

Abstract
Objectives
Two consanguineous families, one of Sudanese ethnicity presenting progressive neuromuscular disease, severe cognitive impairment, muscle weakness, upper motor neuron lesion, anhydrosis, facial dysmorphism, and recurrent seizures and the other of Egyptian ethnicity presenting with neonatal hypotonia, bradycardia, and recurrent seizures, were evaluated for the causative gene mutation.
Methods and Results
Homozygosity mapping and whole exome sequencing (WES) identified damaging homozygous variants in SCN10A, namely c.4514C>T; p.Thr1505Met in the first family and c.4735C>T; p.Arg1579* in the second family. A third family, of Western European descent, included a child with febrile infection‐related epilepsy syndrome (FIRES) who also had compound heterozygous missense mutations in SCN10A, namely, c.3482T>C; p.Met1161Thr and c.4709C>A; p.Thr1570Lys. A search for SCN10A variants in three consortia datasets (EuroEPINOMICS, Epi4K/EPGP, Autism/dbGaP) identified an additional five individuals with compound heterozygous variants. A Hispanic male with infantile spasms [c.2842G>C; p.Val948Leu and c.1453C>T; p.Arg485Cys], and a Caucasian female with Lennox–Gastaut syndrome [c.1529C>T; p.Pro510Leu and c.4984G>A; p.Gly1662Ser] in the epilepsy databases and three in the autism databases with [c.4009T>A; p.Ser1337Thr and c.1141A>G; p.Ile381Val], [c.2972C>T; p.Pro991Leu and c.2470C>T; p.His824Tyr], and [c.4009T>A; p.Ser1337Thr and c.2052G>A; p.Met684Ile].
Interpretation
SCN10A is a member of the voltage‐gated sodium channel (VGSC) gene family. Sodium channels are responsible for the instigation and proliferation of action potentials in central and peripheral nervous systems. Heterozygous mutations in VGSC genes cause a wide range of epileptic and peripheral nervous system disorders. This report presents autosomal recessive mutations in SCN10A that may be linked to epilepsy‐related phenotypes, Lennox–Gastaut syndrome, infantile spasms, and Autism Spectrum Disorder.
Introduction
Four patients from three different families with epilepsy and biallelic SCN10A variants were identified. Family 1 (Fig. 1A) is Sudanese, seen at the Shafallah Medical Genetics Center, Doha, Qatar; two female siblings (II‐6, 18 years and II‐7, 17 years) presented with progressive neuromuscular symptoms and impaired intellectual development. The parents, first cousins, previously had one second‐trimester spontaneous abortion and a daughter who died 4 days after birth. Shortly after birth, patient II‐6 had recurrent attacks of high‐grade fever and absent tearing. She was recognized to be hypotonic at 4 months of age and she was able to walk only at 20 months of age. At the age of 10 years, she developed progressive weakness and difficulty swallowing that led to aspiration pneumonia and frequent hospitalizations. She stopped being ambulatory at the age of 14 years, and her speech became slurred and had notable intellectual impairment. Dysmorphic features included a prominent forehead, long face, and broad nasal bridge. The muscle tone was decreased but with retained deep tendon reflexes and bilateral up‐going plantar reflex. Patient II‐7 had a clinical presentation similar to that of her older sister, but with the onset of seizures at the age of 15 years, described as drop attacks with neck extension, eyes rolling back, and intermittent myoclonus and poor response to combinations of anti‐epileptic drugs, with a modest response to lamotrigine and topiramate. Both sisters had normal Creatine Phosphokinase (CPK), liver function tests, serum lactate, serum amino acids, ammonia, brain MRI, electromyogram (EMG), and nerve conduction studies (NCS). For II‐6, ophthalmologic evaluation did not show any retinal abnormalities and echocardiography was unremarkable. Electron microscopy of a skin biopsy ruled out anhidrotic ectodermal dysplasia. For II‐7 the electroencephalogram (EEG) showed generalized subcortical epileptiform discharges. Chromosomal analysis and molecular karyotyping did not reveal any copy number variants (CNVs) of clinical significance, especially within the homozygosity regions.
Figure 1.
