

In this study, Tang et al. investigated the mechanisms underlying alternate splicing. They show that hsh155 mutant alleles in S. cerevisiae, counterparts of SF3B1 mutations frequently found in cancers, specifically change splicing of suboptimal branch site pre-mRNA substrates, thus providing a mechanistic framework to explain the consequences of intron recognition and splicing of SF3B1 mutations found in disease.
Keywords: SF3B1/Hsh155, disease mutation, HEAT motif, Prp5, pre-mRNA splicing fidelity
Abstract
Mutations in the U2 snRNP component SF3B1 are prominent in myelodysplastic syndromes (MDSs) and other cancers and have been shown recently to alter branch site (BS) or 3′ splice site selection in splicing. However, the molecular mechanism of altered splicing is not known. We show here that hsh155 mutant alleles in Saccharomyces cerevisiae, counterparts of SF3B1 mutations frequently found in cancers, specifically change splicing of suboptimal BS pre-mRNA substrates. We found that Hsh155p interacts directly with Prp5p, the first ATPase that acts during spliceosome assembly, and localized the interacting regions to HEAT (Huntingtin, EF3, PP2A, and TOR1) motifs in SF3B1 associated with disease mutations. Furthermore, we show that mutations in these motifs from both human disease and yeast genetic screens alter the physical interaction with Prp5p, alter branch region specification, and phenocopy mutations in Prp5p. These and other data demonstrate that mutations in Hsh155p and Prp5p alter splicing because they change the direct physical interaction between Hsh155p and Prp5p. This altered physical interaction results in altered loading (i.e., “fidelity”) of the BS–U2 duplex into the SF3B complex during prespliceosome formation. These results provide a mechanistic framework to explain the consequences of intron recognition and splicing of SF3B1 mutations found in disease.
Over the past several years, a large number of mutations in human splicing factors have been identified that correlate with a subset of myelodysplastic syndromes (MDSs) characterized by increased ring sideroblasts (Abdel-Wahab and Levine 2011; Malcovati et al. 2011; Papaemmanuil et al. 2011; Yoshida et al. 2011), chronic lymphocytic leukemia (CLL) (Rossi et al. 2011; Wang et al. 2011; Quesada et al. 2012; Landau et al. 2013; Strefford et al. 2013; Wan and Wu 2013), and breast (Ellis et al. 2012), pancreatic (Biankin et al. 2012), uveal (Furney et al. 2013; Harbour et al. 2013), and other cancers (Hahn and Scott 2012; Kong et al. 2014). The accumulation of disease-related mutations in the spliceosome, the machinery for intron recognition and removal, correlates with aberrations in alternative splicing that have been suggested to contribute to tumorigenesis (Tazi et al. 2009; Zhang and Manley 2013; Sveen et al. 2016). Disease-related mutations cluster in proteins involved in spliceosome assembly, particularly ones important for intron recognition, such as SF3B1 (also known as SAP155, SF3b155, and Hsh155p), SRSF1, and U2AF (Yoshida et al. 2011; Quesada et al. 2012). In disease-related SF3B1 mutants, changes in branch site (BS) usage have been identified (Darman et al. 2015; Alsafadi et al. 2016; Kesarwani et al. 2016); however, the mechanisms by which these mutations affect splicing are largely unknown.
The spliceosome is a dynamic RNA–protein complex, highly conserved from yeast to humans, composed of five snRNAs and >150 proteins that recognize three intronic consensus sequences—5′ splice site (SS), BS, and 3′SS—for their removal and subsequent exon ligation (Will and Luhrmann 2011; Hoskins and Moore 2012). Intron recognition initiates with the recruitment of U1 snRNP to the 5′SS, SF1 (BBP) recognition of the BS, and U2AF binding to the 3′SS. Subsequent replacement of SF1 by U2 snRNP allows base pairing between the BS and U2 snRNA, forming the prespliceosome and defining the BS region or BS–U2 duplex (Wahl et al. 2009). The U4/U5/U6 tri-snRNP complex joins and major RNA–RNA and RNA–protein rearrangements occur to form an activated spliceosome (Bact) and, subsequently, the catalytically active spliceosome (C complex) (Staley and Guthrie 1998; Wahl et al. 2009). Eight spliceosomal DExD/H ATPases remodel RNA and RNA–protein complexes and are essential for the progression of spliceosomal assembly and disassembly. In addition, they enhance splicing fidelity through kinetic proofreading (Burgess et al. 1990; Mayas et al. 2006; Xu and Query 2007; Koodathingal and Staley 2013).
SF3B1 is the largest component of the 450-kDa heteroheptameric SF3B complex, a subunit of 17S U2 snRNP and the analogous minor spliceosomal U12 snRNP (Will and Luhrmann 2011). SF3B1 binds and cross-links to pre-mRNA on both sides of the intron BS region (Gozani et al. 1998; Will et al. 2001; McPheeters and Muhlenkamp 2003; Lardelli et al. 2010). It additionally binds SF3B6 (a protein that directly contacts the BS adenosine) and to U2AF (a dimeric protein complex that binds polypyrimidine tract and 3′SS) through its N-terminal domain (Will et al. 2001; Schellenberg et al. 2006; Spadaccini et al. 2006). The C-terminal domain, where the most common disease-related mutations lie, contains 22 HEAT repeats (domains originally found in Huntingtin, EF3, PP2A, and TOR1), each of which consists of two anti-parallel α helices and which together are thought to represent flexible protein interaction surfaces (Fig. 1A; for review, see Andrade et al. 2001). This suggests that mutations in SF3B1 may have effects during spliceosomal assembly and fidelity. In the cryo-electron microscropy (cryo-EM) structures of the Bact complex (before first-step catalysis), the SF3B1 HEAT motifs adopt a torus or ring-like structure through which the 3′ end of the intron is threaded; in addition, the first and last HEATs cradle the BS–U2 duplex (Rauhut et al. 2016; Yan et al. 2016). The SF3B complex is displaced upon conversion to the active spliceosomal C complex by ATPase Prp2 (Warkocki et al. 2009; Lardelli et al. 2010) and is not present in the cryo-EM structures of complexes immediately after first-step catalysis (Galej et al. 2016; Wan et al. 2016).
Figure 1.
