

Abstract
High mobility group box 1 (HMGB1) protein is a non-histone architectural protein that is involved in regulating many important functions in the genome, such as transcription, DNA replication, and DNA repair. HMGB1 binds to structurally distorted DNA with higher affinity than to canonical B-DNA. For example, we found that HMGB1 binds to DNA interstrand crosslinks (ICLs), which covalently link the two strands of the DNA, cause distortion of the helix, and if left unrepaired can cause cell death. Due to their cytotoxic potential, several ICL-inducing agents are currently used as chemotherapeutic agents in the clinic. While ICL-forming agents show preferences for certain base sequences (e.g., 5'-TA-3' is the preferred crosslinking site for psoralen), they largely induce DNA damage in an indiscriminate fashion. However, by covalently coupling the ICL-inducing agent to a triplex-forming oligonucleotide (TFO), which binds to DNA in a sequence-specific manner, targeted DNA damage can be achieved. Here, we use a TFO covalently conjugated on the 5' end to a 4'-hydroxymethyl-4,5',8-trimethylpsoralen (HMT) psoralen to generate a site-specific ICL on a mutation-reporter plasmid to use as a tool to study the architectural modification, processing, and repair of complex DNA lesions by HMGB1 in human cells. We describe experimental techniques to prepare TFO-directed ICLs on reporter plasmids, and to interrogate the association of HMGB1 with the TFO-directed ICLs in a cellular context using chromatin immunoprecipitation assays. In addition, we describe DNA supercoiling assays to assess specific architectural modification of the damaged DNA by measuring the amount of superhelical turns introduced on the psoralen-crosslinked plasmid by HMGB1. These techniques can be used to study the roles of other proteins involved in the processing and repair of TFO-directed ICLs or other targeted DNA damage in any cell line of interest.
Keywords: Genetics, Issue 117, triplex-forming oligonucleotide, DNA repair, DNA interstrand crosslinks, plasmid ChIP assay, 2D supercoiling assay, HMGB1
Introduction
Triplex-forming oligonucleotides (TFOs) bind duplex DNA in a sequence-specific fashion via Hoogsteen-hydrogen bonding to form triple-helical structures1-5. Triplex technology has been used to interrogate a variety of biomolecular mechanisms, such as transcription, DNA damage repair, and gene targeting (reviewed in references6-8). TFOs have been used extensively to induce site-specific damage on reporter plasmids9,10. Our lab and others have previously used a TFO, AG30, tethered to a psoralen molecule to induce site-specific DNA interstrand crosslinks (ICLs) in the supF gene on the plasmid pSupFG15,10-12. ICLs are highly cytotoxic as these lesions covalently crosslink the two DNA strands, and if left unrepaired, can block gene transcription and impede the DNA replication machinery13,14. Because of their cytotoxic potential, ICL-inducing agents have been used as chemotherapeutic drugs in the treatment of cancer and other diseases15. However, the processing and repair of ICLs in human cells is not well understood. Thus, a better understanding of the mechanisms involved in the processing of ICLs in human cells may help to improve the efficacy of ICL-based chemotherapeutic regimens. TFO-induced ICLs and their repair intermediates have the potential to cause significant structural distortions to the DNA helix. Such distortions are probable targets for architectural proteins, which bind to distorted DNA with higher affinity than to canonical B-form duplex DNA16-20. Here, we studied the association of a highly abundant architectural protein, HMGB1 with ICLs in human cells via chromatin immunoprecipitation (ChIP) assays on psoralen-crosslinked plasmids and identified a role for HMGB1 in modulating the topology of the psoralen-crosslinked plasmid DNA in human cancer cell lysates.
HMGB1 is a highly abundant and ubiquitously expressed non-histone architectural protein that binds to damaged DNA and alternatively structured DNA substrates with higher affinity than canonical B-form DNA17-20. HMGB1 is involved in several DNA metabolic processes, such as transcription, DNA replication, and DNA repair16,21-23. We have previously demonstrated that HMGB1 binds to TFO-directed ICLs in vitro with high affinity20. Further, we have demonstrated that lack of HMGB1 increased the mutagenic processing of TFO-directed ICLs and identified HMGB1 as a nucleotide excision repair (NER) co-factor23,24. Recently, we have found that HMGB1 is associated with TFO-directed ICLs in human cells and its recruitment to such lesions is dependent upon the NER protein, XPA16. Negative supercoiling of DNA has been shown to promote the efficient removal of DNA lesions by NER25, and we have found that HMGB1 induces negative supercoiling preferentially on TFO-directed ICL-containing plasmid substrates (relative to non-damaged plasmids substrates)16, providing a better understanding of the potential role(s) of HMGB1 as an NER co-factor. The processing of ICLs is not fully understood in human cells; thus, the techniques and assays developed based on the molecular tools described herein could lead to the identification of additional proteins involved in ICL repair, which in turn may serve as pharmacological targets that can be exploited to improve the efficacy of cancer chemotherapy regimens.
Here, an effective approach to assess the efficiency of TFO-directed ICL formation in plasmid DNA by denaturing agarose gel electrophoresis has been discussed. Further, using the plasmids containing the TFO-directed ICLs, techniques to determine the association of HMGB1 with ICL-damaged plasmids in a cellular context using modified ChIP assays have been described. Additionally, a facile method to study topological modifications introduced by the architectural protein HMGB1, specifically on ICL-damaged plasmid substrates in human cell lysates has been determined by performing supercoiling assays via two-dimensional agarose gel electrophoresis. The techniques described can be used to further the understanding of the involvement of DNA repair and architectural proteins in the processing of targeted DNA damage on plasmids in human cells.