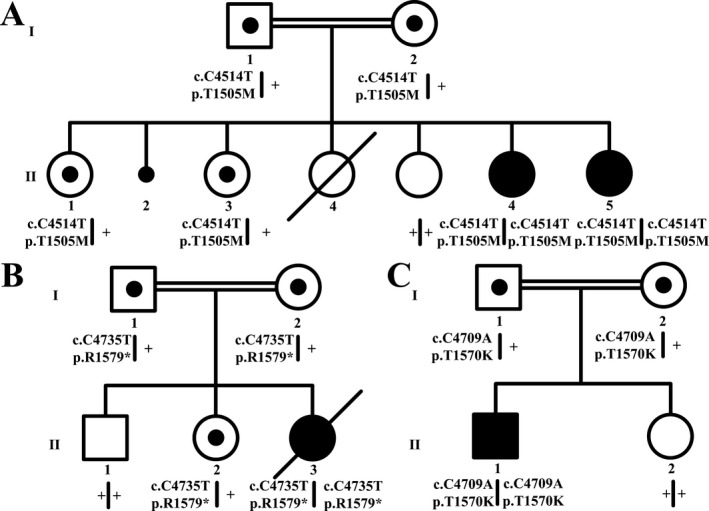
Pedigree of families 1 (A), 2 (B), and 3 (C) with the seggregating SCN10A mutations. Genotypes are indicated below each participating individual. Double lines represent first cousin marriage.
Family 2 (Fig. 1B) represented a consanguineous marriage of Egyptian first cousins seen at Hôpital d'Enfant, Dijon, France. The family had several infantile deaths previously. In utero, the proband (II‐3) had third trimester fetal sinus bradycardia without maternal lupus erythematosis. She was born at 37 weeks with normal length, weight and head circumference, and profound hypotonia, absent deep tendon reflexes but no tongue fasciculation. The only facial dysmorphism was a tented upper lip. She was admitted to the intensive care unit for recurrent apnea, bradycardia and poor feeding. The echocardiography showed a patent foramen ovale. Brain MRI, ophthalmological examination, abdominal ultrasound, bone survey, electroretinogram (ERG), EMG, EEG, visual, and auditory evoked potentials, metabolic screen, mitochondrial screen, muscle enzymes, cerebrospinal fluid investigations, including neurotransmitter assays, and muscle, and liver biopsies were all normal. At 3 months of age, the patient remained hypotonic with feeding difficulties. There was no eye contact or head control, but there was plastic rigidity and abnormal ocular movements. The EEG showed modifications in basal rhythm with rare and bilateral inner temporal spikes. Several weeks later, she had tonic spasms and tonic‐clonic seizures. A follow‐up EEG revealed generalized spike‐wave discharges without hypsarrhythmia. The seizures were difficult to control with three different anti‐epileptic drugs (phenobarbital, diazepam and dilantin), and progressed to recurrent episodes of status epilepticus. Chromosomal and molecular karyotyping were unremarkable. DNA testing for SMN, CDKL5, and FOXG1 was negative, thus ruling out spinal muscular atrophy, X‐linked infantile spasms and FOXG1 syndromes. The patient died at 6 months of age.
The third family (Fig. 1C) is of Portuguese and Italian descent. The male proband (II‐1) was developmentally appropriate without medical issues until the abrupt onset of febrile flu‐like prodrome at the age of 6 years, and 5 days after the onset of the fever he was found unresponsive; this immediately progressed to intractable multifocal epilepsy requiring prolonged placement in a medically induced coma with intravenous midazolam, ketamine, paraldehyde, and phenobarbital titrated to burst‐suppression such that he required inotropic support with other treatment attempts such as hypothermia and ketogenic diet. Brain MRI showed abnormal T2 signal with restricted diffusion in both hippocampi, right greater that left. MRS showed an increased glutamine‐glutamate peak possibly representing excitotoxic neuronal changes. A PET scan showed markedly abnormal focal uptake in the left parieto‐occipital region, moderately diffused increased uptake in the left hemisphere, multiple foci of FDG accumulation, and diffusely decreased uptake in the anterior right temporal lobe. The CSF showed 7 WBCs, 2 RBCs, glucose 3.8 mmol/L, protein 0.28 mg/dL. Extensive infectious testing was negative and diagnostic brain biopsy was unremarkable. Visual evoked potentials showed no clearly reproducible cortical response. Somatosensory‐evoked potential of the median nerve showed very low cortical response. Brainstem auditory evoked potential responses were normal. 3 months into the illness, he had choreiform movements. He remains neurologically devastated with frequent seizures requiring intravenous anti‐epileptic drugs, vagal nerve stimulation, and chronic ventilation via tracheostomy.
Material and Methods
The study was conducted in accordance with the provisions of the Declaration of Helsinki and a signed informed consent was obtained from each participant or his/her legal guardian. Blood samples from all available family members of the three families were obtained and genomic DNA was extracted with Gentra Puregene Blood kit [Qiagen, Valencia, CA] according to the manufacturer's specifications and guidelines.