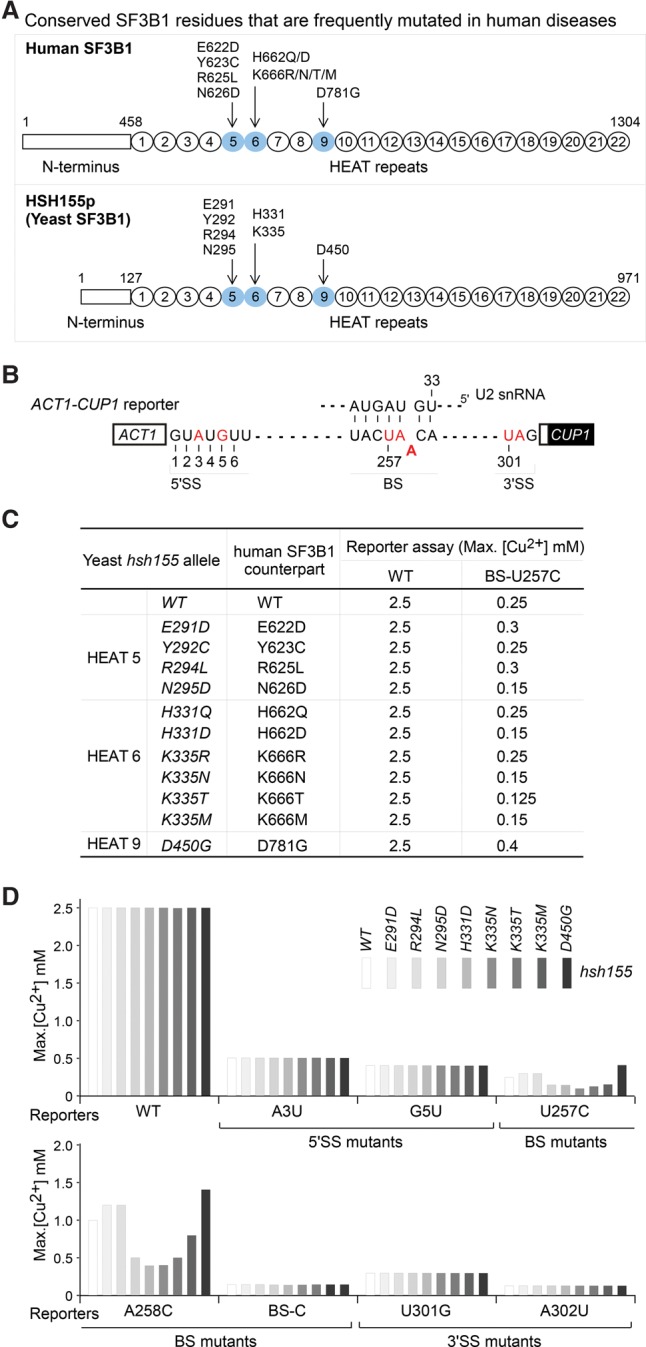
SF3B1 mutations found in human diseases cause altered branch region splicing fidelity in Saccharomyces cerevisiae. (A) Seven conserved residues of human SF3B1 that are frequently mutated in human MDS and CLL diseases (Yoshida et al. 2011; Quesada et al. 2012) and their counterparts in S. cerevisiae Hsh155p. (B) Scheme of ACT1-CUP1 splicing reporters. Mutant reporters used in this study are indicated in red; the BS–U2 RNA duplex is indicated. (C) ACT1-CUP1 copper reporter assays indicate that most disease mutation-containing hsh155 alleles either improved or exacerbated splicing activities of the BS-U257C reporter but not of the wild-type reporter. Multiple mutations at residues H331 and K335 were tested; other residues were based on human disease mutations. (D) Intron mutations that reduce pairing with U2 snRNA (U257C and A258C) are sensitive to hsh155 mutations analogous to those found in human disease, whereas the 5′SS, 3′SS, and branch nucleophile mutations are not.
The ATPase Prp5 is involved in the formation of U1:U2:intron complexes (prespliceosomes, also known as complex A) (Ruby et al. 1993; O'Day et al. 1996; Xu et al. 2004). Prp5 interacts with U1 snRNP through its N-terminal SR-like domain and with the U2 snRNP SF3B complex through its DPLD motif, providing a U1:Prp5:U2 platform for prespliceosome formation (Xu et al. 2004; Shao et al. 2012). Mutations in both the ATPase domain and the DPLD motif of Prp5 modulate splicing fidelity, specifically favoring the recognition and use of suboptimal substrates at the BS region (Xu and Query 2007; Liang and Cheng 2015). SF3B1 mutations affect alternative splicing by promoting the use of alternate branch points (Furney et al. 2013; Darman et al. 2015; DeBoever et al. 2015; Alsafadi et al. 2016), similar to prp5 mutants in yeast (Xu and Query 2007).
Here, we show that disease-related SF3B1 mutations alter splicing of suboptimal introns in yeast. We demonstrate that SF3B1/Hsh155p interacts directly with Prp5p through its HEAT repeat domain, specifically HEAT repeat fragments (HEATs) 1–6 and 9–12. These HEATs contain the sites of predominant disease mutations, and these mutations alter SF3B1/Hsh155p–Prp5p interaction, which we demonstrate by protein interaction assays using purified proteins and yeast lysates. Hsh155p mutations exhibit altered branch region fidelity similarly to Prp5p mutations. Together, these results lead to a model in which mutations in either SF3B1/Hsh155p or Prp5p alter the kinetics of BS–U2 duplex loading into the SF3B complex.
Results
MDS and CLL mutations in SF3B1 alter splicing of suboptimal introns in Saccharomyces cerevisiae
Dozens of SF3B1 mutations have been identified from patients with MDSs (Papaemmanuil et al. 2011; Yoshida et al. 2011) and chronic lymphocytic leukemia (Wang et al. 2011; Quesada et al. 2012; Landau et al. 2013) clustered within the 22 highly conserved HEAT repeats at the C terminus. To address how SF3B1 disease mutations mechanistically affect splicing, we selected seven residues that are conserved between humans and yeast and are frequently mutated in human diseases, including E622, Y623, R625, and N626 in HEAT motif 5; H662 and K666 in HEAT motif 6; and D781 in HEAT motif 9 (Fig. 1A; Supplemental Fig. S1). The K700 residue, most frequently mutated in disease, is not conserved in budding yeast and thus was not included in this analysis. We constructed analogous mutations in the S. cerevisiae HSH155 gene (the homolog of human SF3B1) and tested their splicing activities in vivo using the well-characterized ACT1-CUP1 reporter, which allows cell growth on copper-containing media in proportion to the splicing of the reporter gene (Fig. 1B; Lesser and Guthrie 1993). To test a variety of defects in the splicing pathway, we used mutations at the 5′SS region, branch region, and 3′SS region that produce suboptimal introns that are defective at different stages in the splicing pathway. None of these hsh155 alleles changed the efficiency of the 5′SS, BS nucleophile, or 3′SS mutants in comparison with wild-type HSH155 (Fig. 1C,D), although all of the tested mutant reporters can be improved or exacerbated by other known spliceosomal mutations, such as prp28 for the 5′SS (Yang et al. 2013), prp16 for the BS nucleophile (Burgess et al. 1990), or prp8 for all the three sites (Query and Konarska 2004). Of the eight hsh155 alleles in Figure 1D, some improved and some inhibited splicing of the BS-U257C and BS-A258C reporters (which reduce base pairing with U2 snRNA) (Fig. 1B), suggestive of altered proofreading at the BS region and phenocopying the effects of prp5 ATPase mutants (Xu and Query 2007). We thus focused on branch-flanking mutations and the potential for interaction with Prp5p.
