence of fluctuations. From the probability density of the reverse ET-related decay times (Fig. 4B, gray histogram) and assuming an average torsional angle θav = 75°, we next compute the probability density of θ, P(θ) (Fig. 4E). P(θ) can be used to build the potential of the mean force according to H(θ) = -kBTloge[P(θ)] (Fig. 4F). Fitting H(θ) with a harmonic model, i.e., H(θ) = kBT(θ - θav)2/2αθ, results in a variance αθ = 0.63°2, a value larger than the error introduced by the MLE analysis (here 0.18°2) and that reflects changes in θ as large as 4° (Fig. 4E).
Although indicative of through-bond D-A coupling fluctuations, H(θ) provides only a static picture with no information on the time scale on which fluctuations occur. Dynamic information can be obtained by monitoring the reverse ET-related decay time. Using bins of 500 photons, we can access lifetime fluctuations as fast as hundreds of milliseconds. Recently, Yang et al. (13) showed that in single flavin reductase/FAD complexes lifetime fluctuations related to photoinduced ET can span a broad time range, from milliseconds to seconds. To unravel such dynamic changes, they applied a photon-by-photon analysis (25, 26) that can retrieve dynamic changes with high temporal resolution (see below).
We next use the photon-by-photon approach on our single-molecule fluorescence trajectories. Data are collected such that for each photon we register both the delay time (tn) with respect to the excitation pulse and the time lag (Tn) with respect to the previously detected photon, i.e., the chronological time. For a single molecule, if the lifetime does not fluctuate, the probability distribution of tn is single exponential with a mean value equal to the fluorescence lifetime (τ0 ≡ k-1) of the molecule. If the lifetime does fluctuate, the probability distribution of tn becomes multiexponential. The dynamics of lifetime fluctuations can be retrieved from the second-order correlation of lifetimes C2(t) = 〈Δk-1(t)Δk-1(0)〉 with Δk-1(t) = k-1(t) -〈k-1〉 and calculated on a photon-by-photon basis (25). For a single molecule not showing lifetime fluctuations, C2(t) = 0 (Fig. 4D). If the single molecule shows two conformations, i.e., two states with different lifetimes 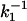 and
and 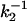 the correlation of lifetimes becomes
the correlation of lifetimes becomes 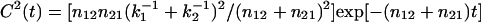 with n12, n21 interconversion rates (25). For a single molecule probing multiple conformations, C2(t) becomes multiexponential, reflecting the time range of k-1 fluctuations. C2(0) gives the variance of k-1. In the assumption that for a single molecule the fluorescence intensity and the lifetime are correlated, C2(t) can be constructed on a photon-by-photon basis (see Supporting Text). Shown in Fig. 4D is the C2(t) from a single PN16 molecule in ZE. As expected, when no ET is present, the lifetime shows no correlation, i.e., C2(t) = 0. Fig. 4C is the C2(t) of the single PN16 molecule in PS accounting for the data shown in Fig. 3. Here, C2(t) was constructed only with delayed photons related to reverse ET. The decay of C2(t) spans several decades, from tens of milliseconds, where it shows the highest amplitude, up to seconds. A single-exponential decay model cannot characterize the decay of C2(t) and hence we used a stretch exponential M(t) = M(0)exp(-(t/τ0)γ) with τ0 = 1.1 ms and γ = 0.17, which reflects a distribution of lifetimes covering a broad time range (Fig. 4C).
with n12, n21 interconversion rates (25). For a single molecule probing multiple conformations, C2(t) becomes multiexponential, reflecting the time range of k-1 fluctuations. C2(0) gives the variance of k-1. In the assumption that for a single molecule the fluorescence intensity and the lifetime are correlated, C2(t) can be constructed on a photon-by-photon basis (see Supporting Text). Shown in Fig. 4D is the C2(t) from a single PN16 molecule in ZE. As expected, when no ET is present, the lifetime shows no correlation, i.e., C2(t) = 0. Fig. 4C is the C2(t) of the single PN16 molecule in PS accounting for the data shown in Fig. 3. Here, C2(t) was constructed only with delayed photons related to reverse ET. The decay of C2(t) spans several decades, from tens of milliseconds, where it shows the highest amplitude, up to seconds. A single-exponential decay model cannot characterize the decay of C2(t) and hence we used a stretch exponential M(t) = M(0)exp(-(t/τ0)γ) with τ0 = 1.1 ms and γ = 0.17, which reflects a distribution of lifetimes covering a broad time range (Fig. 4C).