We describe detailed protocols for the formation of TFO-directed site-specific psoralen ICLs on plasmid DNA, and subsequent plasmid ChIP and supercoiling assays to identify proteins that associate with the lesions, and proteins that alter the DNA topology, respectively. These assays can be modified to perform with other DNA damaging agents, TFOs, plasmid substrates, and mammalian cell lines of interest. In fact, we have shown that there is at least one potential unique and high affinity TFO-binding site within every annotated gene in the human genome26. However, for clarity, we described these techniques for the use of a specific psoralen-conjugated TFO (pAG30) on a specific mutation-reporter plasmid (pSupFG1) in human U2OS cells as we have utilized in Mukherjee & Vasquez, 201616.
Protocol
1. Preparation of TFO-directed ICLs on Plasmid Substrates
Incubate equimolar amounts of plasmid pSupFG110,11 DNA (5 µg plasmid DNA is a good starting point) with psoralen-conjugated TFO AG30 in 8 µl of triplex binding buffer [50% glycerol, 10 mM Tris (pH 7.6), 10 mM MgCl2] and dH2O to a final volume of 40 µl in an amber tube. Mix thoroughly by pipetting up and down. Incubate the reaction at 37 °C water bath for 12 hr.
Warm up the UVA lamp to ensure full power output. Place the triplex reaction on paraffin film, and place the paraffin film on ice under the UVA (365 nM) lamp. Place a Mylar filter between the lamp and the reaction.
UVA irradiate for a total dose of 1.8 J/cm2. Store the samples at 4 °C for further use. Caution: Wear proper protective eyewear and clothing with UVA irradiation.
Linearize 200 ng of the above prepared plasmid with the restriction enzyme EcoR1 in a 20 µl total volume using the manufacturer supplied 10x buffer. Heat denature the enzyme for 20 min at 65 °C. Spin the samples for 10 min at 10,000 x g and store at 4 °C.
Prepare 50x alkaline buffer [1.5 M NaOH, 50 mM Ethylenediaminetetraacetic acid (EDTA)]. Add 0.5 mg of agarose to 50 ml dH2O. Boil to dissolve the agarose and place it in a 50 °C water bath.
After the temperature of the dissolved agarose is down to 50 °C, add 1 ml of the 50x alkaline buffer, mix it and then pour the gel into the gel tray. Allow time for the gel to solidify at room temperature prior to use.
Add 4 µl of 0.5 M EDTA to the 20 µl of linearized DNA sample and incubate for 10 min at room temperature.
Add 6x alkaline gel loading buffer (300 mM NaOH, 6 mM EDTA, 18% (w/v) high molecular weight polysaccharide, 0.06% Bromocresol Green in dH2O) to the samples and to a 1 kb DNA ladder. NOTE: It is important to use 6x alkaline gel loading buffer, please see discussion.
Load samples onto the gel in the cold room and run overnight in 1x alkaline buffer (12-16 hr) at 3.2 volts per cm. Once the samples have entered the gel, place a glass plate on top of the gel to prevent it from floating.
Neutralize the samples by soaking the gel in neutralization buffer [1 M Tris-Cl (pH 7.6), 1.5 M NaCl, 3 M sodium acetate (pH 5.2)] for 45 min at room temperature.
Stain the gel for DNA with 4 µl of 1% ethidium bromide (EtBr) in 50 ml water for 1 hr at room temperature. Destain with water for 15 min. Visualize the DNA by using an imaging system. Caution: EtBr is a potent mutagen and can be absorbed through the skin. Avoid direct contact with EtBr.
2. Transfection and Immunoprecipitation of TFO-directed ICL-containing Plasmids in Human Cells
Plate 400,000 mammalian cells (e.g., U2OS osteosarcoma cells) per 60 mm dish in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) without antibiotics 24 hr before transfection. Incubate cells overnight at 37 °C with 5% CO2.
Prior to transfection, warm growth media and phosphate buffered saline (PBS) to 37 °C in a water bath. Warm the transfection reagent to room temperature.
Incubate 30 µl of transfection reagent with 500 µl of 1x growth media (mix 1) and 2 µg of TFO-directed ICL-containing plasmids in 500 µl of 1x growth media (mix 2) in separate 14 ml round bottom tubes for 10 min at room temperature.
Add mix 1 to mix 2, mix well by pipetting up and down. Incubate for 25-30 min at room temperature.
Wash cells twice with warm PBS. Add the transfection mix in a drop-wise fashion distributing evenly throughout the plate of cells. Add 1 ml of growth media to the plate and bring the final volume to 2 ml. Mix well. Place the culture plates in the 37 °C incubator with 5% CO2 for 4 hr.
Replace the media with 3 ml growth media supplemented with 10% FBS, and incubate further for 16 hr. Do not use antibiotics.
Treat cells with 80 µl of 37% fresh formaldehyde per plate to a final concentration of approximately 1% and incubate for 10 min at room temperature in the absence of light. Caution: Formaldehyde is toxic and should be handled according to its safety instructions. Formaldehyde exposure can cause harm to skin, eye and respiratory tract.
Quench formaldehyde crosslinking by adding 300 µl of chilled glycine (supplied in commercially available kits) per plate and incubating for 5 min. Remove the media and wash twice with chilled PBS, and then collect cells by scraping in 1 ml chilled (4 °C) PBS into a microcentrifuge tube. Keep the samples on ice at all times.
Prepare cell pellets by centrifuging at 4 °C for 5 min at 13,400 x g using a tabletop refrigerated centrifuge. Pipet out the supernatant and place the cell pellet on ice.
