
Figure 1. Prediction and explanation of molecular energies with a deep tensor neural network.

(a) Molecules are encoded as input for the neural network by a vector of nuclear charges and an inter-atomic distance matrix. This description is complete and invariant to rotation and translation. (b) Illustration of the network architecture. Each atom type corresponds to a vector of coefficients 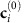 , which is repeatedly refined by interactions vij. The interactions depend on the current representation
, which is repeatedly refined by interactions vij. The interactions depend on the current representation 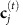 , as well as the distance Dij to an atom j. After T iterations, an energy contribution Ei is predicted for the final coefficient vector
, as well as the distance Dij to an atom j. After T iterations, an energy contribution Ei is predicted for the final coefficient vector 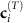 . The molecular energy E is the sum over these atomic contributions. (c) Mean absolute errors of predictions for the GDB-9 dataset of 133,885 molecules as a function of the number of atoms. The employed neural network uses two interaction passes (T=2) and 50,000 reference calculation during training. The inset shows the error of an equivalent network trained on 5,000 GDB-9 molecules with 20 or more atoms, as small molecules with 15 or less atoms are added to the training set. (d) Extract from the calculated (black) and predicted (orange) molecular dynamics trajectory of toluene. The curve on the right shows the agreement of the predicted and calculated energy distributions. (e) Energy contribution Eprobe (or local chemical potential
. The molecular energy E is the sum over these atomic contributions. (c) Mean absolute errors of predictions for the GDB-9 dataset of 133,885 molecules as a function of the number of atoms. The employed neural network uses two interaction passes (T=2) and 50,000 reference calculation during training. The inset shows the error of an equivalent network trained on 5,000 GDB-9 molecules with 20 or more atoms, as small molecules with 15 or less atoms are added to the training set. (d) Extract from the calculated (black) and predicted (orange) molecular dynamics trajectory of toluene. The curve on the right shows the agreement of the predicted and calculated energy distributions. (e) Energy contribution Eprobe (or local chemical potential  , see text) of a hydrogen test charge on a
, see text) of a hydrogen test charge on a 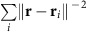 isosurface for various molecules from the GDB-9 dataset for a DTNN model with T=2.
isosurface for various molecules from the GDB-9 dataset for a DTNN model with T=2.