A screen for factors that modulate branch region selectivity yields Hsh155p/SF3B1 and other U2 snRNP components
Because we identified suboptimal branch region mutations as specifically affected by disease-related hsh155 mutations, we wanted to broadly identify factors that could modulate this intron defect. To screen genome-wide, we used a strain containing the BS-U257C reporter and URA plasmid-borne PRP5 UV-mutagenized and replica-plated onto a variety of copper concentrations (Fig. 2A). One-hundred-ninety-four colonies were selected; those containing ACT1-CUP1 recombinants (i.e., recreating wild-type intron) were discarded, and the remainder were investigated by a variety of traditional genetics and genome sequencing (Fig. 2B). (1) By plasmid shuffling that replaced the wild-type PRP5-URA with a wild-type PRP5-TRP plasmid (using the chemical reagent 5-FOA), we identified several strains carrying mutated prp5-K379G-URA that, when isolated, improved U257C splicing, as expected (Fig. 2B). (2) We tested all strains for synthetic lethality with the prp5-SAT-to-GAR allele, which has severely reduced ATPase activity (Xu and Query 2007), identifying seven such strains. We then used a genomic library (Hvorecny and Prelich 2010) to rescue the synthetic lethality. All genomic plasmids that rescued viability carried PRP9 (a U2 snRNP SF3a component), and subsequent sequencing of the PRP9 locus in these strains revealed that all carried an R341K mutation. (3) The remaining strains were subjected to whole-genome DNA sequencing followed by SNP analysis compared with the starting strain; this approach identified mutations within hsh155 (SF3B1), cus1 (SF3B2), and rse1 (SF3B3), which were confirmed by Sanger sequencing. Thus, when mutated, many components of U2 snRNP can result in improved splicing of the U257C reporter; however, the most abundant source of mutants from this open screen was hsh155 (SF3B1) (Fig. 2B).
Figure 2.
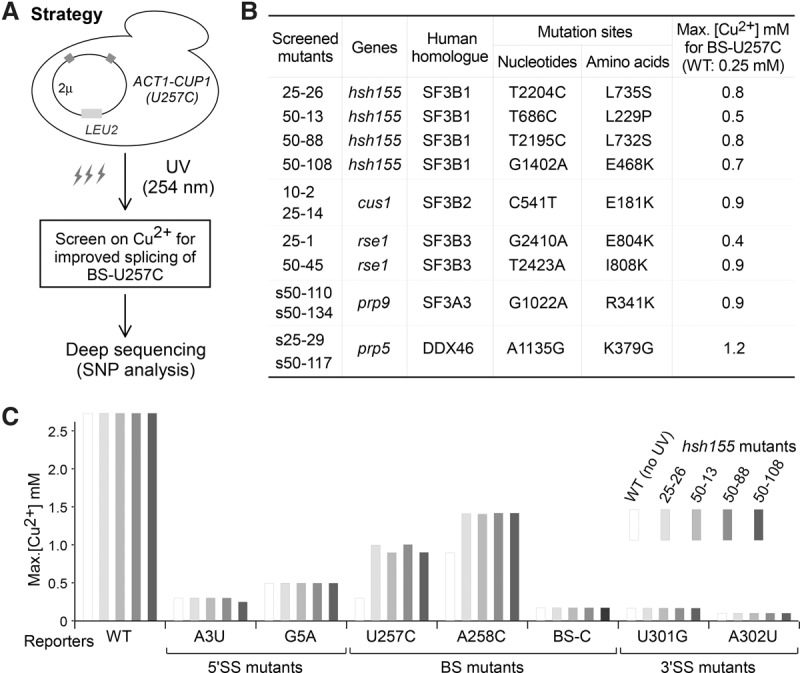
Screen for factors that improve splicing of the BS-U257C suboptimal BS–U2 duplex substrate. (A) Strategy for the UV mutagenesis screen of S. cerevisiae mutant alleles that improve splicing of the BS-U257C mutant reporter. Mutations were identified by a combination of traditional genetics and genome sequencing techniques (see the Results). (B) List of identified yeast alleles with enhanced splicing activity of BS-U257C. (C) Only intron mutations reducing the BS–U2 duplex stability (U257C and A258C) are sensitive to screened alleles, whereas others are not.
We further tested the four obtained hsh155 alleles with other suboptimal ACT1-CUP1 splicing reporters and confirmed that all alleles modulated substrate selectivity only at branch-flanking positions (Fig. 2C), analogous to the above-tested human disease hsh155 alleles.
Hsh155p/SF3B1 HEATs 1–6 and 9–12 interact with Prp5p
As stated above, hsh155 mutants phenocopied the effects on sensitive splicing reporters that prp5 mutants had shown previously (Xu and Query 2007). This suggested the possibility of a direct mechanistic contribution between Hsh155p/SF3B1 and Prp5p. To test for direct Hsh155p–Prp5p binding and identify interaction domains/motifs, we purified a number of truncated recombinant GST-Hsh155p proteins (representing a tiled array) and His-Prp5p protein from Escherichia coli (Supplemental Table S1) and performed in vitro binding and pull-down assays using Ni agarose beads (Fig. 3A). Full-length Prp5p (FL) efficiently pulled down HEATs of Hsh155p, including HEATs 1–8, 5–12, and 9–16 (Fig. 3B, lanes 2–4), but not other fragments (the N-terminal domain, HEATs 13–20, and HEATs 17–22) (Fig. 3B, lanes 1,5–6).
Figure 3.

Identification of interacting motifs between Hsh155p and Prp5p by in vitro protein interaction assays. (A) Schematic of domains/motifs in Hsh155p and Prp5p. (B) HEATs 1–8, 5–12, and 9–16 of Hsh155p are pulled down by Prp5p, whereas other regions are not. (C) HEATs 9–12 of Hsh155p are sufficient for binding to Prp5p. (D) The integrity of HEATs 1–6 is important for their interaction with Prp5p. (E) The N terminus of Prp5p is required for Hsh155p–Prp5p interaction. GST-tagged Hsh155p and 6xHis-tagged Prp5p were expressed and purified from E. coli. Pull-down assays were performed using Ni-NTA agarose beads, and GST alone was used as negative control.
Next, we prepared shorter Hsh155p fragments with only four HEATs for pull-down assays. Within HEATs 9–16, HEATs 9–12 were sufficient for binding to Prp5p, whereas HEATs 13–16 were not (Fig. 3C, lanes 3–4). Within HEATs 1-8, neither HEATs 1–4 nor HEATs 5–8 could be pulled down by Prp5p (Fig. 3C, lanes 1–2). However, HEATs 1–6 retained binding to Prp5p (Fig. 3D). These results identify two distinct Prp5p-interacting regions in Hsh155p: One is HEATs 1–6, and the other is HEATs 9–12.