Homozygosity mapping was performed utilizing the Human Mapping OmniExpress‐12 v1‐1 SNP genotyping array [Illumina, San Diego, CA]. Data iltration and genotyping were done with GenomeStudio v2011.1. For the determination of linkage intervals, data were analyzed by the “HomozygosityMapper” software.1, 2
For families 1 and 2, whole exome target enrichment was performed on ABI SOLiD™ 4 platform (Applied Biosystems, Foster City, CA) according to the manufacturer's specifications. DNA library preparation was with TargetSeq™ Exome Enrichment system (Applied Biosystems) as multiplex fragment libraries utilizing both the SOLiD™ Fragment Library construction Kits and SOLiD™ Fragment Library Barcoding Kit Module 1–16 for the ABI SOLiD™ 4 system. Bead preparation and enriching was done on an EZ Bead Emulsifier, Amplifier and Enricher utilizing E80 scale (Applied Biosystems). Sequencing modality was with multiplex fragment paired‐end.
The bioinformatics of the whole exome NGS data started with the analysis of the raw data files (in a proprietary XSQ format) with the Life Technologies LifeScope v2.5.4 software (Life Technologies, Carlsbad, CA) on a dedicated cluster to align the reads produced by the SOLiD™ to the hg19 reference sequence, sourced from the University of California at Santa Cruz Genome Informatics Group (UCSC). The aligned (BAM) files were validated, duplicate sequences were identified and removed and incorrectly identified Mate‐Pairs were corrected, using the Picard v1.87 software. The Genome Analysis Tool Kit (GATK) v.3.0.0 (Boston, MA, USA) was applied to the “corrected” output files to recalibrate the base quality scores, using machine learning to model any systematic errors in the data, to carry out localized realignments around possible insertion/deletion sequences to ensure mapping accuracy, to identify viable variants from the sequence reads and to recalibrate the variants to ensure accuracy of the variant calling in a variant‐specific manner. Once a suitable list of variants was produced, it was filtered using in‐house scripts to confirm variant zygosity and identify those variants that conform to the inheritance model. These variants were annotated using an in‐house script in conjunction with the software, ANNOVAR [http://www.openbioinformatics.org/annovar/] to produce an annotated variant list with the most recent available information from a number of reference Websites.
Whole Exome sequencing for Family 3 was performed on Illumina HiSeq2000 (Illumina) with 101‐bp paired‐end read sequencing. Image analyses and base calling were with the Illumina Genome Analyzer Pipeline software v.1.13.48.0 with default parameters. Reads were aligned to a human reference sequence (UCSC hg19; NCBI build 37) with the “Efficient Large‐scale Alignment of Nucleotide Databases” software (Illumina). Genotypes were called at all positions with high‐quality sequence bases using the Bayesian algorithm “Most Probable Genotype.” The BAM files were visualized with Integrative Genomics Viewer (The Broad Institute, Cambridge, MA). Variants were filtered using VarSifter v1.5 tool by considering minor allele frequency (MAF) of <0.05 and the availability of variants in public databases of dbSNP, ClinSeq and EXAC (The Broad Institute). Filtered variants were annotated with CADD score using online tools [http://cadd.gs.washington.edu/score].
Targeted DNA resequencing was done by Sanger Big‐dye terminator v3.1 cycle sequencing (Applied Biosystems) on an ABI 3730 automatic sequencer (Applied Biosystems) to screen for mutations in candidate genes, perform allele frequency studies in related populations, and determine co‐segregation of variants with the phenotype within each family.
VCF files were obtained from the dbGaP entry for the ARRA Autism Sequencing Collaboration (phs000298). Only those consented for autism research (AO) were downloaded. The dataset(s) were deposited by the ARRA Autism Sequencing Collaborative, an ARRA funded research initiative. Exome VCF files from the Epi4K Epilepsy Phenome/Genome Project (EPGP) were requested and downloaded from the dbGaP (phs000653 v2p1).3, 4, 5
Results
The SNP genotyping and homozygosity mapping for Family 1 identified four intervals spanning about 70 Mb and containing about 850 protein‐coding genes (Table 1). The WES for II‐6 showed no significant de novo or X‐linked variants but identified two exonic nonsynonymous homozygous variants within the homozygosity intervals, DYNC2H: c.12254G>A; p.Arg4085His and SCN10A: c.4514C>T; p.Thr1505Met.
Table 1.
Homozygosity intervals for families 1 & 2
| Family | Chromosome | From coordinates | To coordinates | Length [Mb] | Protein‐Coding gene number |
|---|---|---|---|---|---|
| Family 1 | |||||
| 3p |
32,417,644 rs4364205 |
51,874,275 rs9853511 |
19.5 | 235 | |
| 11p |
41,180,155 rs10768669 |
66,262,606 rs2511224 |
25.1 | 391 | |
| 11q |
102,385,738 rs7118775 |
123,074,915 rs485345 |
20.7 | 171 | |
| 14q |
101,679,885 rs8015515 |
107,246,846 rs2078693 |
5.6 | 57 | |
| Family 2 | |||||
| 1p |
117,010,232 rs17575616 |
164,541,977 rs2792251 |
47.5 | 456 | |
| 3p |
8,734,471 rs6766036 |
57,499,769 rs6445905 |
48.8 | 419 | |
| 5q |
111,893,837 rs7703562 |
115,949,578 rs1873853 |
4 | 22 | |
The SNP genotyping and homozygosity mapping for Family 2 identified three intervals spanning about 100 Mb and containing about 880 protein‐coding genes (Table 1). The WES for II‐3 showed no significant de novo or X‐linked variants but identified two exonic nonsynonymous homozygous variants within the homozygosity intervals, LAMB2: c.4981C>T; p.Arg1661Trp and SCN10A: c.4735C>T; p.Arg1579*.