Libration of adjacent phenyl rings, a process we invoke as responsible for the changes in through-bond D-A coupling, is a vibrational motion known to occur in solution in the picosecond time range (27). Immobilization of PN16 in polymer matrix definitely will slow down such a motion, up to milliseconds (see below). However, the fluctuations we observe span a broad time range, from milliseconds to seconds (Fig. 4C).
Dynamics of Decay Time Fluctuations in Single PN8 Molecules. For PN8, forward ET in toluene is highly efficient and as fast as 0.07 ns. For SMD experiments, forward ET in NP8 is too fast to be resolved. Hence fluorescence decays detected from single PN8 molecules undergoing reversible ET are apparently free of fast components, i.e., faster than the lifetime of the unquenched acceptor (6.5 ns). Nevertheless, they are multiexponential (Fig. 5A), indicating fluctuating decay times during the measurement. With a forward ET that is too fast to be resolved by SMD, we can unravel dynamics of decay time fluctuations in single PN8 molecules only by focusing on the analysis of the delayed fluorescence photons. We can do so only by assuming that for PN8, as for PN16, forward and reverse ET rate constants correlate and hence originate from a similar cause. Shown in Fig. 5 A and B are fluorescence decays and decay time trajectories (500 photons per bin), respectively, of single PN8 molecules in PS. They feature two particular cases that we observed within the population of single PN8 molecules undergoing reversible ET. These examples help us to understand the origin of the decay time fluctuations in both compounds (see below).
The first example, fluorescence decay and decay time trajectory in Fig. 5 A and B, respectively, refers to a single PN8 molecule switching its reverse ET-related decay time between two distinct values, on average between 10 and 14 ns. The C2(t) of this molecule decays with a single exponential at times longer than hundreds of milliseconds (Fig. 5C). Additionally, we detect fluctuations in the time range of tens of milliseconds. This is a clear example of a single molecule switching between two distinct “states,” denoted here as 1 (decay time around 10 ns) and 2 (decay time around 14 ns), in a time scale of seconds. They resemble two distinct conformational states that are reversibly explored by the single molecule (see below). We next build the probability density of θ, P(θ) (Fig. 5E), using the decay times from Fig. 5B and assuming an average torsional angle of 75°. P(θ) features two modes peaking at θ1av = 75° (higher probability) and θ2av = 76.4° (lower probability). The bimodal P(θ) we use to build the potential of mean force H(θ) (Fig. 5G), which can be formally modeled by a combination of two harmonic potentials of the form Hj(θ) = kBT(θ - θj,av)2/2αjθ with j = 1, 2. The best fits of H1(θ) and H2(θ) give variances α1θ = 0.26°2 (high-probability mode) and α2θ = 0.32°2 (low-probability mode), respectively, that reflect overall changes in the torsion angle of 2° and 2.5°, respectively. H1(θ) and H2(θ) have energetic minima that, on the reaction coordinate scale, are displaced at θ2av - θ1av = 1.4°, and they apparently cross each other at θcross = 75.9°. From an energetically point of view, switching from state 2 to state 1 requires apparently an energy of only 0.1kBT, whereas from state 1 to state 2 the required energy is larger, 2kBT (Fig. 5G). Such small barriers are incompatible with the time scale of the dynamics we detect here (see below). The potential from Fig. 5G is just a 2D projection of the multidimensional energy landscape probed by the single molecule. Hence, the two states should not be regarded as two “connected conformations” related to an adiabatic process. More probably, they relate to two distinct donor moieties that alternate for the same acceptor in a nonadiabatic process (see below). There is no doubt that the single exponential decay of C2(t) in the time scale of seconds (Fig. 5C) reflects the reversible passage of the single molecule between states 1 and 2. We argue that interconversion between the two states relates to a reversible switch between two different donors within the same molecule, a switch that is induced by polymer chain motion. Recalling the expression for the rate constant for ET, both H and FCWD play a role in the value and behavior of kET. For the specific polymer we use (PS, molecular weight Mr = 106, glass transition temperature TG = 100°C), polymer chain reorientation can change the polarity of the