

Abstract
In the past decades, numerous types of nanomedicines have been developed for efficient and safe delivery of nucleic acid-based drugs for cancer therapy. Given that the destination sites for nucleic acid based drugs are inside the cancer cells, delivery systems need to be both targeted and shielded in order to overcome the extracellular and intracellular barriers. One of the major obstacles that has hindered the translation of nanotechnology-based gene delivery systems into the clinic has been the complexity of the design and assembly process resulting in non-uniform nancarriers with unpredictable surface property and efficiency. Consequently, no product has reached the clinic yet. In order to address this shortcoming, we genetically engineered a multifunctional targeted biopolymer in one step; therefore, eliminating the need for multiple chemical conjugations. Then by systematic modulation of the ratios of the targeted recombinant vector to PEGylated peptides of different sizes, a library of targeted-shielded viral-mimetic nanoparticles (VMNs) with diverse surface properties was assembled. Through use of physico-chemical and biological assays, targeted-shielded VMNs with remarkably high transfection efficiency (>95%) were screened. In addition, the batch-to-batch variability of the assembled targeted-shielded VMNs in terms of uniformity and efficiency were examined and in both cases the coefficient of variation was calculated to be below 20%, indicating a highly reproducible and uniform system. Our results provide design parameters for engineering uniform targeted-shielded VMNs with very high cell transfection rate that exhibit the important characteristics for in vivo translation. These design parameters and principles could be used to tailor-make and assemble targeted-shielded VMNs that could deliver any nucleic acid payload to any mammalian cell type.
Keywords: Gene delivery, Nanomedicine, viral mimetic, nanoparticles, cancer gene therapy
Graphical Abstract
1. Introduction
In cancer gene therapy, for nucleic acid based drugs such as siRNA and plasmid DNA (pDNA) to reach their target sites, they must overcome several extracellular as well as intracellular barriers. In attempts to overcome these obstacles, multifunctional polymeric and lipid-based non-viral gene delivery systems (vectors) have been developed. These vectors are designed to protect nucleic acids from endonucleases by condensation into nanosize carriers, exploit the leakiness of tumor vessels and facilitate their accumulation in the tumor environment, enhance their internalization into the cancer cells through use of targeting ligands and ultimately mediate efficient gene expression or knockdown. In order to extend the half-life and blood circulation time, the surfaces of gene carriers are usually decorated with highly hydrophilic polymers such as polyethylene glycol (PEG). The role of PEG is to sterically stabilize the surface of the nanoparticles, minimize interaction with plasma proteins (opsonization) and enhance the probability for accumulation in tumors via enhanced permeation and retention (EPR) effect.[1, 2] PEG helps to achieve this goal by reducing the surface positive charge of the nanoparticles; thereby, minimizing the interaction with negatively charged blood components such as albumin and erythrocytes. Consequently, clearance by the mononuclear phagocyte system is significantly reduced.[3] Because the target sites of the nucleic acid delivery systems (e.g., siRNA and pDNA) are inside the cells, mere accumulation in the tumor environment is not enough and it is essential for the nanoparticles to be internalized by the cells. For this purpose, the nanoparticles also need to be equipped with targeting ligands (e.g., antibodies).[4] Therefore, an optimum balance of PEG to targeting ligand must be achieved on the surface of nanoparticles in order to reach maximum shielding without compromising internalization activity.
A number of PEGylated (shielded but non-targeted) drug delivery systems for small molecules (e.g., Doxil™, Oncaspar™) have reached the clinic, but no shielded and targeted delivery system for nucleic acid-based drugs has yielded such success. In fact, to date only one PEGylated and targeted formulation has reached Phase I clinical trials, namely CALAA-01.[5, 6] One of the major reasons that has hampered the translation of nanotechnology-based cancer therapy into the clinic is the lack of delivery systems (vector) that are not only clinically safe and efficient but from manufacturing standpoint cost-effective and compliant with criteria for batch-to-batch uniformity.[7] A critically important consideration with regard to the design of PEGylated-targeted nanomedicines relates to the fact that many promising nanomedicines reported in the literature are multi-component and quite complex, and therefore difficult to synthesize and standardize by the pharmaceutical industry. Production of these multi-component systems involves several synthetic, purification and assembly steps. This increases the costs, complexity and batch-to-batch variation of such constructs and as a result significantly decreases their commercial attractiveness and clinical application.[8] Recently, during the Image-Guided Drug Delivery Summit organized by the National Institutes of Health (USA) the importance of simplicity in nanoparticle engineering and assembly process and batch-to-batch variation in relation to a final clinical product was notably highlighted as critical by a panel of scientists and physicians from industry and academia (Supplementary Figure 1).[9]
Our lab in the past few years has demonstrated the possibility of creating targeted multifunctional (multicomponent) vectors in one step by using genetic engineering techniques. In this approach, the need for multiple conjugation/purification steps has been eliminated and the number of variables that needs to be optimized in structure/activity correlation studies reduced.[10–12] Utilizing this know-how, the objective of this research was to systematically engineer highly efficient targeted-shielded viral-mimetic nanoparticles (VMNs) with well-defined surface properties and uniform structure that can be made through an uncomplicated self-assembling process. To achieve the objective, we first genetically engineered a single chain multifunctional biopolymer that could overcome intracellular barriers by providing DNA condensation and internalization, endosome membrane disruption, nuclear localization and efficient gene expression. To overcome extracellular barriers and provide shielding, we then synthesized PEGylated histone H2A and adenovirus Mu peptides using a solid phase peptide synthesis approach. A library of VMNs with diverse physico-chemical properties was constructed as a result of complexation of pDNA with the multifunctional biopolymer in combination with PEGylated histone H2A or adenovirus Mu peptides. Through use of a series of physico-chemical and biological assays, the VMN library was screened in order to identify the constructs that are highly efficient, shielded, stable, bear an almost neutral surface charge and can be assembled in a reproducible fashion.