We also investigated the region of Prp5p required to interact with Hsh155p (Fig. 3A). In contrast to full-length Prp5p, two truncated Prp5p proteins without their N-terminal regions (amino acids 1–206), ΔN and ΔN&ΔC, could not pull down Hsh155p HEAT repeats. However, truncated Prp5p that lacked the C-terminal region, ΔC, still efficiently pulled down Hsh155p HEAT repeats (Fig. 3E). Although we were unable to express and test the N-terminal domain directly, these results imply that the N-terminal region of Prp5p is required for interaction with the two regions of Hsh155p.
Directed screen for hsh155 alleles that improve splicing of BS mutant U257C yields disease-prevalent mutations
To stringently test the region of Hsh155p that contributes to BS selectivity and Prp5p interaction, we performed a directed yeast genetic screen for hsh155 alleles at the Hsh155p–Prp5p interaction interface that alter splicing activity of BS-U257C mutant reporter, which is more sensitive and has a greater range for improvement than other BS mutant reporters, using error-prone PCR products of hsh155. To generate randomly mutated hsh155 alleles, PCR products containing the mutated hsh155 region (from the N terminus to HEAT 11; nucleotides 1–1604) (Fig. 4A) were cotransfected with linearized wild-type HSH155-LYS2 plasmid into a yeast shuffle strain that carries HSH155 on a URA plasmid followed by homologous recombination (gap repair) and 5-FOA selection to remove the wild-type HSH155-URA. We obtained >200 hsh155 alleles that improved splicing of the BS-U257C mutant reporter, most of which contained multiple-residue changes. We then chose the strongest alleles and made new hsh155 alleles that isolated each of the mutations. Copper reporter assays of these single-residue mutated hsh155 alleles confirmed 22 alleles that improved splicing of the BS-U257C mutant (listed in Fig. 4B). None of these alleles exhibited growth defects at 16°C, 30°C, or 37°C (Supplemental Fig. S2A), consistent with nearly all yeast introns having canonical BS sequences (UACUAAC) and with changes in copper tolerance being due only to splicing of the reporter gene. All of the mutated residues are located in the HEAT motifs from 3 to 9; none are in the N-terminal domain, correlating with the location of the two Prp5p-interacting regions identified above.
Figure 4.
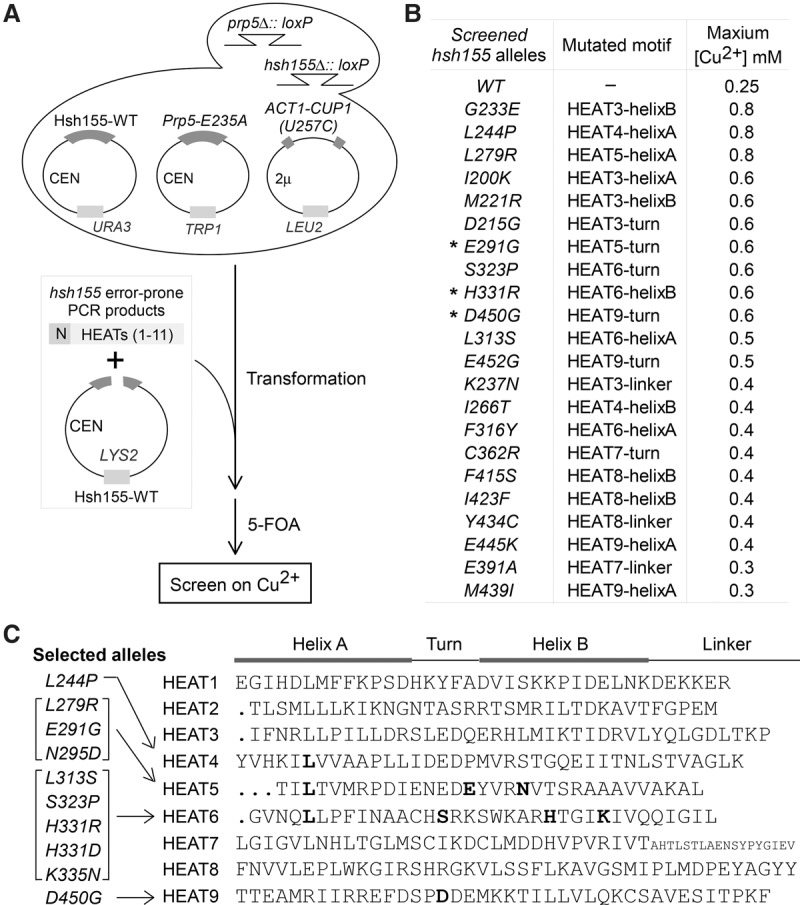
Genetic screen for hsh155 alleles that alter splicing of the BS-U257C reporter and potentially change interaction with Prp5p. (A) Schematic of a genetic screen for hsh155 alleles that were generated by error-prone PCR and improve splicing of BS-U257C. PCR products containing the mutated hsh155 region (N terminus to HEAT 11) were cotransfected with the linearized wild-type HSH155-LYS2 plasmid into a yeast strain carrying the HSH155 gene on URA plasmid followed by homologous recombination (gap repair). (B) Confirmation of 22 isolated single-residue mutated hsh155 alleles that improve splicing of U257C. (C) Ten hsh155 alleles selected for further analysis. Mutated sites in HEAT motifs are listed.
Three alleles—hsh155-E291G, hsh155-H331R, and hsh155-D450G (Fig. 4B, asterisks)—have mutations at the same residues as SF3B1 disease-related mutations (Wang et al. 2011; Yoshida et al. 2011; Quesada et al. 2012). Another three alleles—hsh155-L244P, hsh155-L279R, and hsh155-L313S—significantly improved splicing of BS-U257C up to twofold to threefold in comparison with the wild-type HSH155 allele (maximum growth on copper plates was increased from 0.25 mM to 0.8 mM or 0.5 mM). Located in HEATs 4, 5, and 6, respectively, these three mutated residues are all leucine, which is a conserved residue in the helix A region of most of the 22 HEATs in Hsh155p (Fig. 4C) and other HEAT repeat-containing proteins, such as phosphatase 2A subunit PR65/A and Importin-β (Wang et al. 1998; Cingolani et al. 1999; Groves et al. 1999; Neuwald and Hirano 2000), suggesting that these leucine residues are critical in both the structure and function of Hsh155p. Thus, through a wider screen of hsh155 alleles (from the N terminus to the 11th HEAT), we obtained a number of hsh155 mutants that alter splicing of BS-U257C, including mutations found in human diseases. (We note that the nature of the screen allowed for identification of only alleles that improve splicing and not alleles that exacerbate splicing defects.) These results confirm that disease-related alleles represent prominent mutations that can alter splicing of suboptimal branch regions and identify additional SF3B1 mutations that may similarly contribute to disease.
Selected hsh155 alleles alter splicing fidelity at the BS and in vitro Prp5p interaction
To further address splicing proofreading at the branch region by Prp5p–Hsh155p interaction, we chose 10 representative hsh155 alleles that have mutations in HEAT 4 (L244P), HEAT 5 (L279R, E291G, and N295D), HEAT 6 (L313S, S323P, H331R, H331D, and K335N), and HEAT 9 (D450G) (Fig. 4C). All were obtained from either our genetic screens or human disease mutations (Fig. 4C) and altered splicing activities of suboptimal branch region substrates (Figs. 1D, 4B).