The WES and comparative genome analyses for Family 3 showed no significant de novo, homozygous or X‐linked variants. Compound heterozygous exonic nonsynonymous variants were identified in SASH1: c.2458C>T; p.Arg820Trp and c.2995G>A; p.Gly999Arg, and in SCN10A: c.3482T>C; p.Met1161Thr and c.4709C>A; p.Thr1570Lys.
Following the identification of the SCN10A variants in the three families, three consortia databases were examined for compound heterozygous variants in SCN10A. The Epi4K/EPGP dataset,4 containing WES data on 356 patients, identified SCN10A compound heterozygous variants in two patients; a Caucasian female with Lennox‐Gastaut syndrome (LGS, MIM 606369) [c.1529C>T; p.Pro510Leu and c.4984G>A; p.Gly1662Ser] and a Hispanic male with infantile spasms [c.2842G>C; p.Val948Leu and c.1453C>T; p.Arg485Cys]. The Autism dbGaP dataset, containing WES data from 486 patients with ASD, identified SCN10A compound heterozygous variants in three patients; [c.4009T>A; p.Ser1337Thr and c.1141A>G; p.Ile381Val], [c.2972C>T; p.Pro991Leu and c.2470C>T; p.His824Tyr], and [c.4009T>A; p.Ser1337Thr and c.2052G>A; p.Met684Ile]. The MAFs of all identified variants, the in silico mutation effect predictions and amino‐acid conservation across species are presented in Table 2. The positions of all protein variants are shown in Figure 2.
Table 2.
SCN10A variants
| Patient | dbID | cDNA (NM_006514.3) | PROTEIN | MAF in Exac | POLYPHEN | POLYPHEN SCORE (HumDiv/HumVar) | SIFT | SIFT SCORE | Mutation Taster | PhyloP conservation score | Phastcons conservation score | CONSERV‐ATION OF AMINO ACID | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound heterozygous variants in families included in the study | |||||||||||||
| Family 1 | 1 & 2 | rs184521520 | c.4514C>T | p.Thr1505Met | T: 0.00045 | Probably damaging | 1/0.961 | Deleterious | 0.01 | Polymorphism | 1.397 | 0.864 | Highly conserved |
| Family 2 | 1 & 2 | N/A | c.4735C>T | p.Arg1579a | T: 0.000008242 | N/A | N/A | N/A | N/A | Disease causing | 1.616 | 0.968 | Highly conserved |
| Family 3 | 1 | rs200713724 | c.3482T>C | p.Met1161Thr | C: 0.0002231 | Probably damaging | 0.998/0.993 | Deleterious | 0 | Disease causing | 4.48 | 1 | Highly conserved |
| 2 | N/A | c.4709C>A | p.Thr1570Lys | N/A | Probably damaging | 1.00/0.976 | Deleterious | 0 | Disease causing | 5.94 | 0.99 | Highly conserved | |
| Compound heterozygous variants in various databases included in the study | |||||||||||||
| Epi4K 1 | 1 | rs151090729 | c.4984G>A | p.Gly1662Ser | 0.0013 | Probably damaging | 1/1 | Deleterious | 0 | Disease causing | 6.075 | 1 | Highly conserved |
| 2 | N/A | c.1529C>T | p.Pro510Leu | 0.00001664 | Possibly damaging | 0.899/0.382 | Deleterious | 0.01 | Polymorphism | 0.936 | 0.009 | Moderately conserved | |
| Epi4K 2 | 1 | rs145694222 | c.2842G>C | p.Val948Leu | 0.00043 | Benign | 0.120/0.049 | Tolerated | 0.12 | Polymorphism | 0.128 | 0.009 | Moderately conserved |
| 2 | rs151153639 | c.1453C>T | p.Arg485Cys | 0.0007 | Probably damaging | 0.983/0.405 | Deleterious | 0.01 | Disease causing | 2.17 | 1 | Highly conserved | |
| dbGaP 1 | 1 | ars11711062 | c.4009T>A | p.Ser1337Thr | 0.0035 | Benign | 0.059/0.050 | Tolerated | 0.3 | Polymorphism | −1.929 | 0 | Weakly conserved |
| 2 | rs150923753 | c.1141A>G | p.Ile381Val | 0.0007 | Probably damaging | 0.995/0.992 | Deleterious | 0 | Disease causing | 5.017 | 1 | Highly conserved | |
| dbGaP 2 | 1 | rs138413438 | c.2972C>T | p.Pro991Leu | 0.00062 | Possibly damaging | 0.985/0.710 | Deleterious | 0.01 | Polymorphism | 1.9 | 0.01 | Highly conserved |
| 2 | N/A | c.2470C>T | p.His824Tyr | N/A | Benign | 0.012/0.038 | Tolerated | 0.06 | Polymorphism | −1.277 | 0 | Weakly conserved | |
| dbGaP 3 | 1 | ars11711062 | c.4009T>A | p.Ser1337Thr | 0.0035 | Benign | 0.059/0.050 | Tolerated | 0.3 | Polymorphism | −1.929 | 0 | Weakly conserved |
| 2 | N/A | c.2052G>A | p.Met684Ile | N/A | Possibly damaging | 0.584/0.138 | Tolerated | 0.06 | Disease causing | 5.593 | 1 | Highly conserved | |
Identical variants in different cases.