2. Results and Discussion
Numerous publications have previously explained the advantages of using recombinant techniques to synthesize fusion biopolymers allowing the production of biomacromolecules in a cost-effective manner. [13–15] In comparison to viral vectors and in terms of production costs, the recombinant biopolymers can be produced far cheaper than their viral counterparts. In addition, given the fact that multifunctional biopolymers can be synthesized/purified in one single step and that there is no need for the removal of toxic solvents or un-reacted monomers, such recombinant multifunctional biopolymers could be just as, if not more cost-effective than their synthetic counterparts.[13]
To overcome the intracellular barriers, we genetically engineered a single chain multifunctional fusion biopolymer (vector) composed of a pH responsive fusogenic peptide (G), four repeating units of Histone H2A with an inherent nuclear localization signal (H) and a human epidermal growth factor receptor 2 (HER2) targeting affibody (T) (Figure 1A and B). Affibodies are small antibody mimetics composed of a three-helix bundle based on the scaffold of one of the IgG-binding domains of Protein A.[16, 17] For simplicity, we refer to this vector as THG.
Figure 1.

A) Schematic representation of THG recombinant vector composed of GALA (fusogenic peptide), four repeating units of histone H2A with an inherent nuclear localization signal (4HP/NLS) and a HER2 targeting motif (TM). The 3-D structures of TM and Histone H2A are predicted using SWISS-MODEL program. B) The amino acid sequences of TM (red), 4HP/NLS (blue), and GALA (purple) in the THG vector. C) Particle size and charge analysis of THG in complex with pEGFP at different N:P ratios. The data are shown as mean±s.d. (n=3).
The major reason behind combining all four functional motifs into a single chain vector rather than four separate ones was to drastically reduce the number of variables that needs to be optimized in structure/activity correlation studies. As a result, the development process could be achieved in a shorter period of time. In a series of mechanistic studies, we previously demonstrated that all motifs in the THG vector are functional and it could efficiently transfect SKOV-3 HER2+ cancer cells.[12] As the THG vector could efficiently overcome the intracellular barriers, we used it as a base to formulate highly efficient targeted-shielded VMNs that could also overcome the extracellular barriers.
Firstly, the gene coding for THG was designed and optimized to be synthesized in an E.coli expression system. Western blot analysis and SDS-PAGE confirmed the expression and purity of THG protein (>95%) after Ni-NTA affinity chromatography (Supplementary Figure 2). In the next step, the THG vector was desalted and used to complex with pDNA (pEGFP) to form targeted VMNs which were then characterized in terms of hydrodynamic particle size and charge. The results of this study showed that the sizes of the VMNs formed at N:P ratio of 2 or higher are below 100 nm and not statistically different from each other (p<0.05) (Figure 1 C). The desalting step is highly critical because it helps to remove the ions from the system and stabilize the particles’ sizes by minimizing the potential for salt bridge formation in between nanoparticles and ensuing aggregation during storage time. The results of the zeta potential study revealed that the VMNs surface charge increased to a stable ca. +20mV at N:P ratios 2 or higher (Figure 1C).
To stabilize and reduce the surface charge of targeted VMNs and incorporate shielding, we mixed the THG vector with two different PEGylated peptides (i.e., H2A and Mu). These two peptides were purposely chosen because their efficient DNA condensation capabilities have previously been examined and reported.[18, 19] PEG2K and PEG5K were covalently attached to H2A and Mu peptides to generate H2A-PEG2K, H2A-PEG5K, Mu-PEG2K and Mu-PEG5K. These peptides were used in combination with THG at various ratios to complex with pEGFP and form PEGylated-targeted VMNs (Figure 2). A series of physico-chemical and biological assays were then performed to characterize the assembled VMNs as explained below.
Figure 2.

Schematics of PEGylated-targeted VMNs prepared through mixing of THG with H2A-PEG2K, H2A-PEG5K, Mu-PEG2K, or Mu-PEG5K followed by complexation with pDNA (pEGFP).