First, we confirmed that the hsh155 alleles selected from the error-prone PCR screen altered splicing fidelity only at the branch region. Compared with the wild-type HSH155 allele, none of the selected hsh155 alleles changed splicing activities of the wild-type, 5′SS mutated, or 3′SS mutated reporters (Fig. 5). Among them, hsh155 alleles—L244P, L279R, E291G, L313S, S323P, H331R, and D450G—significantly improved the splicing of BS mutant reporters (Fig. 5A, top; Supplemental Fig. S2C). Importantly, hsh155 alleles—N295D, K335N, and H331D, which are mutations present in human diseases and did not come from our screens—significantly inhibited the splicing of BS mutant reporters (Fig. 5B, top; Supplemental Fig. S2C) while having no obvious growth defects (Supplemental Fig. S2B). These data demonstrate that all of the selected hsh155 alleles specifically alter splicing fidelity at the branch region and fall into two groups: either enhancing or exacerbating splicing of suboptimal BS substrates.
Figure 5.

hsh155 alleles that alter BS splicing fidelity exhibit altered interaction with Prp5p in vitro. (A) hsh155 alleles that improve splicing of BS mutant reporters in vivo (top) exhibit enhanced interaction with Prp5p in vitro (bottom). (B) hsh155 alleles that inhibit splicing of BS mutant reporters in vivo (top) exhibit inhibited interaction with Prp5p in vitro (bottom). (C) Prp5p-DPLD motif mutants that improve splicing of BS mutant reporters (Shao et al. 2012) exhibit enhanced interaction with Hsh155p in vitro. [Relative (%)] Prp5p immunoprecipitation efficiencies (IP/input) were averaged from three independent repeats and normalized to the corresponding wild-type protein.
Second, we asked whether the altered splicing fidelity at the branch region by these hsh155 alleles was due to changed Hsh155p–Prp5p interaction. To address this, we performed in vitro protein–protein interaction assays. For hsh155 alleles that improved splicing of BS mutant reporters, all of the proteins exhibited enhanced in vitro interaction with Prp5p at various levels—in particular mutant proteins Hsh155p-L313S and Hsh155p-H333R (Fig. 5A, bottom). For hsh155 alleles that inhibited splicing of BS mutant reporters (Fig. 5B, top), the Hsh155p proteins showed a decreased in vitro interaction with Prp5p—in particular mutant proteins Hsh155p-H331D and Hsh155p-K335N (Fig. 5B, bottom). Strikingly, hsh155-H331R and hsh155-H331D mutations provided strong opposite effects. The Hsh155p-H331R mutant protein showed enhanced interaction with Prp5p (Fig. 5A, lane 7), whereas the Hsh155p-H331D mutant protein showed decreased interaction (Fig. 5B, lane 3), suggesting that the residue charge at this position is critical for Hsh155p–Prp5p interaction.
In addition, we previously identified prp5-DPLD mutant alleles in the conserved DPLD motif at the N terminus of Prp5p to improve splicing of BS mutant reporters (Shao et al. 2012). In vitro protein–protein interaction assays, as above, revealed that Prp5-DPLD-to-APLD and Prp5-DPLD-to-AAAA mutant proteins pulled down more Hsh155 protein, including HEAT 1–8, 5–12, and 9–16 fragments, at constantly mild levels (Fig. 5C) and did not pull down the HEAT 13–20 fragment (analogous to wild-type Prp5p), demonstrating that the Prp5p DPLD motif is involved in the Prp5p–Hsh155p interaction.
In summary, there is a correlation between the in vitro Hsh155p–Prp5p interaction and in vivo splicing fidelity at the BS region: Hsh155p or Prp5p mutations that increase Hsh155p–Prp5p interaction in vitro enhance splicing of suboptimal BS mutant reporters; in contrast, Hsh155p mutations that decrease the Hsh155p–Prp5p interaction in vitro exacerbate splicing of suboptimal BS mutant reporters.
Increased Prp5p–Hsh155p interaction leads to an increased release of Prp5p from the prespliceosome
Prp5p was demonstrated to be essential for prespliceosome assembly (Xu et al. 2004) and could be released immediately after this assembly to allow recruitment of U4/5/6 tri-snRNP (Liang and Cheng 2015). To investigate the functional contribution of Prp5p–Hsh155p interaction in prespliceosome assembly, we tested in vivo interactions between Hsh155p and Prp5p proteins in budding yeast lysates by coimmunoprecipitation using HA-tagged hsh155 or Flag-tagged prp5 allele strains. In comparison with the wild-type HA-Hsh155p, mutant Hsh155p proteins that previously exhibited increased in vitro interaction with Prp5p (Fig. 5A) coimmunoprecipitated much less wild-type Flag-Prp5p in vivo, such as Hsh155p-L313S, Hsh155p-H331R, and Hsh155p-D450G (Fig. 6A, lanes 2–4) as well as Hsh155p-L244P, Hsh155p-L279R, and Hsh155p-E291R mutant proteins (Supplemental Fig. S3A). Similarly, the DPLD motif mutants of Prp5p, Prp5p-APLD and Prp5p-AAAA that showed increased Hsh155p interaction in vitro (Fig. 5C) were coimmunoprecipitated much less by wild-type HA-Hsh155p in vivo (Fig. 6B, lanes 1–3). However, Hsh155p-H331D and Hsh155p-K335N that showed significantly decreased in vitro Prp5p interaction (Fig. 5B) coimmunoprecipitated amounts of wild-type Flag-Prp5p similar to those of wild-type HA-Hsh155p (Fig. 6A, lanes 7–9).
Figure 6.

Conformational changes of the prespliceosome are affected by interaction between Prp5p and Hsh155p. (A, left) Hsh155p mutants that enhanced in vitro Prp5p interaction had decreased in vivo affinity with Prp5p. (Right) In contrast, Hsh155p mutants that inhibited in vitro Prp5p interaction had no obvious altered in vivo affinity with Prp5p. (B) The DPLD motif mutant Prp5p that enhanced in vitro Hsh155p interaction had decreased in vivo affinity with Hsh155p. (C) Prp5p significantly associates with U1, U2 snRNAs, and pre-mRNA. (D) In the presence of the hsh155-H313S or hsh155-H331R allele, Prp5p's associations with U1, U2 snRNA, and pre-mRNA are significantly decreased but not in the presence of the hsh155-H331D or hsh155-K335N allele. (E) Disruption of complex conformation by high salt restores the interaction between Hsh155p and Prp5p. In vivo pull-down assays in A, B, and E were performed by HA beads against HA-Hsh155 and visualized by Western blotting. RT-qPCR assays in C and D were performed by Flag beads against Flag-Prp5p. [Relative (%)] Prp5 immunoprecipitation efficiencies (IP/Input) were averaged from three independent repeats and normalized to the corresponding wild-type protein. Brown and green labels represent two opposite groups of hsh155 alleles. (*) P < 0.05; (**) P < 0.01.
