

Abstract
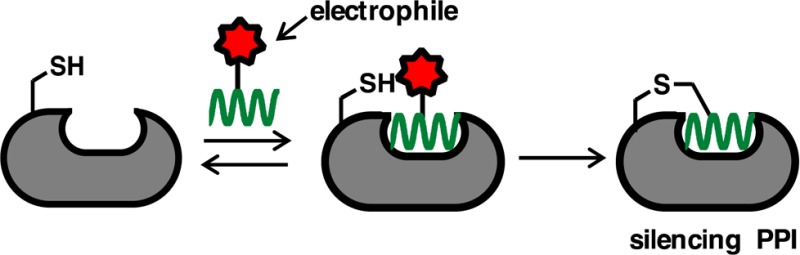
Protein–protein interactions mediate most physiological and disease processes. Helix-constrained peptides potently mimic or inhibit these interactions by making multiple contacts over large surface areas. However, despite high affinities, they typically have short lifetimes bound to the protein. Here we insert both a helix-inducing constraint and an adjacent electrophile into the native peptide ligand BIM to target the oncogenic protein Bcl2A1. The modified BIM peptide bonds covalently and irreversibly to one cysteine within the helix-binding groove of Bcl2A1, but not to two other exposed cysteines on its surface, and shows no covalent bonding to other Bcl2 proteins. It also penetrates cell membranes and bonds covalently to Bcl2A1 inside cells. This innovative approach to increasing receptor residence time of helical peptides demonstrates the potential to selectively silence a PPI inside cells, with selectivity over other nucleophilic sites on proteins.
Keywords: Covalent inhibitor, protein−protein interaction, Bcl2, acrylamide, helix, electrophile
Intracellular protein–protein interactions (PPIs) are emerging as the most prevalent biological targets for developing new therapeutics. However, most PPIs involve large, shallow, solvent-exposed, polar surfaces without hydrophobic pockets for accommodating small drug-like compounds. Conventional small organic molecule drug discovery has consequently met with little success to date in producing effective and selective modulators of PPIs.1 However, peptides present larger protein-like surfaces2 and, especially when helix-constrained, have shown promise for modulating PPIs inside cells due to enhanced potency and some, albeit limited, cell permeability and metabolic stability.3−9 Another drawback of injectable peptide drugs10 is that they often have fast off-rates from their target protein,11 contributing to only moderate cell activity at micromolar concentrations. For some small molecule drugs, duration of drug action has been increased by incorporating an electrophile, producing irreversible inhibitors of enzymes like kinases.12−14 Historically, this approach has been accompanied by off-target side effects due to indiscriminate bonding of the electrophile to endogenous nucleophiles.12 However, in recent years the quest for longer acting drugs with greater clinical efficacy has led to a resurgence of covalent drugs, especially with more discriminating and milder electrophiles like acrylamides with fewer off-target side effects.15−18 There are now 42 approved covalent drugs (3 of 27 approved drugs in 2013 were covalent inhibitors).18 Recently, we outlined a computational and design approach to finding proteins bearing a nucleophile (e.g., Cys or Lys) located in or nearby the binding site of endogenous helical ligands, and to designing synthetic helix-constrained peptides bearing an electrophile appropriately positioned to make an optimal covalent bond.19 This approach is validated here, extending the scope of covalent drugs to peptidomimetics that can modulate PPIs.
We sought to increase the residence time on an oncogenic target protein Bcl2A120 (Figure 1A) of a helix-constrained peptide Bim21,22 (Figure 1B) by innovatively19 introducing an electrophilic warhead positioned carefully to form a putative covalent bond to sulfur in Cys55 in the target protein (Figure 1B,C).
Figure 1.