Firstly, we characterized the nanoparticles formed as a result of complexation of PEGylated peptides (e.g., H2A-PEG2K, H2A-PEG5K, etc.) and pDNA at various N:P ratios to evaluate their DNA condensation ability and particle surface charge. The results of the complexation studies showed that H2A-PEG2K, H2A-PEG5K, Mu-PEG2K and Mu-PEG5K in complexation with pEGFP can form nanoparticles with sizes of less than 100nm and with almost neutral surface charges (Supplementary Figure 3). At high N:P ratios (e.g., 10 or 12) where pDNA is fully condensed, the peptides that were PEGylated with PEG2K had on average significantly smaller sizes (ca. 20nm) than peptides with PEG5K. This was expected as PEG5K has a larger molecular weight than PEG2K and could attribute to the increase in hydrodynamic particle radius. Furthermore, it could be observed that the presence of PEG did not interfere with the electrostatic interactions between peptides and pDNA and the nanoparticle formation process. In terms of zeta potential, the surface charge neutrality of these PEGylated nanoparticles is most likely related to the presence of PEG on the nanoparticles surface. Overall, the nanoparticle surface charge studies show that at one end of the spectrum the targeted non-PEGylated nanoparticles (THG/pEGFP) have a surface charge of ca. +20mV (Figure 1C) while at the other end the PEGylated non-targeted nanoparticles (Supplementary Figure 3) had surface charges of almost zero.
To prepare targeted-shielded VMNs, various amounts of THG were mixed with PEGylated peptides at N:P ratios of 8, 10 and 12. These ratios were selected because this was the range that both targeted nanoparticles and PEGylated nanoparticles displayed maximum pDNA condensation. The results of the particle size and charge characterization study for targeted-PEGylated VMNs revealed that as the PEG content increased and THG content decreased, the nanoparticles surface charge gradually decreased from ca. +20mV to −2mV (Supplementary Figures 4 and 5). This indicated that the PEG molecules could present themselves on the nanoparticles surface thereby reducing the VMNs surface charge. Overall, all the prepared nanoparticles had sizes between 40–100nm which suggests that the presence of PEG2K and PEG5K in the nanoparticles did not interrupt the complexation process and all combinations were efficient in pDNA condensation. Nanoparticles in this size range (<150nm) are known to be suitable for receptor mediated endocytosis as they can fit into clathrin-coated vesicles.[20]
Having a library of nanoparticles with known surface properties at our disposal, we investigated the ability of these PEGylated-targeted VMNs to internalize and transfect SKOV-3 cancer cells. This cell line was used as a model HER2+ mammalian cell line for this study because we have previously demonstrated that the affibody in the THG structure could recognize the HER2 on the surface of the cells and internalize (Supplementary Figure 6).[12] In order to find the formulations of VMNs with the highest transfection efficiency, first we used THG/pEGFP nanoparticles at N:P ratios 1 to 12 to transfect cells. This was to determine the ratio at which the nanoparticles exhibit the maximum transfection efficiency. The results of the transfection studies showed that THG/pEGFP nanoparticles had maximum efficiency at N:P ratios of 8, 10 and 12 (Figure 3).
Figure 3.

Evaluation of transfection efficiency of THG/pEGFP nanoparticles at N:P ratios of 1 to 12. A) Flowcytometry graphs showing the percentage of GFP positive cells. Each graph is an overlay of three independent repeats. B) Fluorescent microscopy images of the transfected SKOV-3 cells illustrating the extent of gene expression. C) A bar chart that quantitatively demonstrates the percentage of transfected cells and total green fluorescent protein expression. The data are shown as mean±s.d. (n=3).
To examine the effect of PEG content in PEGylated-targeted VMNs on transfection efficiency, all the formulations of PEGylated-targeted VMNs that were prepared at N:P ratios of 8, 10 and 12 used to transfect SKOV-3 cells. For example, for N:P 8, all THG/PEGylated peptides combinations at weight/weight ratios of 12:0, 10:2, 8:4, 6:6, 4:8, 2:10 and 0:12 were examined. The weight/weight ratio of 12:0 indicates 12μg of THG and 0μg of PEGylated peptide (e.g., H2A-PEG2K or Mu-PEG5K, etc.), whereas weight/weight ratio of 0:12 indicates 0μg of THG and 12μg of PEGylated peptide. The general pattern of transfection efficiency demonstrated that particles with higher or equal amounts of THG to PEGylated peptide had higher transfection efficiency (weight/weight combinations of 10:2, 8:4, 6:6) in comparison to the nanoparticles with lower amount of THG (4:8, 2:10, 0:12), (Figure 4A–D).
Figure 4.

(A–D) Percentage of GFP-positive cells and total green fluorescent protein expression (TFI) for SKOV-3 cells transfected with vector/pEGFP complexes at various N:P ratios and in different combinations. The data are shown as mean±s.d. (n=10).