To examine whether the altered in vivo Hsh155p–Prp5p interaction was caused by an inefficient formation of the SF3B complex, we tested and found that all Hsh155p proteins coimmunoprecipitated the same amounts of Cus1p, a yeast homolog to the human SF3B3 (Supplemental Fig. S3B), consistent with the interactions between Hsh155p mutant proteins and other SF3B subunits being unaltered, as also found by Cretu et al. (2016).
Furthermore, we used quantitative RT–PCR to assess whether disease-related SF3B1/Hsh155 mutations alter the association of Prp5p with pre-mRNA and snRNAs in vivo. As expected, in the presence of wild-type Hsh155p, wild-type Flag-Prp5p efficiently coimmunoprecipitated pre-mRNA, U1 and U2 snRNAs, and much less U4, U5, or U6 snRNA (Fig. 6C), consistent with the previous report that Prp5p is released before the U4/U6.U5 tri-snRNP recruitment (Liang and Cheng 2015). However, wild-type Flag-Prp5p coimmunoprecipitated much less U1 and U2 snRNAs in the presence of hsh155 mutant allele hsh155-L313S or hsh155-H331R, but similar levels of U1 and U2 snRNAs in the presence of the hsh155-H331D or hsh155-K335N allele (Fig. 6D), consistent with the above analyses of in vivo protein interactions (Fig. 6A); coimmunoprecipitated U4, U5, and U6 snRNAs were not obviously changed (Supplemental Fig. S3C). Since these hsh155 alleles had shown opposite effects on proofreading at the BS region (i.e., hsh155-L313S and hsh155-H331R alleles enhanced splicing of BS mutants, whereas hsh155-H331D and hsh155-K335N alleles inhibited) (Fig. 5), we asked whether the association of pre-mRNA of the BS mutant for Prp5p was affected. Prp5p coimmunoprecipitated much less wild-type ACT1-CUP1 reporter pre-mRNA in the presence of the hsh155-L313S or hsh155-H331R allele than in the presence of the wild-type Hsh155 allele but more wild-type pre-mRNA in the presence of the hsh155-H331D or hsh155-K335N allele (Fig. 6D, right). Furthermore, the association of U257C pre-mRNA, a BS region mutant, exhibited a pattern similar to Prp5p but relatively more than the association with wild-type pre-mRNA (Supplemental Fig. S3D). Taken together, these results suggested that prespliceosome assembly is altered in the presence of mutant hsh155 alleles, in which interaction between Prp5p and Hsh155p is critical and results in different BS selection.
To address the difference between the in vivo and in vitro interaction assays, we hypothesized that direct Prp5p–Hsh155p interaction facilitates prespliceosome formation, and then Prp5p is released from the prespliceosome. The in vitro assay (using purified proteins) would represent the Prp5p–Hsh155p interaction prior to the formation of the prespliceosome, whereas the in vivo assay (using cell lysates) would represent their interaction after prespliceosome formation. To test this hypothesis, we performed a “high-salt disruption” assay in which yeast cell lysate was incubated with glucose followed by high-salt (1 M NaCl) treatment to disrupt pre-existing complexes and then was adjusted back to normal salt before the coimmunoprecipitation (Fig. 6E, left). Interestingly, after the disruption by high salt, we observed stronger interaction between Prp5p and Hsh155p-H331R, whereas a mock treatment still exhibited decreased interaction, as in the previous in vivo assay (Fig. 6E, middle). For the mutant hsh155-K335N representing the opposite class, we observed a decreased interaction with Prp5p after the disruption treatment, whereas the mock still showed similar interaction as in the above in vivo assay (Fig. 6E, right).
Taken together, results from the in vitro assays, the normal-salt lysate, and the high-salt disruption lysate imply that Hsh155p–Prp5p interaction is dynamic before and after prespliceosome assembly. These results are compatible with a model in which strong interaction between Hsh155p and Prp5p allows fast formation of complex A and then a quick or enhanced release of Prp5p. In contrast, in this model, a weakened Hsh155p–Prp5p interaction would slow the release of Prp5p.
Discussion
A better molecular and cellular understanding of the consequences of HEAT repeat mutations on the normal functions of SF3B1 could greatly inform the role of mutant SF3B1 in MDS etiology. Our biochemical data in yeast indicate that the HEAT repeats of Hsh155p/SF3B1 interact with Prp5p, the first ATPase that acts during spliceosome assembly. Prp5p physically bridges between U1 snRNP (which identifies the 5′ end of the intron) and U2 snRNP (which identifies the 3′ end of the intron) and catalyzes a conformational change that locks U2 snRNP onto the intron. As part of this mechanism, we identified physical interactions between Prp5p and the HEAT repeats of Hsh155p/SF3B1 that harbor the most abundant MDS mutations; these mutations alter both interaction with Prp5p and splicing in vivo and lead to a model of the molecular mechanism of altered BS–U2 sequence specificity.
Genome-wide screen for factors that modulate branch region selectivity yields (only) Hsh155p/SF3B1 and other U2 snRNP components
The above data identified a select intron region that is affected by hsh155/SF3B1 mutations (branch-flanking region). We previously observed exactly the same pattern for prp5 mutants (Xu and Query 2007). This suggests that there may be a special (i.e., mechanistic) relationship between Hsh155p/SF3B1 and Prp5p; alternatively, it could be that mutations in many factors produce this splicing phenotype. It is important for clinical prediction to distinguish these possibilities. To interrogate genome-wide what factors exhibit this phenotype, we used a genome-wide UV mutagenesis screen. The selected and confirmed strains were subjected to genomic DNA sequencing followed by SNP analysis compared with the starting strain; this approach identified mutations mostly within hsh155(SF3B1) and prp5, with a few in other SF3 components [cus1(SF3B2), rse1(SF3B3), and prp9(SF3A3)]. Thus, the most abundant source of mutants from this open screen was hsh155(SF3B1) (Fig. 2B). We further tested the four obtained hsh155 alleles with other suboptimal ACT1-CUP1 splicing reporters, confirming that all of them modulate splicing activity only at branch-flanking sites (Fig. 2C). Thus, SF3B1 mutants phenocopy Prp5 mutants, and only mutations in a few other SF3 components produce similar effects.
Direct Prp5p–Hsh155p interaction
The similarity of hsh155/SF3B1 and prp5 mutant phenotypes, together with our previous results that Prp5p binds U2 snRNP, suggested that they might be direct binding partners. We tested this by making recombinant His-Prp5p protein and GST-Hsh155p/SF3B1 proteins and performed in vitro binding and pull-down assays. We tested a set of tiled overlapping fragments and found two regions of Hsh155p/SF3B1 that bind to Prp5p (HEATs 1–6 and 9–12), which are the same HEAT domains that contain the Hsh155p/SF3B1 mutations of interest (Figs. 1, 3).