Homogeneously resuspend the pellet in 1 ml chilled (4 °C) Buffer A (supplied in commercially available kits) and incubate on ice for 10 min. Mix occasionally by inverting the tube. Pellet cells as before (see step 2.9 above).
Homogeneously resuspend the pellet in 1 ml chilled (4 °C) Buffer B (supplied in commercially available kits) and incubate on ice for 10 min with occasional mixing by inverting. Pellet cells by centrifugation as before (see step 2.9 above).
Resuspend the cells again in 200 µl chilled Buffer B. Add 2 µl of Micrococcal nuclease (MNase) and incubate at room temperature for 10 min. Mix reaction occasionally by flicking the tube. Stop the MNase reaction by placing the tube on ice and adding 40 mM EDTA. Pellet cells as before (see step 2.9 above).
Resuspend cells in 100 µl 1x chromatin immunoprecipitation buffer (ChIP) buffer (supplied in commercially available kits) supplemented with protease inhibitor cocktail.
- Place the resuspended cells in a thin-walled microfuge tube. Sonicate the samples by floating them on water bath sonicator for 20 sec followed by 20 sec incubation on ice. Repeat sonication steps 9 times to generate ~800-1,200 bp fragments.
- To 20 µl of the sonicated samples add 3 µl 5 M NaCl and 1 µl RNase A. Mix well by vortexing and incubate at 37 °C for 30 min. Subsequently, add 2 µl Proteinase K and incubate for 2 hr at 65 °C. Purify the samples using a PCR purification kit. Run samples on 1% agarose gel and visualize with EtBr. NOTE: Maximum intensity of the resulting DNA should be at or under 1,000 bp.
In three separate tubes, add 30 µl of lysate in a pre-chilled siliconized tube and then add 70 µl of chilled 1x ChIP buffer with added protease inhibitors. Add 1 µg of anti-immunoglobulin G (IgG), anti-histone H3 (as a positive control), or anti-HMGB1 antibodies to the respective tubes. Incubate overnight in a cold room with continuous rotation.
Resuspend protein G beads homogeneously in the cold room. Add 4 µl of protein G beads per tube and incubate for another 2-4 hr in the cold room with rotation.
Using a magnetic rack, pellet the beads and remove the supernatant. Add 200 µl 1x ChIP buffer mixed with protease inhibitor cocktail to the pellet and wash for 5 min in the cold room with rotation. Repeat wash twice.
Perform one additional wash with high salt 1x ChIP buffer (containing 70 mM NaCl).
Resuspend the pellets with 160 µl elution buffer (supplied in commercially available kits) and incubate at 37 °C with rotation for 30 min.
Pellet the beads and collect the supernatant in a fresh tube. Add 6 µl of 5 M NaCl and 2 µl of proteinase K, and incubate overnight at 65 °C.
Purify the DNA using a PCR product purification kit and elute the DNA using 50 µl dH2O.
Perform PCR16 reactions using primer sets to the region(s) of interest, and resolve the products on a 1% agarose gel stained with EtBr.
3. Supercoiling Assay and 2-dimensional Agarose Gel Electrophoresis
Prepare DNA repair synthesis buffer (45 mM Hepes-KOH, pH 7.8; 70 mM KCl; 7.4 mM MgCl2; 0.9 mM DTT; 0.4 mM EDTA; 2 mM ATP, 20 µM dNTPs, 3.4% glycerol and 18 µg bovine serum albumin). Store at -80 °C. Thaw on ice and prior to use add 40 mM phosphocreatine and 2.5 µg creatine phosphokinase.
Prepare 10x supercoiling buffer [500 mM Tris (pH 8.0), 500 mM NaCl, 25 mM MgCl2, 1 mM EDTA], and store at room temperature.
Linearize 300 ng of supercoiled plasmid pSupFG1 DNA completely using 1 µl Vaccinia Topoisomerase I (5-15 U/µl) with 1x supercoiling buffer in a 10 µl reaction. Incubate the mixture at 37 °C for 1 hr.
Thaw HeLa cell-free extract24 on ice. To the completely relaxed plasmids, add 25 µl HeLa cell-free extract and DNA repair synthesis buffer. Mix well. Add additional 1 µl of Vaccinia Topoisomerase I to the mix, and incubate for 1 hr at 37 °C.
Terminate the reactions by adding sodium dodecyl sulfate (SDS) (1% final concentration) and proteinase K (0.25 mg/ml final concentration). Incubate at 65 °C for 2 hr.
Cast a 1% agarose gel with 1x Tris Borate EDTA (TBE) buffer. Add DNA loading dye and load the reactions directly onto the gel at least 4 lanes apart. Run the gel for 8-10 hr at 2 V/cm in 1x TBE (dimension 1) at room temperature to resolve the topoisomers. NOTE: Prepare a 5x TBE stock solution in 1 L of dH2O by adding 54 g of Tris base. 27.5 g of boric acid and 20 ml of 0.5 M EDTA (pH 8.0).
Prepare 2 L of 1x TBE with 3 µg/ml chloroquine-phosphate. Soak the gel in 200 ml of this solution for 20 min. Cover the gel tray with aluminum foil.
To electrophorese the gel in the second dimension, turn the gel 90° in a clockwise fashion and run it for 4-6 hr at 2 V/cm in chloroquine containing 1x TBE to resolve the positive and negative supercoils.
Stain the gel with EtBr for 1 hr on a rocker. Destain the gel with dH2O for 15 min and subsequently visualize the thermal distribution of the topoisomers using an imager.