The impact of PEG content was to an extent that none of the PEGylated nanoparticles (non-targeted) were able to transfect SKOV-3 cells. This is most likely due to the presence of PEG on nanoparticles surface which inhibits interaction between cell membranes and nanoparticles. Similar observations have previously been reported by other groups.[21] In contrast, a remarkably high rate of transfection efficiency (>%95) was observed with THG/Mu-PEG5K (8:8) at N:P ratio of 12. We believe that at this particular ratio the number of targeting peptides exposed on the nanoparticle surface is at the optimum density providing the ideal opportunity for the nanoparticles to interact with cells very effectively and internalize. It has previously been shown that there are an optimum number of ligands that should be present on the surface of each nanoparticle to achieve maximum efficiency.[22] Increasing ligand number yields a corresponding increase in receptor binding associated with increased avidity. However, above a certain level, this increased avidity could result in increased receptor down regulation due to endosomal sorting to lysosomes for degradation, resulting in a decreased number of receptors available for binding the vector. This concept has been elegantly shown in a study by Wagner et al. (1990) using transferrin conjugated onto polylysine.[23]
To examine whether formulation cytotoxicity was the cause of the low transfection in the cells, for example with THG/Mu-PEG5K (4:12) in comparison to THG/Mu-PEG5K (8:8), a cell toxicity assay was performed. This study was carried out on some of the most efficient nanoparticles prepared as a result of complexation of THG/Mu-PEG5K or THG/H2A-PEG2K with pEGFP. The results of the cell toxicity assay illustrated that none of the tested formulations were toxic (p<0.05); and therefore, transfection efficiency was not negatively affected by the PEGylated-targeted VMN formulations (Figure 5). Previous studies with THG also showed that the vector itself or complexed with pDNA had no significant impact on cellular viability most likely due to its biodegradability and low surface positive charge.[12, 24]
Figure 5.

Evaluation of the viability of SKOV-3 cells after transfection with targeted and PEGylated- targeted VMNs. The data are shown as mean±s.d. (n=3).
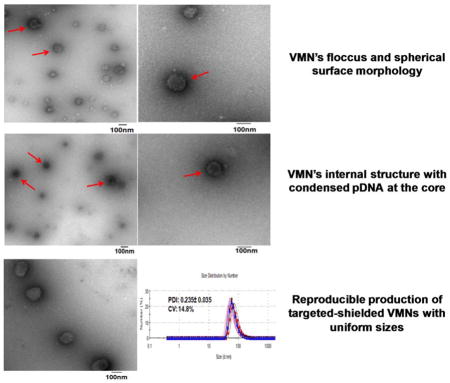
The remarkably high efficiency of the THG/Mu-PEG5K (8:8) VMNs formed at N:P 12 prompted us to characterize them further in terms of shape and surface morphology and examine particle size uniformity. We adapted a previously published method for transmission electron microscopy (TEM) of viruses in order to study the surface morphology and internal structures of our formulated VMNs.[25] Brief staining was used to observe the surface morphology of the particles and the results showed that the prepared VMNs are somewhat floccus, uniform in size and spherical (Figure 6A). To investigate internal structures, the staining time was extended which helped to visualize the packaged pDNA inside VMNs. In this case, the condensed pDNA could be observed at the nanoparticle’s core (Figure 6B).
Figure 6.


Transmission electron microscopy of the negatively stained targeted nanoparticles, PEGylated-targeted nanoparticles, and poxvirus. A) THG/pEGFP targeted nanoparticles stained briefly to emphasize on the representation of the surface structures. B) THG/pEGFP targeted nanoparticles with long staining times revealing internal structures. C) right panel: PEGylated-targeted VMNs formed as a result of complexation of THG/Mu-PEG5K (8:8) with pEGFP. left panel: poxvirus surface structure revealed as a result of negative staining. In this panel, a latex bead with a diameter of 100nm is localized close to poxvirus. The poxvirus image is adapted with permission from reference 25.
For comparison’s sake, we searched the literature to find clear TEM images of a model virus and compare with our VMNs. The results of the side-by-side shape comparison study illustrates that the surface morphology and internal structure of the formulated nanoparticles (Figure 6A–B and 6C right panel) are similar to poxvirus (Figure 6C, left panel). Overall, these observations show that the nanoparticle formation process could produce uniform, highly compact and spherical nanoparticles with similar morphology to viruses.
As mentioned in the introduction, apart from efficiency, a formulator needs to demonstrate that the formulation can be prepared in a reproducible fashion. To measure reproducibility (batch-to-batch uniformity) usually ten independent batches of each formulation is prepared in order to determine the coefficient of variation for polydispersity index (PDI). Given that particle size and particle size distribution directly impact upon gene transfection efficiency, production of nanoparticles with a reproducible PDI allows the formulator to obtain consistent and reliable transfection results. In addition, as no specific limit is set for the associated coefficient of variation (CV) for nanosuspensions by the FDA and it can vary from product to product (ranging from 15–25%), we used Ambisome™ as a control because it is an FDA approved nanotechnology-based product (non-PEGylated liposome) and currently available in the market. Another good control for this study would be Doxil™ (PEGylated liposome) but unfortunately do to the high costs (ca. $1500/vial) and serious shortage in the market this product was not used. To demonstrate reproducibility in VMN assembly process, we prepared ten independent batches of the VMNs through complexation of pEGFP with THG/Mu-PEG5K (8:8) or THG and evaluated the polydispersity index (PDI) and the corresponding CV. The results of the study demonstrated that the assembly process is reproducible because the coefficient of variation remained below 20% (Figure 7). As expected, Ambisome™ met the FDA requirements for batch-to-batch uniformity. It is worth emphasizing that for nanoparticles to be FDA approved they do not have to be perfect spheres or all of the same size. The nanoparticle formation process that is able to produce VMNs with statistically similar size distributions and efficacy is the critical factor. The results of our transfection studies (Figure 4), for example with THG/Mu-PEG5K (8:8) at N:P 12, also shows reproducibility in efficiency as the CV for ten repeats (n=10) remained below 20%.