phyloP (values between −14 and +6) separately measures conservation at individual columns, ignoring the effects of their neighbors. Moreover, phyloP can not only measure conservation (slower evolution than expected under neutral drift) but also acceleration (faster than expected). Sites predicted to be conserved are assigned positive scores, while sites predicted to be fast‐evolving are assigned negative scores.
phastCons values vary between 0 and 1 and reflect the probability that each nucleotide belongs to a conserved element, based on the multiple alignment of genome sequences of 46 different species (the closer the value is to 1, the more probable the nucleotide is conserved). It considers not just each individual alignment column, but also its flanking columns.
Figure 2.
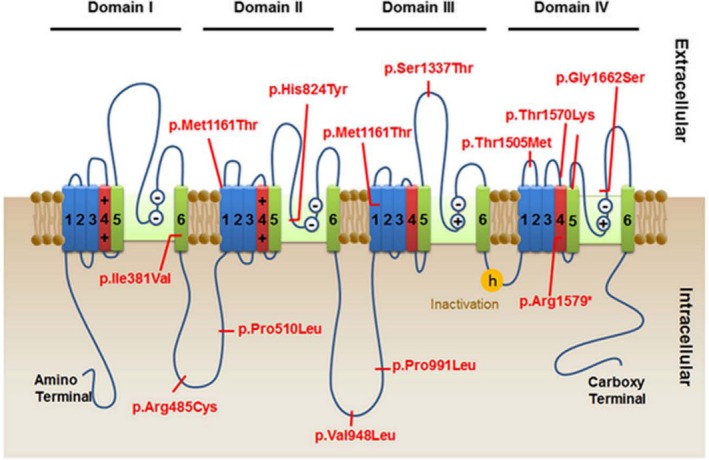
Location of SCN10 variants detected in the nine presented subjects.
Discussion
Two exonic nonsynonymous homozygous variants contained within runs of homozygosity, were identified in Family 1, namely DYNC2H: c.12254G>A; p.Arg4085His and SCN10A: c.4514C>T; p.Thr1505Met. Although the DYNC2H variant co‐segregates with the phenotype in the family and was absent in 400 ethnically matched control chromosomes, mutations in DYNC2H are associated with asphyxiating thoracic dystrophy and short rib‐polydactyly type III,6, 7 the features of which were absent in the two girls. On the other hand, the SCN10A variant segregated with the disease phenotype, was not detected in 600 ethnically matched control chromosomes, changed a highly conserved amino acid8 and was predicted to be damaging9, 10 (Table 2). The variant affects a residue between helices S1 and S2 of repeat domain IV of the sodium channel (Fig. 2), putatively in an extracellular position. Accordingly, the SCN10A variant was considered the best candidate gene responsible for the phenotype in this family.
In Family 2, two exonic nonsynonymous homozygous variants contained within runs of homozygosity, namely LAMB2: c.4981C>T; p.Arg1661Trp and SCN10A: c.4735C>T; p.Arg1579*. Both variants co‐segregated with the phenotype and were absent in 400 ethnically matched control chromosomes and publically available databases. However, the LAMB2 variant is predicted to be benign9, 10 and affects a residue that is not evolutionary conserved.8 Moreover, mutations in LAMB2 are associated autosomal recessive Pierson syndrome (MIM 609409) and nephrotic sydrome type 5, with or without ocular abnormalities (MIM 614199).11 On the other hand, the SCN10A variant affects the voltage‐sensor helix S4 in Domain IV (Fig. 2), causing protein termination at residue 1579 and putatively producing a protein missing 377 amino acids (20%) of its C terminus and is possibly subject to nonsense‐mediated mRNA decay. Accordingly, the SCN10A variant was considered the best candidate gene responsible for the phenotype in this family.