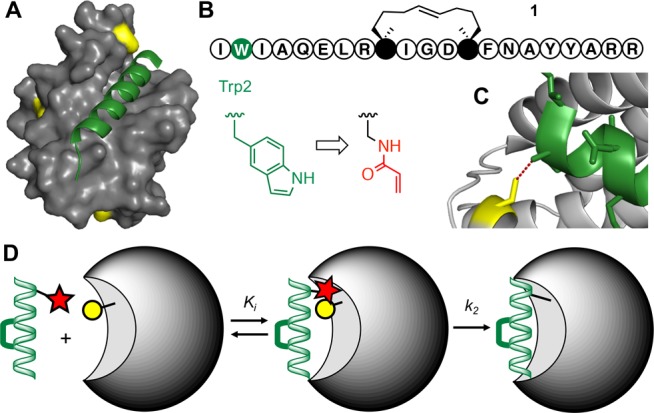
PPI targeting by covalent helical peptides. (A) Bim peptide (green) bound to Bcl2A1 protein (gray) where a Cys residue (yellow) is close to helix-binding site. Bcl2A1 also has two other surface Cys residues (PDB: 2VM6). (B) Sequence of helix-constrained peptide 1 (BimSAHBA, full structure in Figure S1) with an indole of Trp2 (green) replaced by an acrylamide electrophile (red).19 (C) Distance (3.8 Å) between S of Cys55 (yellow) in Bcl2A1 and β-carbon of Bim Trp2 (green) can fit a small electrophile. (D) Helix-constrained peptide (green) first binds noncovalently to protein (gray), then electrophile (red) in peptide bonds covalently to nucleophilic Cys (yellow) in protein.
Bcl2A1 is amplified in ∼30% of melanomas and is necessary for melanoma growth, with suppression of this gene promoting apoptosis.23 Bcl2A1 is also overexpressed in other types of cancer, including leukemias and lymphomas and induces resistance to chemotherapeutic drugs.23−27 Bcl2A1 binds to BH3-only proteins, including pro-apoptotic BimBH3 helix.28 Trp2 of BimBH3 peptide is close to Cys55 in Bcl2A1 (Figure 1A, 1C), enabling potential insertion of an electrophile to make a covalent adduct.20 The idea is that helix-constrained BimBH3 peptide 1 (BimSAHBA, Figure 1B) forms an initial noncovalent and selective interaction with Bcl2 proteins, followed by subsequent slower and more specific covalent bonding of a mild electrophile, such as acrylamide, to the nearby nucleophilic sulfur of Cys55 in the BH3-binding site of Bcl2A1, but not in other Bcl2 proteins. Acrylamides are Michael acceptors previously employed as physiologically compatible electrophiles in covalent inhibitors, including Ibrutinib, an FDA-approved drug acting on B-cell tumors.18
An analogue of 1, with diaminopropionic acid (Dap) replacing Trp2, was synthesized on solid phase using on-resin ring closing metathesis to create the helix-inducing macrocyclic constraint.21 Acrylamide was then attached to the Dap side-chain affording 2 (Figure 2 and Figure S1), which showed similar α-helicity to peptide 1 (Figure S1).
Figure 2.
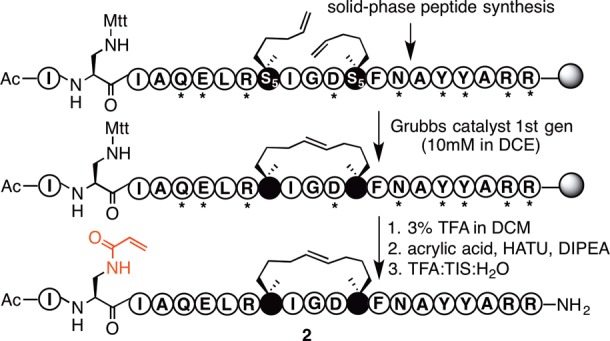
Synthesis of electrophilic peptide 2. Grubbs catalyst = benzylidene-bis(tricyclohexylphosphino)-dichlororuthenium. Mtt = methyltrityl; * = standard protecting group for Fmoc chemistry. S5 = S-2-(4′-pentenyl)alanine (see structure of S5 in Figure S1).
