
Figure 1.
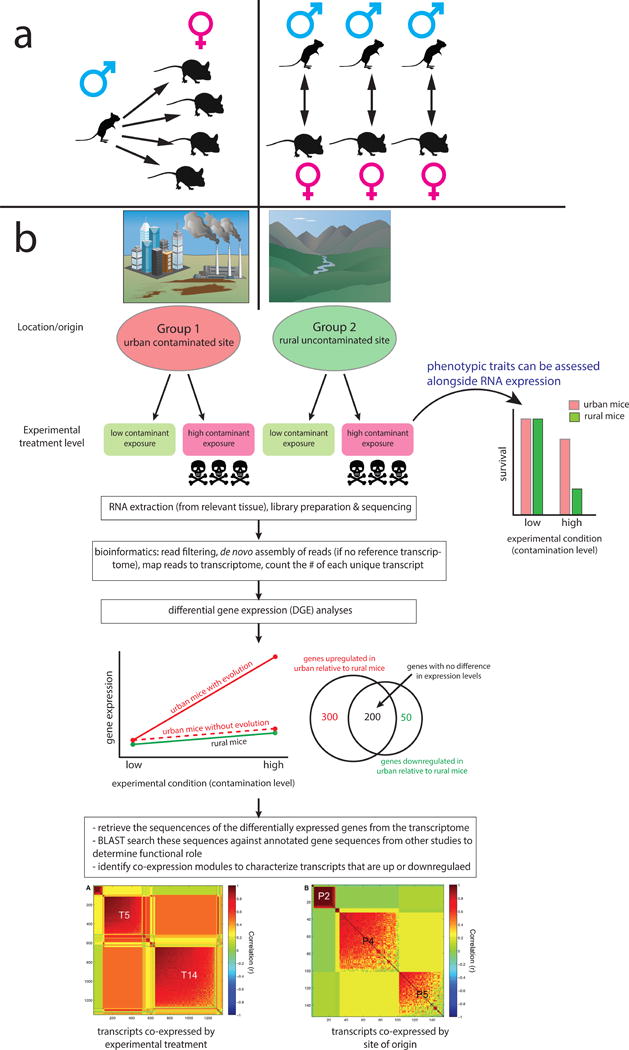
Schematic illustration of the potential for transcriptomic research in Peromyscus. Panel A demonstrates how transcriptomics could used to identify gene expression differences between different species of Peromyscus with distinct mating behavior and parental care (e.g. P. californicus and P. maniculatus). Panel B illustrates the utility of an experimental transcriptomics approach in identifying the genes under selection in divergent habitats. In this example, individuals of the same species are obtained from contrasting habitats (urban vs. rural areas) with divergent natural selection regimes (contaminated vs. uncontaminated soils). Common-garden experiments are performed on individuals acclimated to the common environment, or their F1 offspring. These experiments manipulate environmental variables hypothesized to be agents of natural selection in their natural habitat. Experimental animals are then evaluated for phenotype, tissue is preserved from the tissue where gene expression is hypothesized to be under selection, and RNA is extracted. After sequencing and bioinformatics is performed, differential gene expression (DGE) analyses are performed. The resulting patterns of gene expression can highlight the effects of both experimental treatment and evolutionary history of the population of origin. Finally, the sequences of the up- or down-regulated genes are searched against a database of gene sequences with annotated functions to identify the likely role of differentially expressed genes. Co-expression modules can also be characterized to look for suites of genes with common functions and shared regulatory pathways. Peromyscus vector image from [7]; co-expression module images from [11].