Representative Results
Formation of the TFO-directed ICL is critical for the plasmid based assays, which are used to interrogate the roles of architectural proteins in ICL processing in human cells. Denaturing agarose gel electrophoresis is a facile way to determine the efficiency of TFO-directed ICL formation. The plasmids harboring TFO-directed ICLs migrate with a slower mobility through the agarose gel matrix (Figure 1A, lane 3) when compared to the un-crosslinked control plasmids (Figure 1A, lane 2). Densitometric quantification of the bands in lanes 2 and 3 (Figure 1A) reveals approximately 70% of the plasmid population migrates through the gel with slowed mobility (identified by a black arrow), indicating TFO-directed ICL formation on those plasmids.
The chromatin immunoprecipitation assays performed on extracts from human U2OS cells transfected with either control plasmid (P) or plasmids containing TFO-directed ICLs (P-ICL) show enrichment of HMGB1 at the P-ICL region compared to the control (P) region, when immunoprecipitated with an anti-HMGB1 antibody (Figure 2, top panel, lanes 2 and 3). These results indicate that HMGB1 associates with the ICLs in human cells. When HMGB1 was depleted from the cells via siRNA treatment, the P-ICL-specific enrichment was diminished (Figure 2, top panel, lanes 4 and 5), as expected. When the same samples were immunoprecipitated using an anti-IgG antibody as an antibody specificity control, no PCR amplification was detected, as expected (Figure 2, top panel, Lanes 6-10). Lanes 10-14 (top panel) in Figure 2 were input samples used for normalization. The top panel shows PCR amplification by the primers near the TFO-directed ICL site and the bottom panel shows PCR amplification when primers distal to the TFO-directed ICL site were used.
HMGB1 is an architectural protein, which binds to distorted DNA with higher affinity than it binds undamaged DNA. The DNA supercoiling assays resolved through two-dimensional agarose gel electrophoresis demonstrate architectural modification of the TFO-directed ICL-containing plasmids (P-ICL) compared to control plasmids (P) by HMGB1 in HeLa cell extracts (Figure 3). The architectural modification induced by HMGB1 preferably on P-ICL-containing substrates is detected as increase in negative supercoiling. Supercoiled plasmids migrate faster through agarose gels relative to relaxed or linear plasmids. The distribution of the topoisomers indicates more supercoiling of the P-ICLs compared to P in the presence of HMBG1 (Figure 3). Negatively supercoiled DNA runs as left-handed arc in the second dimension in the presence of chloroquine. The supercoiling induced by HMGB1 on P-ICL seems to consist mostly of topoisomers that form a left-handed arc (~14 topoisomers were identified on the TFO-directed ICL containing plasmids compared to ~7 topoisomers from the control plasmid), indicating formation of negative supercoils. Altogether, these data suggest that plasmids with TFO-directed ICLs can be used as a tool to study the high-affinity association of the architectural protein HMGB1 with DNA lesions in human cells through chromatin immunoprecipitation assays. In addition, specific architectural modifications of the P-ICL substrates by HMGB1 can also be studied using supercoiling assays and two-dimensional agarose gels.
 Figure 1:Alkaline agarose gel electrophoresis assay to quantify TFO-directed ICL formation efficiency on the plasmid pSupFG1. (A) Lane 1, 1 kb DNA ladder; lane 2, plasmid pSupFG1 (P) used as a control; and lane 3, TFO-directed ICL-containing pSupFG1 (P-ICL). The plasmids harboring ICLs migrate more slowly in the gel as demonstrated in lane 3 (indicated by the black arrow on the right hand side of the gel). (B) Densitometric quantification of the bands in lane 2 and lane 3 indicate that approximately 70% of the plasmids are crosslinked. This figure has been modified from reference16. Please click here to view a larger version of this figure.
Figure 1:Alkaline agarose gel electrophoresis assay to quantify TFO-directed ICL formation efficiency on the plasmid pSupFG1. (A) Lane 1, 1 kb DNA ladder; lane 2, plasmid pSupFG1 (P) used as a control; and lane 3, TFO-directed ICL-containing pSupFG1 (P-ICL). The plasmids harboring ICLs migrate more slowly in the gel as demonstrated in lane 3 (indicated by the black arrow on the right hand side of the gel). (B) Densitometric quantification of the bands in lane 2 and lane 3 indicate that approximately 70% of the plasmids are crosslinked. This figure has been modified from reference16. Please click here to view a larger version of this figure.
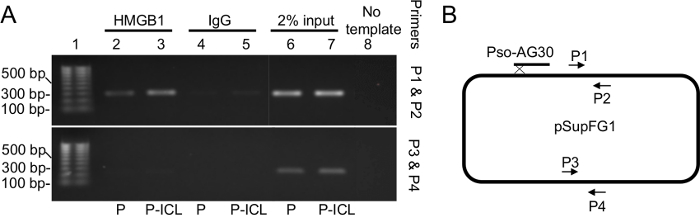 Figure 2: ChIP assay demonstrating enrichment of HMGB1 on the TFO-directed ICL-containing region of the crosslinked plasmid. (A)PCR amplification of the immunoprecipitated fragments were resolved on a 1% agarose gel using a set of proximal primers near the TFO-directed ICL site (B; P1 and P2, top panel) and a second set of primers used as a specificity control that were further (approximately 2,000 bp) from the site-directed ICL (B; P3 and P4, bottom panel). Lanes 2 and 3 indicate enrichment of HMGB1 near the TFO-directed ICL (lane 3) relative to undamaged DNA (lane 2). Lanes 4 and 5 are antibody specificity controls using an IgG antibody. Lanes 6 and 7 are the input samples. Lane 8 is a negative control for the PCR reaction and contains no DNA template. P, plasmid pSupFG1. P-ICL, TFO-directed ICL-containing plasmid pSupFG1. This figure has been modified from reference 16. (B) A schematic diagram of the plasmid pSupFG1 showing the primer sites for P1 and P2 as well as P3 and P4. Please click here to view a larger version of this figure.