Reaction of the electrophilic peptide 2 with the Bcl2A1 protein (Figure 3) was investigated at different concentrations, times, pH, and temperature. Formation of the covalent protein conjugate 3 was first assessed in a dose-dependent manner at pH 7.2 using SDS-PAGE gels (Figure 3B), producing a single higher molecular weight band (>90%) from a ratio as low as 2:1 (peptide/protein) after 2 h at 22 °C. Unsurprisingly, the control peptide (BimSAHBA, 1) did not produce this higher molecular weight adduct (Figure 3B) since it has no electrophile.
Figure 3.
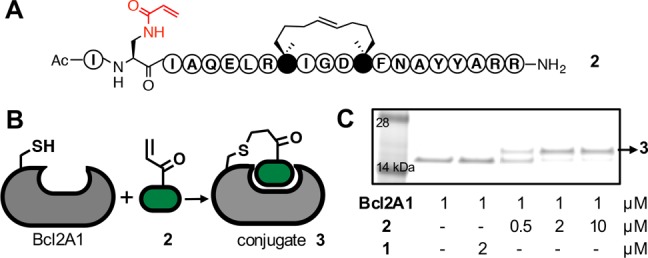
Electrophilic peptide 2 bonds covalently to Bcl2A1. (A) Compound 2 comprises 21 amino acid residues, a helix-constraining hydrophobic linker, and an acrylamide side chain replacing indole of tryptophan. (B) Michael addition of 2 to Cys55 in Bcl2A1. (C) Denaturing SDS-PAGE gel analysis shows conjugation of Bcl2A1 to 2 (0.5, 2, or 10 equiv. after 2 h incubation), but not with 1, to form conjugate 3 (see mass spectrum, Figure 4A).
Bcl2A1 has three free Cys residues, so it was necessary to confirm that the electrophilic peptide 2 covalently bound only to the target Cys55 and not also to one or more of the other two surface-exposed Cys residues. Trypsin digestion of the single covalent adduct band on SDS-PAGE gel (Figure 4A), coupled with MS/MS spectral analysis, showed a 1:1 complex with an expected fragmentation for a single adduct covalently and specifically bound to Cys55 of Bcl2A1 (Figure 4A). Furthermore, we investigated possible reactions of 2 with other proteins of the Bcl2 family (Figure 4B) and with off-target nucleophiles. As anticipated, SDS-PAGE gel analysis showed no covalent reaction of 2 to other Bcl2 proteins (Mcl-1, Bcl-2, Bcl-xL), which do not contain a Cys within the BH3 binding site. Exposed Cys residues in other regions of these other Bcl2 proteins also did not bond covalently to 2 (Figure 4B). Peptide 2 remained intact and mostly unaffected in 1 mM dithiothreitol over several hours (Figure S2) and displayed similar human serum stability as peptide 1, indicating no significant reaction of the electrophile with plasma proteins (Figure S3).
Figure 4.
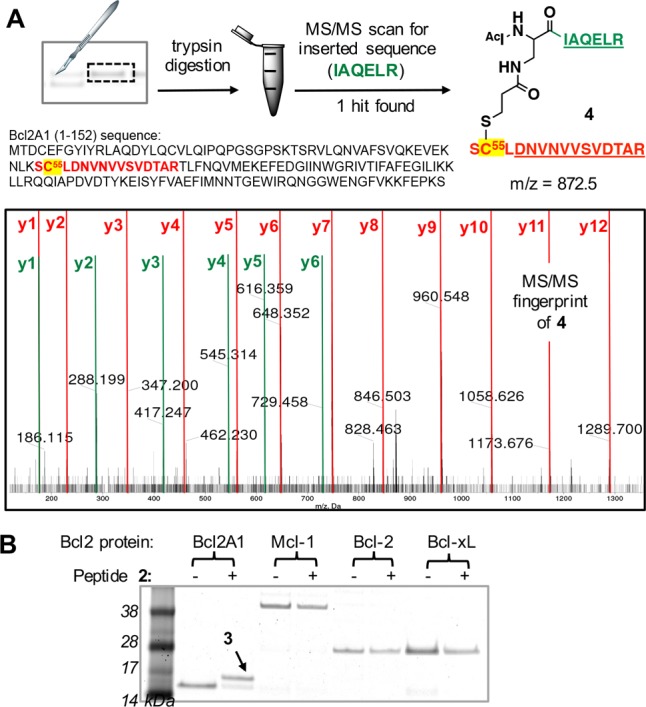
Peptide 2 bonds covalently only to Cys55 in Bcl2A1. (A) MS/MS analysis of trypsin-digested conjugate 3 indicates that 2 bonds covalently and with specificity to Cys55 in Bcl2A1. The digested fragment 4 (expected [M + 3H]+3: 872.5) had a MS/MS fingerprint showing expected y ion fragments for the Bcl2A1-derived sequence DNVNVVSVDTAR (red) and the peptide 2-derived sequence IAQELR (green). No other Cys residue formed a covalent adduct. (B) Peptide 2 covalently bonds to Bcl2A1 but not to other Bcl2 proteins (protein/peptide 1:10 μM, pH 7.2, 5 h).