Since Family 3 does not exhibit consanguinity, WES data could not be filtered for homozygous variants within runs of homozygosity and the analyses were focused on rare damaging variants including compound heterozygous variants. Exonic nonsynonymous compound heterozygous variants were identified in two genes, namely SASH1: c.2458C>T; p.Arg820Trp and c.2995G>A; p.Gly999Arg, and in SCN10A: c.3482T>C; p.Met1161Thr and c.4709C>A; p.Thr1570Lys. SASH1 is a tumor suppressor gene, involved in toll‐like receptor signaling pathway and is significantly down‐regulated in breast tumor tissues and breast cancer cell lines,12 and is thus unlikely to be the offending gene in this patient. On the other hand, both SCN10A variants are damaging according to the prediction tools,9, 10 extremely rare, affect highly conserved residues8 and co‐segregate with the disease phenotype in the family (Table 2). Since seizures and pain disorders occur due to mutations highly homologous sodium channel gene, SCN9A, that is expressed in dorsal root ganglion,13, 14, 15 SCN10A was considered the best candidate gene in this family.
Voltage‐gated sodium channels (VGSC) are responsible for initiating and propagating action potentials in neurons and other excitable cells. Their pore‐forming alpha subunits are integral membrane glycoproteins encoded by a family comprised of nine highly conserved genes, and are sufficient for channel expression.16, 17, 18, 19 Their function includes ion conductance, pore gating and pore regulation. They share a conserved basic structure of about 2000 amino‐acids arranged in four homologous domains (I–IV), each containing six evolutionarily conserved transmembrane alpha helices (S1‐S6) with S4 harboring the voltage sensor20 (Fig. 2). The associated regulatory beta subunits (beta1‐beta4) modulates the properties, kinetics, and trafficking of the pore‐forming alpha subunit.21 SCN10A encodes the alpha subunit of the VGSC, Nav1.8 that is expressed in nociceptors that transmit pain signals, in C‐fibers from dorsal root ganglia, heart muscle, skeletal muscle, and neuronal tissues.22 Mutations in VGSC lead to either hypoactivity or hyperactivity of the sodium channels. Monoallelic VGSC variant are associated with epilepsy,23 cardiac conduction defects,24 skeletal muscle channelopathies,25 and peripheral pain disorders,26 based on their tissue‐specific expression. Inherited mutations cause less severe disease than de novo mutations, and truncating mutations are associated with the most severe phenotypes.27 Most VGSC mutations are heterozygous, with a few exceptions such as, congenital insensitivity to pain due to biallelic SCN9A mutations,26 sick sinus syndrome due to biallelic SCN5A mutations28 and biallelic mutations in SCN4A causing myasthenic syndrome with period paralysis,29 or congenital myopathy.30
To date, no known neurological diseases, particularly seizures, have been associated with biallelic SCN10A mutations. Heterozygous gain‐of‐function SCN10A mutations account for about 5% of small fiber neuropathies, characterized by severe pain attacks and a reduced ability to differentiate between hot and cold (Familial Episodic Pain Syndrome 2, MIM 61551).31, 32 These mutations cause the sodium channels to open more readily, increasing the flow of sodium ions that produce nerve impulses within nociceptors and causing increased sensitivity to pain. SCN10A plays a role in maintaining normal heart rhythm.33, 34 Certain heterozygous SCN10A polymorphisms have been associated with increased risk of cardiac arrhythmia, including Brugada disorder (MIM 601144). These were hypothesized to disrupt the electrical signals that control heartbeat, causing “heart block” arrhythmias due to slowed or interrupted cardiac conduction.35 Loss‐of‐function effects for these heterozygous variants is suspected.36 A recent study suggests that variants in SCN10A are involved in the genesis of atrial fibrillation.37 Interestingly, in neurons derived from a mouse homozygous of Thr790Ala variant, current clamp recordings revealed heightened excitability of the neurons with long‐duration actions potentials, implicating a role of SCN10A in modulating the activity of CNS neurons.
