

Main Text
Substrate transport across the cell membrane relies on conformational changes in transport proteins that may or may not be revealed in their crystal structures. For instance, the conformational state of the outer membrane vitamin B12 transporter BtuB was shown to be influenced by the crystal environment (1), and for the bacterial homolog LeuT of neurotransmitter transporters it was shown that substrate binding does not lead to a clear switch between well-defined conformations, but only to a shift of conformational preferences (2). Such results indicate that crystallography may not necessarily reveal the full truth, even if distinct structures are obtained in the absence and presence of substrate.
A team led by Gregor Hagelueken (Glaenzer et al. (3)) investigated conformational changes in the substrate-binding domain (SBD) of the sialic acid transporter VcSiaP of Vibrio cholerae when binding its substrate. Crystal structures were known for both the substrate-free open (4) and the substrate-bound closed forms (5) of related transporters. However, it was not clear whether in solution the SBD exists in an equilibrium between the two conformations or the conformation switches strictly when the substrate is present. Whereas such information exists for substrate-binding proteins in other classes of transporters, VcSiaP belongs to the family of tripartite ATP-independent periplasmic (TRAP) transporters (6) on which much less information is available than on other transporter families. The SBDs of TRAP transporters are a structurally distinct class (7) and are involved in the capability of TRAP transporters to switch between substrate import and export (8).
As their principal technique, Glaenzer et al. (3) used the electron-paramagnetic-resonance experiment pulsed electron-electron double resonance (PELDOR), which is synonymously known as double electron-electron resonance (DEER), in conjunction with site-directed spin labeling. This technique provides the distribution of distances between spin labels on length scales that fit typical dimensions of proteins (9). Therefore, not only can agreement of a solution structure with a crystal structure be ascertained, one can also observe to what extent the conformation in solution is distributed. In particular, coexistence of several conformations is clearly revealed in the distance distributions (2).
To test conformational change with high sensitivity, the team selected labeling site pairs based on a difference distance matrix obtained from the closed- and open-form crystal structures of the Haemophilus influenzae homolog HiSiaP. They then superimposed the rigid domains from the substrate-free structure of VcSiaP (10) onto the closed-form structure of HiSiaP and predicted the distance distributions between the spin labels for both forms using software that Gregor Hagelueken had previously developed. Lo and behold, the predicted and measured distance distributions agreed quite well both in the absence and presence of substrate, including their width. This suggests that VcSiaP indeed undergoes a clear switch between the two conformations seen in the crystal structures when binding substrate in solution (Fig. 1).
Figure 1.
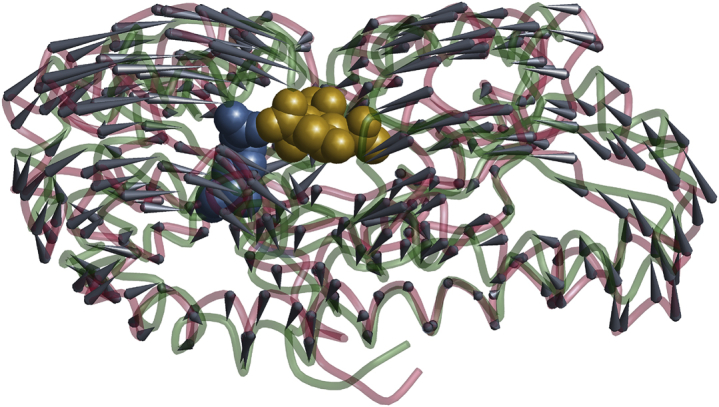
Conformational switch from the substrate-free form of VcSiaP (crimson coil model based on PDB: 4MAG) to the substrate-bound form (green coil model based on the sequence-aligned substrate-bound structure of HiSiaP; PDB: 3B50). (Gray cones) Motion of Cα atoms. (Golden space-filling model) Substrate sialic acid taken from PDB: 3B50; (steel-blue space-filling model) selectivity-filter residue R125 taken from PDB: 4MAG. For the R125A mutant, no conformational switching was observed. For the wild-type, the PELDOR-derived distance distributions from Glaenzer et al. (3) are in full agreement with the simple picture for the conformational switching derived from the crystal structures.
The team further showed that the transition between the two states can be followed by PELDOR/DEER titration. At an intermediate substrate concentration both peaks appear in the distance distribution and an estimate for the dissociation constant Kd can be obtained by analyzing their integral intensity as a function of substrate concentration. This result is still plagued with a rather large uncertainty, because the measurements had to be performed at 25 μM protein concentration for a Kd of ∼0.8 μM. As the sensitivity of PELDOR measurements is still improving by the introduction of new technologies, this limitation may become less serious in the future.