The profile of binding of electrophilic peptide 2 to Bcl2A1 was assessed by fluorescence polarization (FP) experiments. Binding to Bcl2A1 was measured in competition with a known fluorescent ligand Bid (FBID, FITC-βA-DIIRNIARHLAQVGDSMRSI-NH2) that also binds to the same BH3-binding site of the protein.29 Peptide 2 was found to inhibit Bcl2A1-FBID binding (t1/2 28 min) with complete inhibition by ∼90 min (Figure 5A). At the same Bcl2A1/FBID ratio (50:1) where the ligand is saturated with protein, addition of nonelectrophilic peptide 1 did not significantly interfere with the FP signal, suggesting that over this time period peptide 2 bonds covalently to Bcl2A1. The ligand efficiency was compared for 1 versus 2 in a competitive binding assay against the Bcl2A1-FBID complex. A ratio of Bcl2A1/FBID = 3:1 was maintained to allow measurable fluorescence. Figure 5B shows that 2 was 13-fold more potent in blocking Bcl2A1 interaction with FBID than the reversible peptide 1 (IC50 after 2 h incubation: 8.5 nM (2) vs 110 nM (1)). This translated to an apparent Ki < 0.1 nM for 2 and 32 nM for 1 (after 2 h).
Figure 5.
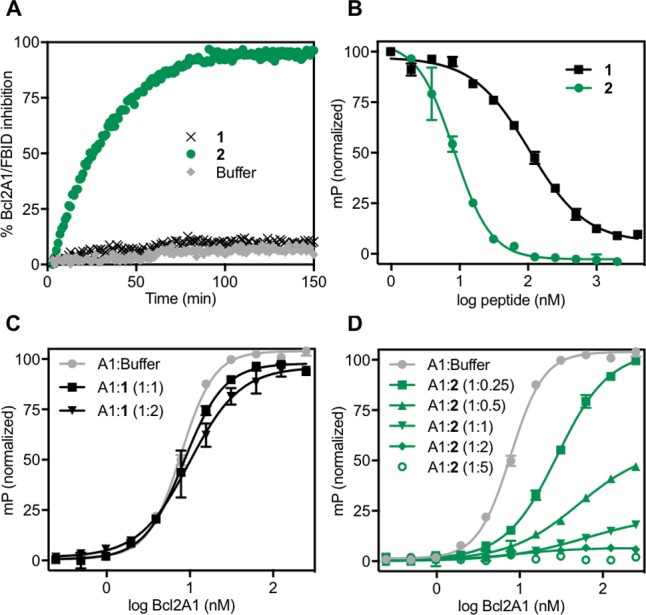
Peptide 2 binds irreversibly to Bcl2A1. Fluorescence polarization (FP) measured for Bcl2A1 (A1) binding to fluorescent Bid (FBID) in competition with 1 (no electrophile, black) or 2 (electrophile, green). (A) Kinetics for inhibiting formation of Bcl2A1-FBID complex measured by FP. FBID (5 nM) was saturated with Bcl2A1 (250 nM) before adding peptide 1 or 2 (500 nM) or buffer. (B) Competitive FP assay showing inhibition of Bcl2A1 (15 nM)-FBID (5 nM) complex by increasing concentrations of 2 or 1 after 2 h incubation. (C,D) Titration binding curves of FBID (5 nM) by Bcl2A1 pretreated with 1 (C) or 2 (D) at different protein/peptide ratios. Addition of buffer instead of peptide was used as control (gray, C,D).