Figure 7.

Polydispersity index analysis and measurement of coefficient of variation. A) Particle size distribution of THG/pEGFP at N:P 12. The data are shown as mean±s.d. (n=10). B) Particle size distribution analysis of nanoparticles formed through complexation of THG/Mu-PEG5K (8:8) at N:P 12 with pEGFP. The data are shown as mean±s.d. (n=10). C) Particle size distribution of Ambisome™. The data for Ambisome™ are shown as mean±s.d. (n=4).
To avoid the complexities and stability problems associated with the long-term storage of nanosuspension formulations (e.g., aggregation), AmBisome™ is formulated as a dry product. A pharmacist or physician can simply add sterile buffer to AmBisome™ vial and reconstitute the formulation prior to usage. Following the same approach, the targeted THG vector and shielded Mu-PEG5K can be stored in one vial while the genetic material (pDNA) can be stored in another. A pharmacist or physician can simply add sterile buffer to each vial and mix the components of the two vials by simple shaking to make ready for injection VMN suspension. As a result, the need for long-term stabilization of the nanosuspension is eliminated and the suspension needs to remain stable and unaggregated only for a short period of time before injection. To examine the VMN stability after reconstitution, a particle stability study over time was performed and the data show that the formulated VMNs are stable for at least three hours with no statistically significant increase in size (Figure 8A).
Figure 8.


Particle size stability studies. A) Stability over time of the nanoparticles prepared through complexation of THG with pEGFP and THG/Mu-PEG5K (8:8) with pEGFP. B) Particle size stability at various salt concentrations. The data are shown as mean±s.d. (n=3).
The approaches that are commonly used in preparation of physically stable suspensions fall into two categories. One is the use of a vehicle to maintain deflocculated particles in suspension and the other is to apply the principles of flocculation to produce flocs that, although they settle rapidly, are easily resuspended with a minimum of agitation. Parenteral suspensions are usually formulated as deflocculated system in order to avoid potential clogging of arteries. The results in Figure 8A indicate that the nanosuspension is a deflocculated system because no significant size increase was observed over three hour storage time.
In addition to storage time stability inside the vial before injection, the stability of the nanoparticles in the presence of salt at physiological concentrations (150mM) and resisting dissociation is also important. Salt stability study is especially important for nanoparticles that are formed predominantly through electrostatic interactions because the presence of ions could easily interfere with the vector/pDNA attractive forces and result in particle dissociation.[26] For this purpose, the impact of salt concentration on the stability of the nanoparticles formed as a result of complexation of pDNA with THG/Mu-PEG5K (N:P 12, 8:8) which had the highest rate of transfection efficiency was studied. Other vectors such as THG (N:P 12), THG/Mu-PEG5K (N:P 4:12), Mu-PEG5K and Mu-PEG2K were also studied as controls. After exposing the nanoparticles to increasing concentrations of the NaCl (up to 150mM), it was observed that the nanoparticles of Mu-PEG5K and Mu-PEG2K were not stable at salt concentrations beyond 10mM and rapidly dissociated at 50mM NaCl or higher concentrations (Figure 8B). Basically, we could not detect any particles at high salt concentrations with these two peptides. In contrast, the THG/pEGFP (N:P 12) and THG/Mu-PEG5K (8:8) nanoparticles were stable in physiological salt concentrations (i.e., 150 mM), although we observed continuous increase in particle size as salt concentration increased. This size increase in the presence of salt was expected because it is known that salt ions could form salt bridges in between nanoparticles and result in formation of loose floccules.[27] It is noteworthy that after injection into the body, the VMNs will get diluted in the blood stream and as long as they don’t dissociate, the potential for aggregation is very low. Nonetheless, there is a possibility for these VMNs to loosely interact with negatively charged plasma proteins because of their slightly positive surface charge. This prompted us to also examine the stability of the VMNs created through complexation of pEGFP with THG/Mu-PEG5K (4:12) which bears an almost neutral surface charge (Supplementary Figure 5, panel F). At this ratio, the nanoparticles contain significant amount of PEG and are expected to resist aggregation. As it can be observed in Figure 8B, these nanoparticles not only resisted aggregation but also dissociation. One factor that may have contributed to the salt stability of the THG-containing VMNs and resisting dissociation is the presence of hydrophobic residues in the THG sequence. These hydrophobic residues could contribute to the stabilization of hydrophobic pockets in the nanoparticle structure and block the penetration of water and ions into the nanoparticle core. As a result, the salt ions may have difficulty interfering with the electrostatic interactions in between pDNA and vector’s cationic residues.