Finally, Glaenzer et al. (3) applied their methodology to mutants that were known from biochemical work to influence substrate transport. With respect to substrate-driven conformational switching, they found that some do it and some do not. In particular, mutation of the selectivity filter residue R125 to alanine almost completely abolishes switching even at high substrate concentrations. The R125A mutant is a substrate trap broken. For some other mutants, partial switching is observed at substrate concentrations where the wild-type switches fully. Analysis of the relative distance peak intensities for the different mutants reveals how the various mutants influence the conformational change required for substrate binding. The switching itself always remains clear cut and in no case is a third conformation or substantial peak broadening observed.
This study not only sheds light onto the mechanism of initial substrate binding in TRAP transporters, it also adds a new twist to studying conformational changes by site-directed spin labeling and distance distribution measurements. Last but not least, the work is notable for the way it complements crystallographic information with structural information in solution and vice versa. Spin labeling always raises the issue whether the labels might have influenced what is observed. Glaenzer et al. (3) crystallized one of their spin-labeled double mutants and found that the backbone conformation is virtually unchanged compared to the wild-type and that the label-to-label distance seen in the crystal nicely falls into the distribution observed in solution. Altogether, this study introduces methodology for experimentally testing models of conformational change of proteins (11) that will certainly be used on other systems in the future.
Editor: David Cafiso.
References
- 1.Freed D.M., Horanyi P.S., Cafiso D.S. Conformational exchange in a membrane transport protein is altered in protein crystals. Biophys. J. 2010;99:1604–1610. doi: 10.1016/j.bpj.2010.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Claxton D.P., Quick M., Mchaourab H.S. Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters. Nat. Struct. Mol. Biol. 2010;17:822–829. doi: 10.1038/nsmb.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glaenzer J., Peter M., Hagelueken G. Dissecting the open-close transition of a sialic acid TRAP transporter substrate binding domain with PELDOR spectroscopy reveals two defined conformational states in solution. Biophys. J. 2016;112:109–120. doi: 10.1016/j.bpj.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Müller A., Severi E., Thomas G.H. Conservation of structure and mechanism in primary and secondary transporters exemplified by SiaP, a sialic acid binding virulence factor from Haemophilus influenzae. J. Biol. Chem. 2006;281:22212–22222. doi: 10.1074/jbc.M603463200. [DOI] [PubMed] [Google Scholar]
- 5.Johnston J.W., Coussens N.P., Apicella M.A. Characterization of the N-acetyl-5-neuraminic acid-binding site of the extracytoplasmic solute receptor (SiaP) of nontypeable Haemophilus influenzae strain 2019. J. Biol. Chem. 2008;283:855–865. doi: 10.1074/jbc.M706603200. [DOI] [PubMed] [Google Scholar]
- 6.Kelly D.J., Thomas G.H. The tripartite ATP-independent periplasmic (TRAP) transporters of bacteria and archaea. FEMS Microbiol. Rev. 2001;25:405–424. doi: 10.1111/j.1574-6976.2001.tb00584.x. [DOI] [PubMed] [Google Scholar]
- 7.Berntsson R.P.A., Smits S.H.J., Poolman B. A structural classification of substrate-binding proteins. FEBS Lett. 2010;584:2606–2617. doi: 10.1016/j.febslet.2010.04.043. [DOI] [PubMed] [Google Scholar]
- 8.Mulligan C., Geertsma E.R., Thomas G.H. The substrate-binding protein imposes directionality on an electrochemical sodium gradient-driven TRAP transporter. Proc. Natl. Acad. Sci. USA. 2009;106:1778–1783. doi: 10.1073/pnas.0809979106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeschke G. DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 2012;63:419–446. doi: 10.1146/annurev-physchem-032511-143716. [DOI] [PubMed] [Google Scholar]
- 10.Setty T.G., Cho C., Ramaswamy S. Bacterial periplasmic sialic acid-binding proteins exhibit a conserved binding site. Acta Crystallogr. D. 2014;70:1801–1811. doi: 10.1107/S139900471400830X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petrone P., Pande V.S. Can conformational change be described by only a few normal modes? Biophys. J. 2006;90:1583–1593. doi: 10.1529/biophysj.105.070045. [DOI] [PMC free article] [PubMed] [Google Scholar]