Reversible (Figure 5C) versus irreversible (Figure 5D) binding of Bcl2A1 to 1 versus 2, respectively, was demonstrated by titration binding curves in the presence of FBID. A 2-fold serial dilution of Bcl2A1 (ranging from 250 to 0.25 nM concentration) was preincubated with each peptide at various protein/peptide ratios (Figure 5C,D). After 1 h, the resulting Bcl2A1/peptide complex was combined with FBID and FP recorded. Peptide 1 did not significantly affect maximum fluorescence measured at high Bcl2A1 concentrations without peptide (Figure 5C), consistent with a reversible inhibitor. In contrast, the covalent inhibitor 2 reduced the amount of FBID binding and fluorescence in a dose-dependent manner (Figure 5D). Full inhibition of Bcl2A1-FBID formation was observed after pretreatment with ≥2-fold 2 for 2 h.
Four electrophiles (acrylamide, chloroacetamide, propiol-amide, cyclopentene-carboxamide) were compared for covalent bonding to Bcl2A1. Incorporated into position 2 of the stapled Bim (via the side-chain of a Dap residue) gave peptides 2, 5, 6, and 7, respectively (Figure 6). Additionally, peptide 8 was prepared by direct coupling to β-chloroalanine at position 2.30 Mass spectra (Figure 6) showed different reactions with Bcl2A1, the more powerful electrophiles (5, 6) adding multiple times to Bcl2A1 instead of the 1:1 complex observed for 2. However, electrophiles in 7 and 8 did not bond covalently to Bcl2A1 under the same conditions, possibly due to unfavorable positioning of the electrophile (8) or reduced electrophilicity (7).
Figure 6.

Mass spectrometry analysis of protein adducts resulting from reacting Bcl2A1 (5 μM) with electrophilic peptides 2 (A), 5 (B), 6 (C), 7 (D), or 8 (E) (25 μM, pH 7.2, 5 h). Structures of electrophilic warheads are shown on left. X = alanine spacer.
Neither the acrylamide electrophile nor an appended fluorophore (Figure 7A) compromised the cell-penetrating capacity of the stapled Bim scaffold according to flow cytometry analysis (Figure 7B). Live cell confocal microscopy of U937 lymphoma cells incubated with FITC-derived peptide 2 (9, Figure 7A) established cellular uptake of the electrophilic peptide and its trafficking to mitochondria, where Bcl2 proteins are predominantly localized (Figure 7A).21,22 Next, we investigated whether peptide 2 also bonds covalently to Bcl2A1 endogenously expressed in U937 lymphoma cells (Figure 7C). Using Western blot, we confirmed high levels of endogenous Bcl2A1 expression in this cell line (Figure S4), which, after lysis, was found to bond covalently to 2 as detected by Western blot (Figure 7C). Additionally, live U937 cells were incubated with 2 (and 1) overnight and the extent of covalent conjugation after peptide internalization was evaluated by measuring Bcl2A1 modification in Western blot assays (Figure 7D). Similar results were observed for HeLa cells overexpressing Bcl2A1 (Figures S4 and S5).
Figure 7.