Figure 2: ChIP assay demonstrating enrichment of HMGB1 on the TFO-directed ICL-containing region of the crosslinked plasmid. (A)PCR amplification of the immunoprecipitated fragments were resolved on a 1% agarose gel using a set of proximal primers near the TFO-directed ICL site (B; P1 and P2, top panel) and a second set of primers used as a specificity control that were further (approximately 2,000 bp) from the site-directed ICL (B; P3 and P4, bottom panel). Lanes 2 and 3 indicate enrichment of HMGB1 near the TFO-directed ICL (lane 3) relative to undamaged DNA (lane 2). Lanes 4 and 5 are antibody specificity controls using an IgG antibody. Lanes 6 and 7 are the input samples. Lane 8 is a negative control for the PCR reaction and contains no DNA template. P, plasmid pSupFG1. P-ICL, TFO-directed ICL-containing plasmid pSupFG1. This figure has been modified from reference 16. (B) A schematic diagram of the plasmid pSupFG1 showing the primer sites for P1 and P2 as well as P3 and P4. Please click here to view a larger version of this figure.
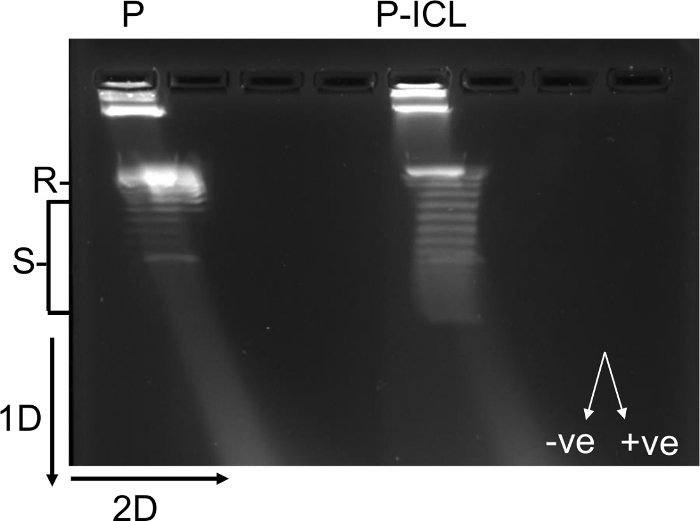 Figure 3: 2D agarose gel electrophoresis showing formation of negative supercoils in TFO-directed ICL-containing plasmids, facilitated by HMGB1. The 1x TBE agarose gel reveals the thermal distribution of the topoisomers generated by the plasmid alone (P) or the psoralen-crosslinked plasmid (P-ICL). The mobility of the supercoiled species is faster compared to the relaxed plasmids. Each band represents a topoisomer that differs from the band below or above by one less or more superhelical turn, respectively. The left-handed arc represents negatively supercoiled (-ve) species and the right-handed arc represents positively supercoiled (+ve) species. The psoralen ICL-containing plasmid population (P-ICL) contains more supercoiled species than the control plasmids (P). R, relaxed plasmids; S, supercoiled plasmids; 1D, first dimension of the gel electrophoresis; 2D, second dimension of the gel electrophoresis. This figure has been modified from16. Please click here to view a larger version of this figure.
Figure 3: 2D agarose gel electrophoresis showing formation of negative supercoils in TFO-directed ICL-containing plasmids, facilitated by HMGB1. The 1x TBE agarose gel reveals the thermal distribution of the topoisomers generated by the plasmid alone (P) or the psoralen-crosslinked plasmid (P-ICL). The mobility of the supercoiled species is faster compared to the relaxed plasmids. Each band represents a topoisomer that differs from the band below or above by one less or more superhelical turn, respectively. The left-handed arc represents negatively supercoiled (-ve) species and the right-handed arc represents positively supercoiled (+ve) species. The psoralen ICL-containing plasmid population (P-ICL) contains more supercoiled species than the control plasmids (P). R, relaxed plasmids; S, supercoiled plasmids; 1D, first dimension of the gel electrophoresis; 2D, second dimension of the gel electrophoresis. This figure has been modified from16. Please click here to view a larger version of this figure.
Discussion
The efficient formation of TFO-directed ICLs is dependent upon two crucial factors: First, proper buffer components (e.g., MgCl2) and time of incubation of the TFO with its target DNA substrate (dependent upon the binding affinity of the TFO to its target duplex, and the concentrations used); and second, the proper dose of UVA (365 nm) irradiation to form psoralen crosslinks efficiently. Optimal triplex formation can be achieved by using HPLC or gel purified TFOs. Impurities contaminating the TFO may lead to unwanted crosslinked products, which can further complicate the experimental outcome. To increase the stability of the TFOs covalently attached to a 5'-HMT psoralen, the TFOs should be stored in aliquots at -80 °C, as multiple freeze thaw of the TFOs may result in degradation of the oligonucleotides. After triplex formation to target the psoralen to a specific site (e.g., 5'-TA-3' and 5'-AT-3' are the preferred psoralen crosslinking sites), psoralen ICL formation requires irradiation with UVA. A dose of 1.8 J/cm2 UVA is sufficient for optimal formation of ICLs on plasmids in vitro. The exposure time should be determined by using a photometer. Depending on the UVA lamp source, in addition to emitting UV at 365 nm, the lamp may also emit shorter wavelengths, which may lead to formation of unintended DNA damage throughout the DNA backbone. Because the TFO-directed ICL-containing plasmids will be used for studies related to DNA repair of an ICL at a specific site, such unintended photoproducts may confound the outcome of the DNA repair studies. Therefore, a Mylar filter should be used to prevent exposure of the samples to shorter wavelengths. Successful ICL formation also depends on the stability of the triplex structure during UVA irradiation. The time of UV irradiation required depends on the wattage of the UVA source. In our case, 20-30 min is required to generate the desired dose of UVA. During this time, heat generated by the lamp may cause evaporative water loss from the samples. Such water loss may alter the buffer concentration and may cause destabilization of the triplex structures. Placing the reactions on ice during UVA irradiation will help to prevent evaporation of the samples.