nanoenvironment, especially that of the donors residing at the rim of the molecule. Polymer chain motion is a process that can take place in the time scale of seconds (28). In a microscopic picture, a decrease in the polarity of the nanoenvironment of the active donor, which can be induced, for example, by the retraction of the polymer chain away from the donor, leads to an increase in ΔG° and hence a less stabilized CSS. Thus, another donor of the molecule can become “active” in ET if, in combination with the acceptor, it leads to a more stabilized CSS. This step is seen as a switch from state 1 to state 2. State 2 involves a less favorable donor in terms of ET and is less energetically stable, such that switching from state 2 to state 1 requires a smaller energy barrier, a barrier that can be passed if, assuming reversible polymer motion, the polymer chain moves closer to the initially active donor. This donor then experiences a higher polarity that, in combination with the acceptor, leads to a better solvation of CSS. Thus, the first donor becomes active again and the molecule switches from state 2 to state 1. The assumption of polymer playing a role in mediating competition between different donors within the same molecule is strongly supported by the absence of a decay of C2(t) in the time scale of seconds for single molecules of PN8 embedded in a PS polymer with a lower molecular weight (Mr = 2,440, TG = 70°C, data not shown). For this polymer, because room temperature is closer to its glass transition temperature, the free volume is larger and the polymer motion happens faster. Hence, the decay of C2(t) within tens of milliseconds time scale that we detect in both low and high molecular weight polymers reflects through-bond D-A coupling changes induced by libration. Conformational changes taking place in millisecond time scale and leading to D-A coupling (distance) changes were previously detected in single flavin reductase/FAD complexes (11). The reversible switch between different donor moieties is a process dictated by the polymer chain motion and contributes to the decaying of C2(t) in the seconds time scale.
The second example we discuss here relates to single PN8 molecules in PS undergoing ET with large fluctuations of the decay time related to reverse ET to the LES. Shown in Fig. 5B (gray) is the decay time trajectory from such a molecule experiencing fluctuations from 10 up to 44 ns. The C2(t) (Fig. 5D) shows a stretched exponential decay spanning a time range from milliseconds to tens of seconds with the highest amplitude in the time scale of seconds (stretch parameters τ0 = 1.6 s, γ = 0.54), suggesting that decay time fluctuations are caused by both libration (milliseconds time scale) and polymer chain reorientation (seconds time scale). If we next construct the corresponding probability density P(θ), we find changes in the torsion angle as large as Δθ = 12° (Fig. 5F). In the assumption that H(θ) from Fig. 5H is described by a harmonic model, a Δθ = 12° corresponds to an αθ = 4.5°2, values far larger than seen for PN16 (see below).
Both αθ and Δθ are computed from decay times whose fluctuations are caused by two processes acting in different time scales. We previously have shown that polymer chain motion can mediate switching between competitive donor moieties within a single molecule. Each D-A pair has its own CSS in terms of energy. Switching between donor moieties within the same molecule is seen here as a change in ΔG° and hence in the FCWD. As a result, the overall potential of mean force becomes a combination of several potentials of the mean force, each of them related to a specific donor and the same acceptor. Within the same PN8 molecule, for a through-bond ET and for symmetry reasons, donors might have similar electronic coupling factors. Thus, it is the FCWD that determines which donor will be, at a given time, the active one in ET. The FCWD will be dictated by the polymer motion, with the latter affecting the local polarity and hence the stabilization of the CSS.
The single-molecule data of PN8 favor the hypothesis of polymer chain motion opening competition between different donor moieties within a single molecule. However, we cannot exclude that fluctuations in decay times might relate only to libration, hence leading to changes of the torsional angle large as Δθ = 12°. If so, polymer motion will still induce fluctuations in the decay time in seconds time scale through changes in the local polarity of the donor involved in ET, as seen for PN16.