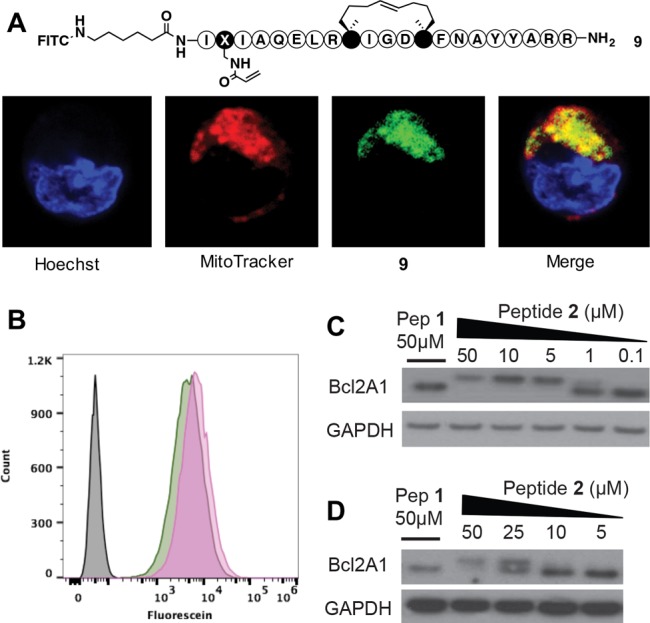
Cell uptake, binding, and localization of peptides to Bcl2A1 in U937 lymphoma cells. (A) Live cell confocal microscopy of U937 cells with FITC-labeled 9 (1 μM) for 4 h stained for nuclei (Hoechst, blue), mitochondria (MitoTracker, red), 9 (green), and colocalization of 9 in mitochondria (merge, yellow). (B) Flow cytometry analysis showing similar cell uptake of 9 (green) compared to a FITC-labeled BimSAHBA1 (pink). (C) Covalent bonding of 2 to Bcl2A1 after 24 h incubation at 37 °C with U937 cell lysates. (D) Live U937 cells were incubated with peptides for 24 h at 37 °C, then lysed and analyzed by Western blot.
In summary, a Bim peptide analogue 2, fitted with a helix-inducing constraint and an acrylamide electrophile, was demonstrated to bond covalently, irreversibly, and specifically to Cys55 within the BH3-binding site of the Bcl2A1 protein. Importantly, 2 (unlike 5 and 6) did not bond covalently and nonspecifically to other surface-exposed cysteine residues, either in Bcl2A1 or in three other Bcl2 proteins that did not have a Cys in the PPI interaction site. Compound 2 was also cell permeable and bound to Bcl2A1 in live cells, indicating the promise for covalent helical peptides as long acting inhibitors of intracellular protein–protein interactions. This irreversible binding of inhibitors confers a number of potential advantages over more conventional reversible inhibitors. A covalent inhibitor–protein complex can more effectively prevent competitive binding by other endogenous ligands, anticipated to be especially beneficial in the case of Bcl2A1, which also interacts with other proteins via its BH3 binding site.24,28 Irreversible binding inhibitors do not readily dissociate, and so their inhibition continues even after the inhibitor leaves the circulation, resulting in less frequent and lower doses of drug to patients.12 This approach is particularly well suited to peptide-based drugs, which are rapidly cleared from the circulation, thereby reducing the chance of nonspecific off-target bonding of electrophilic peptides. Thus, a traditional liability of peptides as drugs can be advantageous in the case of electrophilic drugs. Finally, irreversible covalent bonding peptides and peptidomimetics bring the benefit of pharmacological silencing of a protein target, likely for the lifetime of the protein and only terminating with the synthesis of new protein.
Acknowledgments
We thank Alun Jones (Institute for Molecular Bioscience, Australia) for assistance with mass spectrometry experiments and Yibin Xiang for advice.
Glossary
ABBREVIATIONS
- Bcl2
B-cell lymphoma 2
- Bcl2A1
Bcl2 related protein A1
- Bcl-xL
B-cell lymphoma extra large
- Mcl-1
myeloid cell leukemia 1
- FITC
fluorescein isothiocyanate
- Bid
BH3 interacting domain
Biography
Aline Dantas de Araujo received her BSc in Chemistry in 1999 from the University of Pernambuco (UFPE) in Brazil and soon after worked at Novartis Pharma in Basel, Switzerland, on green synthetic routes for chiral compounds. In 2001, she joined the Max−Planck Institute for Molecular Physiology and University of Dortmund, in Germany, where she performed her PhD studies in Chemical Biology of peptides under supervision of Prof. Herbert Waldmann. Since 2005, she has been working on a number of research projects at the Institute for Molecular Bioscience in Australia focused on the development of peptide molecules for therapeutic drug applications.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00395.