In order to determine the efficiency of psoralen crosslink formation on the plasmid substrates, the samples were resolved in 1% alkaline agarose gels (Figure 1). Most protocols for alkaline gel electrophoresis suggest a 2x alkaline gel loading dye, although some protocols suggest that alkaline loading dye is not necessary, as the denaturing buffer condition of the gel is sufficient to denature the samples. While a 2x dye works well with concentrated DNA samples, this protocol requires loading a large sample volume. Therefore, use a 6x alkaline gel loading dye. To ensure proper denaturation of the psoralen crosslinked plasmid substrates, incubate the samples in the loading buffer for 10 min at room temperature. In addition, Mg2+ can form Mg(OH)2 under alkaline conditions, which entraps the DNA and can cause it to precipitate. The triplex-forming reaction buffer contains Mg2+, and therefore it is important to incubate the samples with EDTA to ensure that the Mg2+ is chelated prior to adding the alkaline loading dye to the samples. After electrophoresis, it is important to neutralize the gel, as EtBr does not intercalate with the DNA under alkaline conditions.
An alternative approach to assess the efficiency of the TFO-directed crosslink formation is the use of denaturing polyacrylamide gel electrophoresis, but this technique is more laborious, and requires restriction digestion of the plasmids followed by radioactive isotope labeling of the fragments. This current approach demonstrates a relatively simple, yet effective method to determine the efficiency of TFO-directed ICL formation on plasmid substrates. However, this technique can be applied to assess the formation of ICLs by other crosslink-forming agents as well.
Successful outcome of the plasmid ChIP assay depends on a number of factors. The critical steps within the protocol and troubleshooting suggestions are discussed here. Critical steps involved in this assay, include formaldehyde crosslinking, micrococcal nuclease digestion, sonication, sample washes, and reversal of the formaldehyde protein-DNA crosslinks in the samples. For efficient protein-DNA crosslinking, it is important to use fresh formaldehyde. Stock bottles of 37% formaldehyde should not be used for more than six months after opening, as exposure to light and oxygen degrades formaldehyde. Therefore, it is also beneficial to perform the formaldehyde crosslinking without light in the cell culture hood. Another important step is micrococcal nuclease (MNase) digestion of the samples. MNase digestion not only cleaves the genomic DNA into smaller fragments but also removes any lingering plasmids that were not delivered into the cells during the transfection method, which can dramatically reduce the background. The incubation time of MNase digestion should be empirically determined. Longer digestion may result in loss of samples, whereas shorter incubation periods may lead to inefficient removal of unwanted DNA. Excellent results can be seen after 10 min incubation at room temperature with occasional mixing of the reactions by inverting the tubes. For efficient immunoprecipitation, the DNA should be fragmented to sizes between 800-1,200 bp. Such fragmentation of DNA can be achieved by thorough sonication of the samples. Efficient sonication of samples can be achieved by re-suspending the pellets in thin-walled PCR tubes and subsequently placing them in a water bath sonicator. Sonic pulses are more effective compared to continuous sonication. In addition, it is important to place the samples on ice between pulses to prevent protein degradation, and it is also important to supplement the 1x ChIP buffer with protease inhibitors, as stable proteins are necessary for the antibody recognition and subsequent precipitation steps. However, during the immunoprecipitation process, due to the hydrophobic nature of the magnetic beads, non-specific DNA will be pulled down. To reduce signals from such non-specific templates, rigorous washing is absolutely necessary. Lastly, proper reversal of the protein-DNA crosslinks of the samples is necessary for PCR amplification of the immunoprecipitated templates. To achieve optimal results, samples should be incubated for at least 12 hr with SDS and proteinase K. There are benefits of using a plasmid-based ChIP assay over genomic ChIP assays. For example, targeted DNA damage can be easily achieved using a plasmid DNA substrate and a TFO, compared to TFO-targeted lesion formation in a genomic locus. The amount of substrate can also be controlled to modulate the intensity of signals when using a TFO-targeted damaged plasmid. Further, such substrates can be used in any cell line of choice and tested for association with various candidate proteins, thus extending the scope of studies on processing and repair of ICLs.