In all three families presented, the obligate heterozygotes had no complaints of episodic pain and there was no family history of sudden death or arhythmia. Thus it appears that the presence of these variants in heterozygosity is not associated with a detectable clinical phenotype. Most of the biallelic variants in the three families are in the ion transport domain of SCN10A. further genotype‐phenotype correlation would require identification of additional cases. Since other sodium channel genes, such as SCN1A and SCN2A have been associated with epilepsy,23, 25 and SCN1A leading to Dravet syndrome is classically precipitated by fever, it is interesting that another refractory epileptic encephalopathy triggered by fever (FIRES) has now been associate with another sodium channel gene.14 In addition, the expression of SCN10A in CNS neurons, as well as in the developing brain provides biological plausibility for the phenotype produced by the biallelic mutations.38, 39, 40, 41
Epileptic seizures affect 7–46% of children with ASD, especially those with intellectual developmental disorder and ASD occurs in 15–35% of those with epilepsy.42 Examination of databases containing whole genome or whole exome data for patients with epilepsy, ASD or both for compound heterozygosity for mutations within genes identified through homozygosity studies would be quite useful. In this study the identification of compound heterozygosity in two patients with LGS and infantile spasms and three patients with ASD further implicates SCN10A in these disorders.
In conclusion, through an international collaboration, we have identified four patients with biallelic SCN10A variants who presented with epileptic seizures. The review of three different publically available disease databases has added several other patients, although the variants may not be responsible for the phenotype. Since epilepsy is observed along with intellectual impairment and peripheral neuropathy, biallelic SCN10A mutations likely interfere with normal functioning of this sodium channel, resulting in aberrant transmission of nerve impulses.
Author Contribution
JAA, MCM and HES conceptulized the sudy and provided guidance and supervision. MK, AS, JS, YAS, WB, JCM, YD, CTR, and JBR analyzed exomes, validated variants and examined seggregation. MK, JSO and CTR performed molecular genetics experiments for biological validation of results. JT, AS, TBO, AMP, CC, JAA, LF, WG and HES ascertained, recruited, and clinically evaluated patients. AC and AB examined datasets. NISC performed NGS data analysis. MK, YAS, and JAA performed homozygosity mapping. MK, JT, AS, AC, JS, TBO, YAS, HB, WB, JCM, AMP, JSO, YD, CC, CTR, LF, JBR, WG, AB, MCM, and HES contributed to the manuscript writing and/or review.
Conflict of Interest
All authors report no conflict of interest.
Acknowledgment
This publication was made possible in part by Qatar National Research Fund (QNRF) NPRP grant 09‐367‐3‐087 (MK and HE) and NPRP grant 6‐359‐3‐095 (HE and AGB) and by the generous support of the Shafallah Center Foundation, Doha, Qatar. It was also supported by NIH 1R01 NS064159‐01A1 (AGB) and 5R21MH100086‐02 and the National Human Genome Research Institute Intramural Research Program. The support for the Autism Sequencing Collaborative was provided by grants R01‐MH089208 awarded to Mark Daly, R01‐MH089175 awarded to Richard Gibbs, R01‐MH089025 awarded to Joseph Buxbaum, R01‐MH089004 awarded to Gerard Schellenberg and R01‐MH089482 awarded to James Sutcliffe. Our appreciation to the Epilepsy Epi4K consortium: Discovery in Epilepsy study (NINDS U01‐NS077303) and the Epilepsy Genome/Phenome Project (EPGP‐NINDS U01‐NS053998). Special thanks to Maryam Al‐Mutawa for her help with the figures and to Hibah Shaath for her valuable contribution to the preparation of this manuscript. This manuscript is dedicated to the memory of Dr. Jamil Al‐Alami who sadly passed away during the work on this project. Jamil was highly regarded as a physician, colleague, friend, and overall a great person.
References
- 1. Seelow D, Schuelke M. HomozygosityMapper2012–bridging the gap between homozygosity mapping and deep sequencing. Nucleic Acids Res 2012;40:W516–W520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seelow D, Schuelke M, Hildebrandt F, Nurnberg P. HomozygosityMapper–an interactive approach to homozygosity mapping. Nucleic Acids Res 2009;37:W593–W599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Epi4K Consortium . Epi4K: gene discovery in 4,000 genomes. Epilepsia. 2012;53:1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Epi4K Consortium , Epilepsy Phenome/Genome Project, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. EuroEPINOMICS‐RES Consortium , Epilepsy Phenome/Genome Project, Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014;95:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dagoneau N, Goulet M, Geneviève D, et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib‐polydactyly syndrome, type III. Am J Hum Genet 2009;84:706–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Merrill AE, Merriman B, Farrington‐Rock C, et al. Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short‐rib polydactyly syndrome. Am J Hum Genet 2009;84:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010;20:110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 10. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc in Human Genet/editorial board, Jonathan L. Haines… [et al.]. 2013;Chapter 7:Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matejas V, Hinkes B, Alkandari F, et al. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat 2010;31:992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zeller C, Hinzmann B, Seitz S, et al. SASH1: a candidate tumor suppressor gene on chromosome 6q24.3 is downregulated in breast cancer. Oncogene 2003;22:2972–2983. [DOI] [PubMed] [Google Scholar]