Experimental procedures, compound synthesis and characterization, CD spectra, in vitro binding and cell-based assays (PDF)
Author Contributions
A.D.A. synthesized all compounds and performed in vitro binding assays. J.L. executed cell-based assays and prepared Figure 7. A.C.G., R.T.S., A.D.A., and D.P.F. designed compounds. All authors contributed to writing or editing the paper.
We thank the National Health and Medical Research Council of Australia (NHMRC) for Senior Principal Research Fellowships to D.F. (1027369, 1117017) and the Australian Research Council for grant support (CE140100011, DP160104442).
The authors declare the following competing financial interest(s): Drs Good and Skerlj are inventors on a patent (Reference 19) directly related to the background of this manuscript.
Supplementary Material
References
- Jin L.; Wang W.; Fang G. Targeting protein-protein interactions by small molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456. 10.1146/annurev-pharmtox-011613-140028. [DOI] [PubMed] [Google Scholar]
- Hill T. A.; Shepherd N. E.; Diness F.; Fairlie D. P. Constraining cyclic peptides to mimic protein structure motifs. Angew. Chem., Int. Ed. 2014, 53, 13020–13041. 10.1002/anie.201401058. [DOI] [PubMed] [Google Scholar]
- Araghi R. R.; Keating A. E. Designing helical peptide inhibitors of protein–protein interactions. Curr. Opin. Struct. Biol. 2016, 39, 27–38. 10.1016/j.sbi.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. Inhibition of [alpha]-helix-mediated protein-protein interactions using designed molecules. Nat. Chem. 2013, 5, 161–173. 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- Cromm P. M.; Spiegel J.; Grossmann T. N. Hydrocarbon stapled peptides as modulators of biological function. ACS Chem. Biol. 2015, 10, 1362–1375. 10.1021/cb501020r. [DOI] [PubMed] [Google Scholar]
- Lau Y. H.; de Andrade P.; Wu Y.; Spring D. R. Peptide stapling techniques based on different macrocyclisation chemistries. Chem. Soc. Rev. 2014, 44, 91–102. 10.1039/C4CS00246F. [DOI] [PubMed] [Google Scholar]
- Walensky L. D.; Bird G. H. Hydrocarbon-stapled peptides: principles, practice, and progress. J. Med. Chem. 2014, 57, 6275–6288. 10.1021/jm4011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo A. D.; Hoang H. N.; Kok W. M.; Diness F.; Gupta P.; Hill T. A.; Driver R. W.; Price D. A.; Liras S.; Fairlie D. P. Comparative α-helicity of cyclic pentapeptides in water. Angew. Chem., Int. Ed. 2014, 53, 7085–7089. 10.1002/ange.201310245. [DOI] [PubMed] [Google Scholar]
- Fairlie D. P.; de Araujo A. D. Stapling peptides using cysteine crosslinking. Biopolymers 2016, 10.1002/bip.22877. [DOI] [PubMed] [Google Scholar]
- Craik D. J.; Fairlie D. P.; Liras S.; Price D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- Miles J. A.; Yeo D. J.; Rowell P.; Rodriguez-Marin S.; Pask C. M.; Warriner S. L.; Edwards T. A.; Wilson A. J. Hydrocarbon constrained peptides - understanding preorganisation and binding affinity. Chem. Sci. 2016, 7, 3694–3702. 10.1039/C5SC04048E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barf T.; Kaptein A. Irreversible Protein kinase inhibitors: balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. 10.1021/jm3003203. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage S. J.; Jones L. H.; Gray N. S. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013, 20, 146–159. 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Kluge A. F. Targeted covalent drugs of the kinase family. Curr. Opin. Chem. Biol. 2010, 14, 475–480. 10.1016/j.cbpa.2010.06.168. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Visscher M.; Arkin M. R.; Dansen T. B. Covalent targeting of acquired cysteines in cancer. Curr. Opin. Chem. Biol. 2016, 30, 61–67. 10.1016/j.cbpa.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Bello C. Designing irreversible inhibitors—worth the effort?. ChemMedChem 2016, 11, 22–30. 10.1002/cmdc.201500469. [DOI] [PubMed] [Google Scholar]
- Bauer R. A. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discovery Today 2015, 20, 1061–1073. 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Skerlj R.; Good A.. WO2014/110420 A1 (PCT, July 17, 2014); U.S. Patent No. 9,493,510 (November 15, 2016).