Within the single molecules in PS we probed, 90% of PN16 and 60% of PN8 do not feature delayed fluorescence, but only lifetimes accounting for the unquenched acceptor (6.5 ns). Recalling the contribution of the short and long decay time components from the ensemble data in toluene, 65% of PN16 molecules and 10% of PN8 molecules do not feature delayed fluorescence. The presence of molecules lacking donor chromophores is excluded because in ethyl acetate all of the molecules undergo ET (Fig. 7). In the stabilization of the CSS, both solvation and coulombic interaction play a role. Solvation relates here to the polarity of the solvent/polymer in which the molecules are dissolved/embedded and coulombic interaction refers to the through-space electrostatic interaction between triphenylamines and perylenediimide. If the through-space coulombic interaction is smaller, the CSS is less stabilized. As a result, the forward ET becomes slower, whereas the reverse ET to the LES becomes faster. For these molecules, delayed fluorescence can no longer be discriminated from the decay time of the unquenched acceptor.
Conclusions
We used single-molecule fluorescence decay times to probe the dynamics of reversible ET occurring in single molecules of the first and second generations of a polyphenylene dendrimer containing several triphenylamine donors and a perylenediimide acceptor. Taking advantage of the reversible ET in individual D-A molecules leading to delayed fluorescence which is emitted with a high photon count rate, we were able to monitor, over a long term, fluctuations in fluorescence decay times. Fluorescence decay times indicate correlated fluctuations in forward and reverse ET spanning a broad time range, i.e., from milliseconds to seconds. Fluctuations are induced, on the one hand, by conformational changes in the dendrimer structure and, on the other hand, by polymer chain reorientation. Conformational changes relate to libration, that is, changes in the torsional angle of adjacent phenyl rings located in the dendritic branches near the donor moiety transferring the charge. This torsional motion has as a net result a change in the through-bond D-A coupling and induces here decay time fluctuations in milliseconds time scale. Polymer chain reorientation leads to changes in the local polarity of the donors and hence to changes in the solvation of CSS. As a result, switching between different donor moieties within a single molecule becomes possible and contributes to fluctuations in decay time in the time scale of seconds. To our knowledge, this is the first report on single-molecule decay time fluctuations induced by through-bond D-A electronic coupling changes. These results may have implications in single-molecule conduction in polyphenylene-based compounds where libration may play a role in leading to fluctuations in the overall transfer of the electronic charge (29).
Supplementary Material
Acknowledgments
M.C. acknowledges the Human Frontier for Science Promotion for a fellowship at Harvard University. This work was supported by the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen, the Flemish Ministry of Education (GOA 2/01), the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie, and the Federal Science Policy of Belgium (IUAP-V-03). A Max Planck Research Award (to F.D.S.) is also acknowledged. X.S.X. acknowledges funding from the U.S. Department of Energy.
Abbreviations: SMD, single-molecule detection; ET, electron transfer; D-A, donor-acceptor; LES, locally excited state; CSS, charge-separated state; GS, ground state; PN, perylenediimide acceptor-triphenylamine donor; ZE, Zeonex; PS, polystyrene; MLE, maximum-likelihood estimator.
References
- 1.Nie, S. M. & Zare, R. N. (1997) Annu. Rev. Biophys. Biomol. Struct. 26, 567-596. [DOI] [PubMed] [Google Scholar]
- 2.Xie, X. S. & Trautman, J. K. (1998) Annu. Rev. Phys. Chem. 49, 441-480. [DOI] [PubMed] [Google Scholar]
- 3.Lu, H. P. & Xie, X. S. (1998) Science 282, 1877-1882. [DOI] [PubMed] [Google Scholar]
- 4.Moerner, W. & Orrit, M. (1999) Science 283, 1670-1676. [DOI] [PubMed] [Google Scholar]
- 5.Weiss, S. (1999) Science 283, 1676-1683. [DOI] [PubMed] [Google Scholar]
- 6.Zhuang, X., Bartley, L. E., Babcock, H. P., Russell, R., Ha, T. J., Herschlag, D. & Chu, S. (2000) Science 288, 2048-2051. [DOI] [PubMed] [Google Scholar]
- 7.Ha, T., Ting, A. Y., Liang, J., Cadwell, W. B., Deniz, A. A., Chemla, D. S. & Weiss, S. (1999) Proc. Natl. Acad. Sci. USA 96, 893-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forster, T. (1959) Discuss. Faraday Soc. 27, 7-17. [Google Scholar]
- 9.Kavarnos, G. J. (1997) Fundamentals of Photoinduced Electron Transfer (VCH, New York), pp. 305-309.