The contribution of Prp5p–Hsh155p interaction to spliceosome assembly
In recent structures of the spliceosome (Rauhut et al. 2016; Yan et al. 2016) and of SF3B alone (Cretu et al. 2016), the SF3B complex forms a “spring-loaded clamp”-like structure in which Hsh155p/SF3B1 is the “spring” and the BS–U2 duplex is “clamped” between the first and last HEAT domains of Hsh155p/SF3B1 (Supplemental Fig. S4). There are two times during spliceosomal assembly when the Hsh155p/SF3B interaction with the BS–U2 duplex changes: (1) In complex A formation, it forms the stable U2 snRNP–BS complex, mediated by Prp5p ATPase activity (Xu and Query 2007). (2) In transition from the Bact to the C complex, this interaction is disrupted, and the SF3B complex is released, mediated by ATPase Prp2p (Warkocki et al. 2009; Lardelli et al. 2010). Both Prp5p and Prp2p interact with similar parts of the Hsh155p/SF3B1 HEAT repeats (Supplemental Fig. S4; Yan et al. 2016). Prp2p is proposed to open the Hsh155p/SF3B1 torus-like structure to release it from the BS–U2 duplex (Warkocki et al. 2009; Rauhut et al. 2016). We propose that Prp5p likewise opens the Hsh155p/SF3B1 torus to allow for binding to the BS–U2 duplex during prespliceosome (complex A) formation. Notably, in the Bact complex, the SF3B component Snu17p binds to the same HEATs as Prp5p (Rauhut et al. 2016), possibly preventing Prp5p interaction subsequent to complex A formation.
Thus, in this model, Prp5p interaction with HEATs 5–12 of Hsh155p/SF3B1 opens the Hsh155p/SF3B1 torus to load the BS–U2 duplex into the clamp, holding it until the remainder of the spliceosomal catalytic core is built. Then, just before the first catalytic step, Prp2p opens the Hsh155p/SF3B1 clamp again, allowing the BS–U2 duplex to move into the catalytic core and participate in first-step catalysis.
Hsh155p/SF3B1 mutations alter BS–U2 duplex fidelity
It is clear that at least four spliceosomal ATPases impact the splicing of suboptimal introns. In each case, mutant alleles that decrease ATPase activity are thought to allow more time for a limiting event preceding a conformational change. prp5 alleles improve splicing of branch region mutants by allowing more time for duplex formation with U2 snRNA (Xu and Query 2007), prp28 alleles improve splicing of 5′SS mutants (Yang et al. 2013), prp16 alleles improve substrates suboptimal for first-step catalysis by allowing a longer dwell time in the first-step conformation (Konarska and Query 2005; Villa and Guthrie 2005), and prp22 alleles improve splicing of substrates suboptimal for second-step catalysis (e.g., 3′SS mutants) by allowing a longer dwell time in the second-step conformation (Mayas et al. 2006). These common features were proposed (Burgess and Guthrie 1993) to similarly affect a preceding event, in each case limiting for different sets of (suboptimal) intron features or other spliceosomal interactions.
Hsh155p interaction with Prp5p is key to understanding the consequences of SF3B1 disease-related mutations on splicing. Competition between the ATPase activity of Prp5p and the stability of the BS–U2 snRNA duplex impacts branch region fidelity: Alteration of Prp5p's ATPase activity can increase (slower ATPase) or decrease (faster ATPase) use of suboptimal BSs (Xu and Query 2007; Shao et al. 2012; Zhang et al. 2013). hsh155 alleles affect splicing of exactly the same suboptimal BS–U2 pairings as prp5 alleles (cf. Figs. 1,2,5; Supplemental Fig. S2 with Xu and Query 2007). We previously observed in fission yeast S. pombe that Prp5p binds U2 snRNP through the SF3B complex (Xu et al. 2004; Shao et al. 2012). Here, we show that Hsh155p/SF3B1 HEAT repeats are the interface for the Prp5–U2 snRNP interaction (Fig. 3). SF3B1 HEAT repeat mutations alter interaction with Prp5 (Figs. 5,6), and we conclude that this altered SF3B1–Prp5 interaction causes changes in the use of suboptimal BSs (fidelity).
The meaning of mutations and changes in BS fidelity
In the above model, Prp5p–Hsh155p/SF3B1 interaction promotes prespliceosome formation (complex A) and then release of Prp5p. These findings lead us to propose that mutations in Hsh155p or Prp5p that increase Prp5p–Hsh155p interaction accelerate prespliceosome formation and, subsequently, a fast release of Prp5p; this specifically suppresses splicing defects caused by suboptimal BS region substrates. Conversely, mutations that decrease Prp5p–Hsh155p interaction slow prespliceosome formation and lead to slowed release of Prp5p; this exacerbates splicing defects of suboptimal BS substrates (Fig. 7).
Figure 7.

Hsh155p/SF3B1 HEAT motif mutations alter its interaction with Prp5p and result in changes to splicing fidelity at the BS region. (A) Summary of phenotypes of selected Hsh155p/SF3B1 mutations from both cancers and yeast genetic screens. (B) Interaction between Prp5p and Hsh155p/SF3B1 promotes prespliceosome formation (complex A) and then leads to the release of Prp5p. In this model, mutations in Hsh155p or Prp5p that increase the Prp5p–Hsh155p interaction accelerate prespliceosome formation and, subsequently, fast release of Prp5p and also specifically suppress the splicing defects caused by suboptimal BS region substrates. Conversely, mutations that decrease the Prp5p–Hsh155p interaction slow down the formation of prespliceosomes and slow the release of Prp5p, thereby exacerbating the splicing defects of suboptimal BS substrates.
These observations are consistent with recent studies of human SF3B1 mutants. For example, an intron from the human ZDHHC16 gene has the BS sequence AAACUAC (CU is equal to the combination of BS-U257C and BS-A258U mutant reporters used here), and wild-type SF3B1 uses this as the branch point. However, in the presence of mutant SF3B1-K666N (hsh155-K335N or hsh155-K700E), an upstream cryptic BS sequence (CUAAC) was used in which the UA is exactly our yeast wild-type reporter sequence (Darman et al. 2015). Similarly, the weak SF3B1-K666T (hsh155-K335T) and SF3B1-R625G mutants prefer cryptic branchpoint sequences that have stronger pairing with U2 snRNA (Alsafadi et al. 2016). Together, these examples demonstrate that weaker Prp5–SF3B1 interaction resulted in higher fidelity at the BS region in the human system.