- Herman M. D.; Nyman T.; Welin M.; Lehtiö L.; Flodin S.; Trésaugues L.; Kotenyova T.; Flores A.; Nordlund P. Completing the family portrait of the anti-apoptotic Bcl-2 proteins: Crystal structure of human Bfl-1 in complex with Bim. FEBS Lett. 2008, 582, 3590–3594. 10.1016/j.febslet.2008.09.028. [DOI] [PubMed] [Google Scholar]
- LaBelle J. L.; Katz S. G.; Bird G. H.; Gavathiotis E.; Stewart M. L.; Lawrence C.; Fisher J. K.; Godes M.; Pitter K.; Kung A. L.; Walensky L. D. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J. Clin. Invest. 2012, 122, 2018–2031. 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A. L.; Wachter F.; Lammert M.; Huhn A. J.; Luccarelli J.; Bird G. H.; Walensky L. D. Cellular uptake and ultrastructural localization underlie the pro-apoptotic activity of a hydrocarbon-stapled BIM BH3 peptide. ACS Chem. Biol. 2015, 10, 2149–2157. 10.1021/acschembio.5b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R.; Yokoyama S.; Hawryluk E. B.; Jönsson G. B.; Frederick D. T.; McHenry K.; Porter D.; Tran T.-N.; Love K. T.; Langer R.; Anderson D. G.; Garraway L. A.; Duncan L. M.; Morton D. L.; Hoon D. S. B.; Wargo J. A.; Song J. S.; Fisher D. E. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 4321–4326. 10.1073/pnas.1205575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler M. BCL2A1: the underdog in the BCL2 family. Cell Death Differ. 2012, 19, 67–74. 10.1038/cdd.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hind C. K.; Carter M. J.; Harris C. L.; Chan H. T. C.; James S.; Cragg M. S. Role of the pro-survival molecule Bfl-1 in melanoma. Int. J. Biochem. Cell Biol. 2015, 59, 94–102. 10.1016/j.biocel.2014.11.015. [DOI] [PubMed] [Google Scholar]
- Brien G.; Trescol-Biemont M. C.; Bonnefoy-Berard N. Downregulation of Bfl-1 protein expression sensitizes malignant B cells to apoptosis. Oncogene 2007, 26, 5828–5832. 10.1038/sj.onc.1210363. [DOI] [PubMed] [Google Scholar]
- Olsson A.; Norberg M.; Okvist A.; Derkow K.; Choudhury A.; Tobin G.; Celsing F.; Osterborg A.; Rosenquist R.; Jondal M.; Osorio L. M. Upregulation of Bfl-1 is a potential mechanism of chemoresistance in B-cell chronic lymphocytic leukaemia. Br. J. Cancer 2007, 97, 769. 10.1038/sj.bjc.6603951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Willis S. N.; Wei A.; Smith B. J.; Fletcher J. I.; Hinds M. G.; Colman P. M.; Day C. L.; Adams J. M.; Huang D. C. S. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Zhai D.; Godoi P.; Sergienko E.; Dahl R.; Chan X.; Brown B.; Rascon J.; Hurder A.; Su Y.; Chung T. D. Y.; Jin C.; Diaz P.; Reed J. C. High-throughput fluorescence polarization assay for chemical library screening against anti-apoptotic Bcl-2 family member Bfl-1. J. Biomol. Screening 2012, 17, 350–360. 10.1177/1087057111429372. [DOI] [PubMed] [Google Scholar]
- de Araujo A. D.; Mobli M.; King G. F.; Alewood P. F. Cyclization of peptides by using selenolanthionine bridges. Angew. Chem., Int. Ed. 2012, 51, 10298–10302. 10.1002/anie.201204229. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.