
Fig. 1.
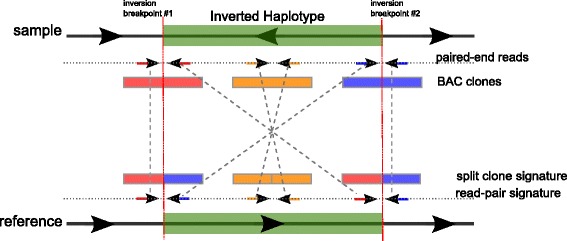
Sequence signatures used by the Valor algorithm. In the presence of an inverted haplotype in the sequenced genome, we look for both read pair and split clone signatures. Paired-end reads that span the inversion breakpoints will be mapped to the same strand with a large distance between them, instead of the concordant read pairs that map to opposing strands [1, 20]. Large insert clones will show mapping properties similar to the split read sequence signature [36], but since we do not have the full clone sequence, or sufficient coverage to assemble clones, we interrogate lengths of contiguous read mapping (Methods)