Based on all the observed results, VMN formulations with different physicochemical properties are at hand which could potentially demonstrate different pharmacokinetic profiles in vivo. Although the in vitro results suggest that THG/Mu-PEG5K (8:8) at N:P ratio of 12 may be the most efficient formulation, ultimately it is in vivo studies that will determine which formulation is the most effective at reaching tumors and transfecting cancer cells. This is due to the fact that in tumor targeting, at first the targeting is achieved via the EPR effect which is a form of passive targeting.[28]
3. Conclusion
Recent discussions among scientists in the academic and pharmaceutical industry indicate that pharmaceutical biotechnology companies face a significant challenge in making multicomponent targeted and shielded nanomedicines that are cost effective, stable and compliant with batch-to-batch uniformity.[9] This non-compliance appears to stem from the excessive number of chemical conjugation steps and the limited control over the reactions involved in the attachment of various functional moieties such as targeting ligands (antibodies) and shielding motifs (PEG) and subsequent formulation development for long-term stabilization of the nanosuspensions.
Overall, our results provide design parameters for engineering uniform targeted-shielded VMNs with high cell transfection rate that exhibit the important characteristics for in vivo translation. These design parameters and principles could be used to tailor-make and assemble targeted-shielded VMNs that could deliver any nucleic acid payload to any mammalian cell type.
In an attempt to ensure biodegradability and homogeneity, all components in VMN construct are amino acid based. As we utilized recombinant DNA technology to create a multifunctional biopolymer (THG) in a single step, the need for conjugation of various biological motifs to the vector backbone in multiple steps was eliminated. Here, we tried diligently to embrace the phrase coined by Prestwich which states “embrace complexity, engineer versatility, and deliver simplicity”.[29]
4. Experimental Section
Production of THG Recombinant vector
The gene encoding the THG vector was synthesized by IDT® integrated DNA technologies with N-terminal NdeI and C-terminal XhoI restriction sites. A C-terminal 6×histidine tag was also designed in the vector sequence to facilitate purification. The THG gene was then cloned into a pET21b expression vector (Novagen®) and transformed into E.coli BL21 (DE3) pLysS. One BL21(DE3) plysS colony bearing THG-hisx6:pET21b was inoculated in 5 ml Circlegrow® medium supplemented with Carbenicillin (50 ug/ml). The culture was incubated overnight at 30°C. The day after, 5 ml overnight culture was added to 500 ml of Circlegrow® medium and was shaken to reach OD600>0.5. IPTG was added to final concentration of 0.4 mM and the incubation was continued for 6 h at 30°C. Under these conditions the THG vector is expressed in soluble form. Cells were then collected at 5000xg for 20 minutes. For the purification, 20 ml lysis buffer (5 M Urea, 1 M NaCl, 100 mM NaH2PO4, 10 mM Tris, 1% Triton X-100 and 10 mM imidazole, pH 8) per gram of cell pellet was added and the solution was stirred for 1 h followed by centrifugation at 37000 g for 1 h at 4°C. Addition of 5M urea to the lysis buffer increases the yield of the purification process by enhancing the exposure of histag to the nickle resins. The supernatant was collected and added to 1 ml Ni-NTA agarose (Qiagen) pre-equilibrated with lysis buffer. The slurry was shaken for 1 h on ice then added to the column. The column was washed with 100 ml of lysis buffer and 40 ml of wash buffer (5 M Urea, 1 M NaCl, 100 mM NaH2PO4, 10 mM Tris and 40 mM imidazole, pH 8). The protein was eluted by Elution buffer (3 M Urea, 500 mM NaCl, 100 mM NaH2PO4, 10 mM Tris and 200 mM imidazole, pH 8) and stored at −20 °C.
Desalting and preparation of THG stock solution
A working stock of THG solution was prepared by passing the purified protein through Sephadex G-25 prepacked column (PD-10, GE Heathcare). The column was first conditioned with bis-tris propane buffer (10mM, pH 7) plus NaCl (5 mM). Then the THG solution was loaded onto the column followed by washing the column with bis-tris propane buffer (10mM, pH 7) plus NaCl (5 mM). The desalted THG solution was collected and the concentration of protein was measured by Nanodrop 2000 spectrophotometer (Thermoscientific) using molecular weight (27.62 kDa) and extinction coefficient (i.e., 13980) of the peptide.
Preparation of targeted and PEGylated-targeted VMNs
To prepare VMNs, total amounts of vector needed for a given N:P ratio was calculated. To prepare targeted VMNs, predetermined amounts of THG vector in HEPES buffer (100 mM, pH 7.4) were flash mixed with 1 μg of pDNA (pEGFP) to form complexes at different N:P ratios (1 to 12) in a total volume of 100μl. Flash mixing (flash nanoprecipitation) means addition of peptide solution to pDNA solution in an instant. For example, to prepare N:P ratio of 1, 1.3 μg of THG vector was used to complex with 1μg of pEGFP.
To prepare PEGylated-targeted VMNs, first truncated Histone H2A (H2A) with the amino acid sequence of RGKQGGKARAKAKTRSSRAGLQFPVGRVHRLLRKGG and adenovirus Mu peptide with amino acid sequence of MRRAHHRRRRASHRRMRGG with >98% purity were synthesized by American Peptide Company (Sunnyvale, CA). Then, the synthesized peptides were PEGylated by using 2000 and 5000 daltons PEGs to make: H2A-PEG2K, H2A-PEG5K, Mu-PEG2K and Mu-PEG5K. The covalent conjugation of PEG to C-terminus of the Mu and H2A peptides were conducted by the American Peptide Company. A mix of THG with H2A-PEG2K, H2A-PEG5K, Mu-PEG2K and Mu-PEG5K at different weight/weight (μg/μg) ratios was prepared and complexed with pEGFP to make PEGylated-targeted VMNs. For example, at N:P 12, THG and PEGylated peptides were mixed at weight/weight ratios of 16:0, 14:2, 12:4, 10:6, 8:8, 6:10, 4:12, 2:14 and 0:16, respectively. The schematics of the method are shown in Figure 2.