- 10.Lu, H. P. & Xie, X. S. (1997) J. Phys. Chem. B 101, 2753-2757. [Google Scholar]
- 11.Knemeyer, J. P., Marme, N. & Sauer, M. (2000) Anal. Chem. 72, 3717-3724. [DOI] [PubMed] [Google Scholar]
- 12.Zang, L., Liu, R., Holman, M. W., Nguyen, K. T. & Adams, D. M. (2002) J. Am. Chem. Soc. 124, 10640-10641. [DOI] [PubMed] [Google Scholar]
- 13.Yang, H., Luo, G., Karnchanaphanurach, P., Louie, T.-M., Rech, I., Cova, S., Xun, L. & Xie, X. S. (2003) Science 302, 262-266. [DOI] [PubMed] [Google Scholar]
- 14.Gronheid, R., Stefan, A., Cotlet, M., Hofkens, J., Qu, J., Müllen, K., Van der Auweraer, M., Verhoeven, J. W. & De Schryver, F. C. (2003) Angew. Chem. Int. Ed. 42, 4209-4214. [DOI] [PubMed] [Google Scholar]
- 15.Lor, M., Thielemans, J., Viaene, L., Cotlet, M., Hofkens, J., Weil, T., Hampel, C., Müllen, K., Verhoeven, J. W., Van der Auweraer, M. & De Schryver, F. C. (2002) J. Am. Chem. Soc. 124, 9918-9925. [DOI] [PubMed] [Google Scholar]
- 16.Liu, D., De Feyter, S., Cotlet, M., Stefan, A., Wiesler, U.-M., Herrmann, A., Grebel-Koehler, D., Müllen, K. & De Schryver, F. C. (2003) Macromolecules 36, 5918-5925. [Google Scholar]
- 17.Cotlet, M., Hofkens, J., Habuchi, S., Dirix, G., Michiels, J., Vanderleyden, J. & De Schryver, F. C. (2001) Proc. Natl. Acad. Sci. USA 98, 14398-14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goes, M., de Groot, M., Koeberg, M., Verhoeven, J. W., Lokan, N. R., Shephard, M. J. & Paddon-Row, M. N. (2002) J. Phys. Chem. A 106, 2129-2134. [Google Scholar]
- 19.Verhey, H. J., Gebben, B., Hofstraat, J. W. & Verhoeven, J. W. (1995) J. Polym. Sci. A Polym. Chem. 33, 399-405. [Google Scholar]
- 20.Ambrose, W. P., Goodwin, P. M., Martin, J. C. & Keller, R. A. (1994) Science 265, 364-367. [DOI] [PubMed] [Google Scholar]
- 21.Xie, X. S. & Dunn, R. C. (1994) Science 265, 361-364. [DOI] [PubMed] [Google Scholar]
- 22.Hofkens, J., Vosch, T., Maus, M., Kohn, F., Cotlet, M., Weil, T., Hermann, A., Müllen, K. & De Schryver, F. C. (2001) Chem. Phys. Lett. 333, 255-263. [Google Scholar]
- 23.Maus, M., Cotlet, M., Hofkens, J., Gensch, T., De Schryver, F. C., Schaffer, J. & Seidel, C. A. M. (2001) Anal. Chem. 73, 2078-2086. [DOI] [PubMed] [Google Scholar]
- 24.Davis, W. B., Ratner, M. A. & Wasielewski, M. R. (2001) J. Am. Chem. Soc. 123, 7877-7886. [DOI] [PubMed] [Google Scholar]
- 25.Yang, H. & Xie, X. S. (2002) J. Chem. Phys. 117, 10965-10979. [Google Scholar]
- 26.Xie, X. S. (2002) J. Chem. Phys. 117, 11024-11032. [Google Scholar]
- 27.Neto, N., Muniz-Miranda, M., Angeloni, L. & Castelucci, E. (1983) Spectrochim. Acta A 39, 97-106. [Google Scholar]
- 28.Ediger, M. D. (2000) Annu. Rev. Phys. Chem. 51, 99-128. [DOI] [PubMed] [Google Scholar]
- 29.Reichert, J., Ochs, R. Beckmann, D., Weber, H. B., Mayor, M. & von Löhneysen, H. (2002) Phys. Rev. Lett. 88, 176804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}