- 13. Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006;444:894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Singh NA, Pappas C, Dahle EJ, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet 2009;5:e1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cox JJ, Sheynin J, Shorer Z, et al. Congenital insensitivity to pain: novel SCN9A missense and in‐frame deletion mutations. Hum Mutat 2010;31:E1670–E1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Catterall WA. Voltage‐gated sodium channels at 60: structure, function and pathophysiology. J Physiol 2012;590:2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Catterall WA. Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu Rev Pharmacol Toxicol. 2014;54:317–338. [DOI] [PubMed] [Google Scholar]
- 18. Catterall WA. Signaling complexes of voltage‐gated sodium and calcium channels. Neurosci Lett 2010;486:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Catterall WA. Structure and function of voltage‐gated sodium channels at atomic resolution. Exp Physiol 2014;99:35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage‐gated sodium channels. Neuron 2000;26:13–25. [DOI] [PubMed] [Google Scholar]
- 21. Eijkelkamp N, Linley JE, Baker MD, et al. Neurological perspectives on voltage‐gated sodium channels. Brain 2012;135:2585–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moldovan M, Alvarez S, Pinchenko V, et al. Na(v)1.8 channelopathy in mutant mice deficient for myelin protein zero is detrimental to motor axons. Brain 2011;134:585–601. [DOI] [PubMed] [Google Scholar]
- 23. Harkin LA, McMahon JM, Iona X, et al. The spectrum of SCN1A‐related infantile epileptic encephalopathies. Brain 2007;130:843–852. [DOI] [PubMed] [Google Scholar]
- 24. Amin AS, Asghari‐Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflugers Arch 2010;460:223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jurkat‐Rott K, Holzherr B, Fauler M, Lehmann‐Horn F. Sodium channelopathies of skeletal muscle result from gain or loss of function. Pflugers Arch 2010;460:239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lampert A, O'Reilly AO, Reeh P, Leffler A. Sodium channelopathies and pain. Pflugers Arch 2010;460:249–263. [DOI] [PubMed] [Google Scholar]
- 27. Brunklaus A, Ellis R, Reavey E, et al. Genotype phenotype associations across the voltage‐gated sodium channel family. J Med Genet 2014;51:650–658. [DOI] [PubMed] [Google Scholar]
- 28. Benson DW, Wang DW, Dyment M, et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest 2003;112:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Habbout K, Poulin H, Rivier F, et al. A recessive Nav1.4 mutation underlies congenital myasthenic syndrome with periodic paralysis. Neurology. 2016;86:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zaharieva IT, Thor MG, Oates EC, et al. Loss‐of‐function mutations in SCN4A cause severe foetal hypokinesia or ‘classical’ congenital myopathy. Brain. 2016;139:674–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Faber CG, Lauria G, Merkies ISJ, et al. Gain‐of‐function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci USA. 2012;109:19444–19449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Waxman SG, Merkies ISJ, Gerrits MM, et al. Sodium channel genes in pain‐related disorders: phenotype‐genotype associations and recommendations for clinical use. Lancet Neurol 2014;13:1152–1160. [DOI] [PubMed] [Google Scholar]
- 33. Facer P, Punjabi PP, Abrari A, et al. Localisation of SCN10A gene product Na(v)1.8 and novel pain‐related ion channels in human heart. Int Heart J 2011;52:146–152. [DOI] [PubMed] [Google Scholar]
- 34. Chambers JC, Zhao J, Terracciano CMN, et al. Genetic variation in SCN10A influences cardiac conduction. Nat Genet 2010;42:149–152. [DOI] [PubMed] [Google Scholar]
- 35. Savio‐Galimberti E, Weeke P, Muhammad R, et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc Res 2014;104:355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu D, Barajas‐Martínez H, Pfeiffer R, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol 2014;64:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jabbari J, Olesen MS, Yuan L, et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ Cardiovas Genet 2015;8:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hawrylycz MJ, Lein ES, Guillozet‐Bongaarts AL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012;489:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jones AR, Overly CC, Sunkin SM. The Allen Brain Atlas: 5 years and beyond. Nat Rev Neurosci 2009;10:821–828. [DOI] [PubMed] [Google Scholar]
- 40. Shen EH, Overly CC, Jones AR. The allen human brain Atlas: comprehensive gene expression mapping of the human brain. Trends Neurosci 2012;35:711–714. [DOI] [PubMed] [Google Scholar]
- 41. Sunkin SM, Ng L, Lau C, et al. Allen Brain Atlas: an integrated spatio‐temporal portal for exploring the central nervous system. Nucleic Acids Res 2013;41:D996–D1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lo‐Castro A, Curatolo P. Epilepsy associated with autism and attention deficit hyperactivity disorder: is there a genetic link? Brain Dev 2014;36:185–193. [DOI] [PubMed] [Google Scholar]