Particle size and charge analysis and evaluation of reproducibility
Targeted and PEGylated-targeted VMNs were prepared as described above and the mean hydrodynamic particle size and zeta potential of the particles were measured at room temperature using Nano-ZS Zetasizer (Malvern Instruments, U.K). The data are presented as mean ± s.d (n=3). While for routine particle size and charge measurements the number of prepared independent batches was set at 3, for reproducibility measurements the number of samples was set at 10 (n=10). Ambisome™ vials (n=4) were kindly provided as a gift by the pharmacy store at the Cancer Institute of New Jersey (New Brunswick, NJ). To measure reproducibility, the average and standard deviation of polydispersity index (PDI) of ten samples was determined by the zetasizer and from that we calculated the coefficient of variation (CV) using the following formula: CV= standard deviation/mean × 100
Particle shape analysis by transmission electron microscopy
To study the shape of the VMNs, one drop of sample was put on a carbon type B coated copper grid (Ted Pella, USA) for 5 minutes. The sample was dried and the grid was stained for 1–3 minutes depending on the need with 1% sodium phosphotungstate solution. The grids were imaged using transmission electron microscope (1200EX electron microscope, JEOL®, USA) at RUMDNJ TEM core imaging facility. This method was adapted with slight modifications from a previously published method for imaging viruses.[25]
Particle stability over time and in the presence of salt
To measure the stability of the VMNs over time, the particle size measurements were performed every 30 minutes on each sample for 180 minutes. For particle stability studies in the presence of salt (NaCl), VMNs were prepared in HEPES buffer (pH 7.4) and considered as zero molar salt. From a 2M NaCl stock solution, aliquots were taken and added to the VMNs in HEPES buffer until the desired NaCl concentration was obtained. The particle sizes at each salt concentration was measured and reported as mean±s.d. (n=3).
Cell transfection studies
The above mentioned VMNs were used to transfect SKOV-3 ovarian cancer cells using the previously reported methods.[24] The transfection protocol is similar to what is set as a standard for transfecting cells with viruses which are targeted nanoparticles with sizes less than 100 nm. For more information please see the standard protocol of cell transfection with adenovirus from MP Biomedicals (Solon, Ohio). In brief, SKOV-3 cells were seeded in 96-well plates. Cells were transfected with vector/pEGFP complexes at various N:P ratios (equivalent of 1 μg pDNA). The green fluorescent protein (GFP) was visualized using an epifluorescent microscope to evaluate GFP gene expression. To quantify transfection efficiency, percent GFP+ cells and total green fluorescence intensity was measured using F500 Flow Cytometer (Beckman Coulter®, USA). Each time 10,000 cells were counted and the total fluorescence intensity of GFP+ cells was normalized against the total fluorescence intensity of untransfected cells (background control). The data are presented as mean ± s.d., (n=10). Percentage of GFP+ cells was determined by Kaluza flow analysis software (Beckman Coulter®, USA ) using 99% gating. Total green fluorescence intensity (TFI) which is a measure of green fluorescent protein expression was calculated using the following formula: TFI= mean fluorescence value of each GFP+ cell (measured by flowcytometer) × total number of transfected GFP+ cells.
Cell viability study
SKOV-3 cells (4 × 104 per well) were seeded in a 96-well plate and incubated in McCoy’s 5A full media. 24 hours later, 100 μl of freshly prepared vector/pEGFP was added to each well. The cells were incubated with the VMNs for 2 hours. Then the medium was replaced with fresh McCoy’s 5A media. The cells were incubated at 37°C for another 48 hours, before WST-1 cell proliferation reagent (Roche Applied Science, Indianapolis, Indiana) was added to each well. The absorbance at 450 nm was measured 2 hours after adding the reagent. Considering the viability of untreated cells as 100%, the viability of other samples was reported accordingly. The data are reported as mean ± s.d. (n =3).
Supplementary Material
Acknowledgments
This work was funded in part by the Department of Defense Cancer program (W81XWH-09-1-0303) and the startup funds from the Rutgers University to A. Hatefi; and a Royal Society International Travel Grant (2010/R3) to H.O. McCarthy.