The supercoiling assay is based on the difference in shape between linear and supercoiled plasmid DNA. Supercoiled DNA plasmids are more compact and migrate faster through the gel matrix compared to relaxed plasmids. The critical steps within the protocol and troubleshooting suggestions are discussed here. Because architectural proteins facilitate topological modifications on damaged DNA substrates, measured through induction of supercoiling of the relaxed plasmids, it is critical to completely relax the plasmids with Topoisomerase I. It is necessary to examine the extent of DNA relaxation caused by Topoisomerase I via agarose gel electrophoresis. The MgCl2 present in the supercoiling buffer can precipitate over time, and the presence of MgCl2 affects the thermal distribution of the positive and negative supercoils by facilitating the formation of positive supercoils. Therefore, it is beneficial to add MgCl2 separately to the mix or prepare fresh buffer each time. To separate the topoisomers of the plasmid populations, it is important to perform the gel electrophoresis at a very low voltage (~2 V/cm) so that migration of the DNA is predominantly based on the structure/shape of the molecules. If relaxation of the plasmids by Topoisomerase I is not complete, then the incubation time and/or the amount of enzyme used should be increased. Complete relaxation has to be ensured before moving on to the subsequent DNA supercoiling step because use of the supercoiling assay with incompletely relaxed plasmids may lead to misinterpretation of the data and the results may be difficult to replicate. Once complete relaxation has been achieved, the supercoiling assay is relatively easy to perform. One important component of this assay is the use of a DNA repair synthesis buffer. The phosphocreatine and the creatine phosphokinase are required to be added to the mix each time to generate ATP. Subsequent to the supercoiling step, it is important to remove proteins from the plasmid DNA by incubating with SDS and proteinase K. SDS assists in dissociating the proteins from the DNA and proteinase K degrades the protein, which leaves the DNA intact with various levels of supercoiling. If the protein is not removed completely, the thermal distribution of the topoisomers will not be clearly visible. The DNA cannot be purified by phenol:chloroform:isoamyl alcohol and ethanol precipitated because this method (and others) can nick the DNA, resulting in the loss of supercoiling. Another important step is addition of chloroquine to the buffer. Chloroquine is an intercalating agent and unwinds the DNA. Upon intercalation, it relaxes the negatively supercoiled DNA and introduces positive supercoiling to relaxed DNA which facilitates separation of the topoisomers containing a high density of supercoiling. Thus positive and negative supercoils can be differentiated. This is generally a facile experiment and is highly reproducible when all points above are considered.
However, if no supercoiling is obvious, then it is necessary to determine the activity of the protein of interest. The 2D supercoiling assay has been used to study topological modifications facilitated by architectural proteins27. This assay has been used here in the context of DNA damage processing of TFO-directed ICLs in human cells. HMGB1 binds to TFO-directed ICLs with high affinity and specificity, as previously demonstrated via in vitro gel electrophoretic mobility-shift assays20 and in human cells using the plasmid ChIP assay described within16. Here, using the 2D supercoiling assay it has been demonstrated that upon binding to the TFO-directed ICLs in HeLa cell-free extracts, HMGB1 assists in the formation of negative supercoiling on the substrates (Figure 3), which may be an important step in repair of the lesion because negative supercoiling is known to facilitate the removal of DNA damage via the NER pathway25. There are many architectural proteins that have been implicated in DNA damage processing that could be studied using this assay. This approach is limited to in vitro studies, nevertheless, it is a simple and useful assay to interrogate the structure-function relationship of architectural proteins in the context of DNA damage repair. However, the ChIP protocol can be easily modified to study DNA-protein interactions in the context of the genome, while assessing the effects on DNA supercoiling by such interactions may prove to be quite difficult. We and other groups have indeed performed triplex-based studies 1) by stable transfection of the plasmid DNA into the genome28-30 2) by targeting a genomic locus31-33 and 3) by incorporating the psoralen-modified TFO (and UVA irradiation) into cells after plasmid transfection10.
Disclosures
The authors declare no conflict of interest.
Acknowledgments
The authors would like to thank the members of Vasquez laboratory for helpful discussions. This work was supported by the National Institutes of Health/National Cancer Institute [CA097175, CA093279 to K.M.V.]; and the Cancer Prevention and Research Institute of Texas [RP101501]. Funding for open access charge: National Institutes of Health/National Cancer Institute [CA093279 to K.M.V.].
References
- Moser HE, Dervan PB. Sequence-specific cleavage of double helical DNA by triple helix formation. Science. 1987;238:645–650. doi: 10.1126/science.3118463. [DOI] [PubMed] [Google Scholar]
- Cooney M, Czernuszewicz G, Postel EH, Flint SJ, Hogan ME. Site-specific oligonucleotide binding represses transcription of the human c-myc gene in vitro. Science. 1988;241:456–459. doi: 10.1126/science.3293213. [DOI] [PubMed] [Google Scholar]
- Beal PA, Dervan PB. Second structural motif for recognition of DNA by oligonucleotide-directed triple-helix formation. Science. 1991;251:1360–1363. doi: 10.1126/science.2003222. [DOI] [PubMed] [Google Scholar]
- Vasquez KM, Dagle JM, Weeks DL, Glazer PM. Chromosome targeting at short polypurine sites by cationic triplex-forming oligonucleotides. J Biol Chem. 2001;276:38536–38541. doi: 10.1074/jbc.M101797200. [DOI] [PubMed] [Google Scholar]