Footnotes
Supporting Information is available on the WWW under http://www.small-journal.com
References
- 1.Chekhonin VP, Zhirkov YA, Gurina OI, Ryabukhin IA, Lebedev SV, Kashparov IA, Dmitriyeva TB. Drug Deliv. 2005;12(1):1–6. doi: 10.1080/10717540590889556. [DOI] [PubMed] [Google Scholar]
- 2.Senior J, Delgado C, Fisher D, Tilcock C, Gregoriadis G. Biochim Biophys Acta. 1991;1062(1):77–82. doi: 10.1016/0005-2736(91)90337-8. [DOI] [PubMed] [Google Scholar]
- 3.Verbaan F, van Dam I, Takakura Y, Hashida M, Hennink W, Storm G, Oussoren C. Eur J Pharm Sci. 2003;20(4–5):419–27. doi: 10.1016/j.ejps.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Gao J, Sun J, Li H, Liu W, Zhang Y, Li B, Qian W, Wang H, Chen J, Guo Y. Biomaterials. 2010;31(9):2655–64. doi: 10.1016/j.biomaterials.2009.11.112. [DOI] [PubMed] [Google Scholar]
- 5.Davis ME. Mol Pharmaceut. 2009;6(3):659–68. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 6.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Nature. 2010;464(7291):1067–70. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen TM, Cullis PR. Advanced drug delivery reviews. 2012. [DOI] [PubMed] [Google Scholar]
- 8.Lammers T, Kiessling F, Hennink WE, Storm G. Journal of controlled release: official journal of the Controlled Release Society. 2012;161(2):175–87. doi: 10.1016/j.jconrel.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 9.Tandon P, Farahani K. Cancer Res. 2011;71(2):314–7. doi: 10.1158/0008-5472.CAN-10-2629. 0008-5472.CAN-10-2629 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canine BF, Wang Y, Hatefi A. J Control Release. 2009;138(3):188–96. doi: 10.1016/j.jconrel.2009.04.017. S0168-3659(09)00233-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mangipudi SS, Canine BF, Wang Y, Hatefi A. Mol Pharm. 2009;6(4):1100–9. doi: 10.1021/mp800251x. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Mangipudi SS, Canine BF, Hatefi A. J Control Release. 2009;137:46–53. doi: 10.1016/j.jconrel.2009.03.005. S0168-3659(09)00156-4 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canine BF, Hatefi A. Adv Drug Deliv Rev. 2010;62(15):1524–9. doi: 10.1016/j.addr.2010.04.001. S0169-409X(10)00082-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghandehari H, Hatefi A. Adv Drug Deliv Rev. 2010;62(15):1403. doi: 10.1016/j.addr.2010.07.007. S0169-409X(10)00150-X [pii] [DOI] [PubMed] [Google Scholar]
- 15.Megeed Z, Ghandehari H. Genetically engineered protein-based polymers: potential in gene delivery. In: Amiji M, editor. Polymeric gene delivery: principles and applications. CRC Press; Boca Raton, FL, USA: 2005. pp. 489–507. [Google Scholar]
- 16.Nygren PA. The FEBS journal. 2008;275(11):2668–76. doi: 10.1111/j.1742-4658.2008.06438.x. [DOI] [PubMed] [Google Scholar]
- 17.Orlova A, Magnusson M, Eriksson TL, Nilsson M, Larsson B, Hoiden-Guthenberg I, Widstrom C, Carlsson J, Tolmachev V, Stahl S, Nilsson FY. Cancer Res. 2006;66(8):4339–48. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 18.Balicki D, Putnam CD, Scaria PV, Beutler E. Proc Natl Acad Sci U S A. 2002;99(11):7467–71. doi: 10.1073/pnas.102168299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keller M, Tagawa T, Preuss M, Miller AD. Biochemistry. 2002;41(2):652–9. doi: 10.1021/bi0156299. [DOI] [PubMed] [Google Scholar]
- 20.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Biochem J. 2004;377(Pt 1):159–69. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deshpande MC, Davies MC, Garnett MC, Williams PM, Armitage D, Bailey L, Vamvakaki M, Armes SP, Stolnik S. Journal of controlled release: official journal of the Controlled Release Society. 2004;97(1):143–56. doi: 10.1016/j.jconrel.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 22.Varga CM, Wickham TJ, Lauffenburger DA. Biotechnol Bioeng. 2000;70(6):593–605. doi: 10.1002/1097-0290(20001220)70:6<593::aid-bit1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 23.Wagner E, Zenke M, Cotten M, Beug H, Birnstiel ML. Proc Natl Acad Sci USA. 1990;87:3410–3414. doi: 10.1073/pnas.87.9.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Canine BF, Hatefi A. Nanomedicine. 2011;7(2):193–200. doi: 10.1016/j.nano.2010.08.003. S1549-9634(10)00246-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laue M. Methods Cell Biol. 2010;96:1–20. doi: 10.1016/S0091-679X(10)96001-9. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Servos MR, Liu J. Journal of the American Chemical Society. 2012;134(24):9910–3. doi: 10.1021/ja303787e. [DOI] [PubMed] [Google Scholar]
- 27.Fall AB, Lindstrom SB, Sundman O, Odberg L, Wagberg L. Langmuir. 2011;27(18):11332–8. doi: 10.1021/la201947x. [DOI] [PubMed] [Google Scholar]
- 28.Yokoyama M. J Artif Organs. 2005;8(2):77–84. doi: 10.1007/s10047-005-0285-0. [DOI] [PubMed] [Google Scholar]
- 29.Prestwich GD. Organogenesis. 2008;4(1):42–7. doi: 10.4161/org.6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.