- Vasquez KM, Christensen J, Li L, Finch RA, Glazer PM. Human XPA and RPA DNA repair proteins participate in specific recognition of triplex-induced helical distortions. Proc Natl Acad Sci U S A. 2002;99:5848–5853. doi: 10.1073/pnas.082193799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Vasquez KM. Triplex technology in studies of DNA damage, DNA repair, and mutagenesis. Biochimie. 2011;93:1197–1208. doi: 10.1016/j.biochi.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin JY, Glazer PM. Repair of DNA lesions associated with triplex-forming oligonucleotides. Mol Carcinog. 2009;48:389–399. doi: 10.1002/mc.20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman MM, Glazer PM. The potential for gene repair via triple helix formation. J Clin Invest. 2003;112:487–494. doi: 10.1172/JCI19552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez KM. Targeting and processing of site-specific DNA interstrand crosslinks. Environ Mol Mutagen. 2010;51:527–539. doi: 10.1002/em.20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Levy DD, Seidman MM, Glazer PM. Targeted mutagenesis in mammalian cells mediated by intracellular triple helix formation. Mol Cell Biol. 1995;15:1759–1768. doi: 10.1128/mcb.15.3.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Seidman MM, Glazer PM. Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science. 1996;271:802–805. doi: 10.1126/science.271.5250.802. [DOI] [PubMed] [Google Scholar]
- Chen Z, Xu XS, Yang J, Wang G. Defining the function of XPC protein in psoralen and cisplatin-mediated DNA repair and mutagenesis. Carcinogenesis. 2003;24:1111–1121. doi: 10.1093/carcin/bgg051. [DOI] [PubMed] [Google Scholar]
- Derheimer FA, Hicks JK, Paulsen MT, Canman CE, Ljungman M. Psoralen-induced DNA interstrand cross-links block transcription and induce p53 in an ataxia-telangiectasia and rad3-related-dependent manner. Mol Pharmacol. 2009;75:599–607. doi: 10.1124/mol.108.051698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vare D, et al. DNA interstrand crosslinks induce a potent replication block followed by formation and repair of double strand breaks in intact mammalian cells. DNA repair. 2012;11:976–985. doi: 10.1016/j.dnarep.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Huang Y, Li L. DNA crosslinking damage and cancer - a tale of friend and foe. Transl Cancer Res. 2013;2:144–154. doi: 10.3978/j.issn.2218-676X.2013.03.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Vasquez KM. HMGB1 interacts with XPA to facilitate the processing of DNA interstrand crosslinks in human cells. Nucleic Acids Res. 2016;44:1151–1160. doi: 10.1093/nar/gkv1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidney DL, Reeck GR. Purification from cultured hepatoma cells of two nonhistone chromatin proteins with preferential affinity for single-stranded DNA: apparent analogy with calf thymus HMG proteins. Biochem Biophys Res Commun. 1978;85:1211–1218. doi: 10.1016/0006-291x(78)90671-x. [DOI] [PubMed] [Google Scholar]
- Bianchi ME, Beltrame M, Paonessa G. Specific recognition of cruciform DNA by nuclear protein HMG1. Science. 1989;243:1056–1059. doi: 10.1126/science.2922595. [DOI] [PubMed] [Google Scholar]
- Jung Y, Lippard SJ. Nature of full-length HMGB1 binding to cisplatin-modified DNA. Biochemistry. 2003;42:2664–2671. doi: 10.1021/bi026972w. [DOI] [PubMed] [Google Scholar]
- Reddy MC, Christensen J, Vasquez KM. Interplay between human high mobility group protein 1 and replication protein A on psoralen-cross-linked DNA. Biochemistry. 2005;44:4188–4195. doi: 10.1021/bi047902n. [DOI] [PubMed] [Google Scholar]
- Das D, Scovell WM. The binding interaction of HMG-1 with the TATA-binding protein/TATA complex. J Biol Chem. 2001;276:32597–32605. doi: 10.1074/jbc.M011792200. [DOI] [PubMed] [Google Scholar]
- Little AJ, Corbett E, Ortega F, Schatz DG. Cooperative recruitment of HMGB1 during V(D)J recombination through interactions with RAG1 and DNA. Nucleic Acids Res. 2013;41:3289–3301. doi: 10.1093/nar/gks1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SS, Mitchell DL, Vasquez KM. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc Natl Acad Sci U S A. 2008;105:10320–10325. doi: 10.1073/pnas.0803181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SS, Reddy MC, Vasquez KM. Human HMGB1 directly facilitates interactions between nucleotide excision repair proteins on triplex-directed psoralen interstrand crosslinks. DNA repair. 2009;8:865–872. doi: 10.1016/j.dnarep.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Ahn B. Effect of DNA topology on plasmid DNA repair in vivo. FEBS letters. 2000. pp. 174–178. [DOI] [PubMed]
- Wu Q, et al. High-affinity triplex-forming oligonucleotide target sequences in mammalian genomes. Mol Carcinog. 2007;46:15–23. doi: 10.1002/mc.20261. [DOI] [PubMed] [Google Scholar]
- Sheflin LG, Spaulding SW. High mobility group protein 1 preferentially conserves torsion in negatively supercoiled DNA. Biochemistry. 1989;28:5658–5664. doi: 10.1021/bi00439a048. [DOI] [PubMed] [Google Scholar]
- Betous R, et al. Role of TLS DNA polymerases eta and kappa in processing naturally occurring structured DNA in human cells. Mol Carcinog. 2009;48:369–378. doi: 10.1002/mc.20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Macris MA, Faruqi AF, Glazer PM. High-frequency intrachromosomal gene conversion induced by triplex-forming oligonucleotides microinjected into mouse cells. Proc Natl Acad Sci U S A. 2000;97:9003–9008. doi: 10.1073/pnas.160004997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez KM, Wang G, Havre PA, Glazer PM. Chromosomal mutations induced by triplex-forming oligonucleotides in mammalian cells. Nucleic Acids Res. 1999;27:1176–1181. doi: 10.1093/nar/27.4.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri N, et al. Minimum number of 2'-O-(2-aminoethyl) residues required for gene knockout activity by triple helix forming oligonucleotides. Biochemistry. 2002;41:7716–7724. doi: 10.1021/bi025734y. [DOI] [PubMed] [Google Scholar]
- Christensen LA, Finch RA, Booker AJ, Vasquez KM. Targeting oncogenes to improve breast cancer chemotherapy. Cancer Res. 2006;66:4089–4094. doi: 10.1158/0008-5472.CAN-05-4288. [DOI] [PubMed] [Google Scholar]
- Boulware SB, et al. Triplex-forming oligonucleotides targeting c-MYC potentiate the anti-tumor activity of gemcitabine in a mouse model of human cancer. Mol Carcinog. 2014;53:744–752. doi: 10.1002/mc.22026. [DOI] [PMC free article] [PubMed] [Google Scholar]