Mutations found in human diseases (such as SF3B1-H662R from uveal melanoma and its yeast homolog, Hsh155p-H331R) were also found in our screen and increased splicing of suboptimal BS substrates. In contrast, SF3B1-H662D and SF3B1-K666N were found in patients with MDS or CLL; their yeast homologs resulted in an opposite impact by increasing splicing fidelity (decreased splicing) at the BS region. Thus, not all SF3B1 mutations found in disease impact splicing in the same way,
The most highly mutated SF3B1 position in MDS and other cancers, K700, resides within a loop that connects two α helices at HEAT repeat 7 (Quesada et al. 2012; Visconte et al. 2012). Recently, it was reported that the U2 snRNP SF3B complex has conformational changes from a closed stage to an open stage to allow interactions with other spliceosomal components and the BS (Golas et al. 2003). Specifically, SF3B1 HEAT repeats 1–12 are on the surface of the complex at the closed stage, whereas they are covered by other components during the open stage (Golas et al. 2005). This suggests that SF3B1 C-terminal HEAT repeats present an additional interface between U2 snRNP and other spliceosomal components and that its conformations might regulate splicing activity. The conformational changes that occur during spliceosomal assembly, besides being dependent on the action of nucleic acid-dependent ATPases, confer kinetic proofreading for splice site selection (Wahl et al. 2009; Semlow et al. 2016). The early entry of U2 snRNP in the spliceosomal assembly process and its interaction with Prp5 (Xu and Query 2007; Shao et al. 2012), an ATPase essential for prespliceosomal formation and fidelity, lead us to hypothesize that the consequences of SF3B1 disease-related mutations were at this stage.
An alternative model could be based on the observations that Prp5 bridges U2 to the substrate through Prp5 binding to U1 (Xu et al. 2004; Shao et al. 2012) and that this bridging function depends on the stability of tethering U2 to U1 via the Prp5p–Hsh155p interaction. In this model, strong interactions would facilitate recruitment of U2 snRNP and thereby binding to weaker BSs.
Disease and yeast genetic screen mutations of SF3B1/hsh155
In this study, we performed two genetic screens in yeast and obtained a number of hsh155 mutants with various abilities to alter splicing activity on suboptimal BS reporters. Many of these screened yeast mutations have conserved counterparts in human diseases, including MDS, CLL, and other diseases or tumor cell lines such as uveal melanoma, prostate cancer, breast cancer, and colorectal cancer (Supplemental Table S4). This result demonstrates the functional conservation of SF3B1/Hsh155p between humans and budding yeast. Of note is that all human disease-related mutants rank in this list at the middle or low levels to alter splicing of suboptimal BS mutants, implying that strong mutations of SF3B1 in humans may be lethal.
Materials and methods
Strains and plasmids
S. cerevisiae strains used in this study were derived from yTQ01 (MATa, ade2, cup1Δ::ura3, his3, hsh155Δ::loxP, leu2, lys2, prp5Δ::loxP, trp1, pRS314-PRP5 [PRP5 TRP1 CEN ARS], pRS316-HSH155 [HSH155 URA2 CEN ARS]) and are listed in Supplemental Table S2. Plasmid-borne alleles of prp5, hsh155, and ACT1-CUP1 reporters were prepared by either in vivo gap repair cloning or traditional cloning using E. coli.
Identification of mutants by genomic sequencing and SNP analysis
DNA libraries were prepared by spheroblast formation and lysis followed by phenol extractions, ethanol precipitation, DNA fragmentation, ligation of Illumina adaptors, and sequencing using an Illumina HiSeq2000 instrument at the Einstein Epigenomics Core facility. Raw FASTQ reads from each mutant yeast strain were quality-trimmed for adapter sequences using QUaRT (quality score aware read trimming) and then mapped to the sacCer3 genome using BWA mem version 0.7.5a (Li and Durbin 2009) with default parameters. Locations of SNPS/indels relative to the wild-type strain were identified using Unified Genotyper from the GATK toolkit (McKenna et al. 2010), which was then queried for known spliceosomal protein genes (Cvitkovic and Jurica 2013). Data were deposited into BioProject under accession number PRJNA356455.
Screening of hsh155 alleles in S. cerevisiae
Wild-type HSH155 on pRS317(LYS2) plasmid was digested by AfeI and SphI and transformed with a pool of error-prone PCR products covering nucleotides +1 to +1604 of the HSH155 CDS for gap repair in the yTQ01 strain that contained ACT1-CUP1 reporter U257C. Transformants were replicated to 5-FOA plates to lose the URA3-marked wild-type HSH155 plasmid and then replicated to select for higher resistance to copper. Subsequent steps for identification and confirmation of hsh155 alleles were carried out as described (Xu and Query 2007). Error-prone PCR was performed using buffer containing 0.15 mM MnCl2, 0.05 U/µL Taq DNA polymerase, and 80 pg/µL template for 15 cycles to introduce three to 10 mutants per product (Wilson and Keefe 2001), which were then gel-purified and used for gap repair cloning.
Copper reporter assay
Copper assays were carried out as described (Lesser and Guthrie 1993; Xu and Query 2007). Plates were scored by the maximum copper concentrations of strain growth and photographed after 4 d.
Recombinant protein expression and purification
To generate GST-tagged Hsh155p proteins and 6xHis-tagged Prp5p proteins, sequences of HSH155 or PRP5 wild type and their mutant fragments were cloned into pGEX-4T-1 and pET-14b vectors, respectively (Supplemental Table S1). Recombinant proteins were induced by 1.0 mM IPTG and expressed in E. coli (Rosetta) for 20 h at 16°C and then purified by either glutathione-sepharose (GE) or Ni agarose (Qiagen) chromatography under standard conditions followed by dialysis against buffer D (20 mM HEPES-KOH at pH 7.9, 0.2 mM EDTA, 100 mM KCl, 0.5 mM dithiothreitol [DTT], 1 mM phenylmethylsulfonyl fluoride [PMSF], 20% glycerol).
In vitro protein–protein interaction
Purified Hsh155p (10 pmol) and Prp5p (40 pmol) proteins were incubated in 600 µL with binding buffer (20 mM Tris-Cl at pH 8.0, 150 mM NaCl, 1mM EDTA, 0.2% Triton X-100, 0.5 mM PMSF) for 4 h at 4°C with Ni agarose. Bead pellets were washed four times with washing buffer (50 mM Tris-Cl at pH 8.0, 140 mM NaCl, 1 mM EDTA, 0.1% Triton X-100) and then resuspended in 50 µL of sample loading buffer for SDS-PAGE electrophoresis. Hsh155p and Prp5p were detected by Western blotting using anti-GST (GST-2) and anti-polyhistidine (His-1) monoclonal antibodies from Sigma.
Coimmunoprecipitation and RT-qPCR
Sequences encoding 3xFlag and 3xHA epitopes were inserted into the 5′ ends of PRP5 and HSH155 ORFs on pRS plasmids, respectively, which were then transformed into strain yTQ01 followed by growth to lose wild-type sequence-containing plasmids. One-hundred milliliters of each cell culture at OD 1.7 was harvested and broken by glass beads to generate a clear lysate in 700 µL of lysis buffer (20 mM Tris-Cl at pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM PMSF, 1× proteinase inhibitor [Roche]), and an additional 200 U/mL RNase inhibitor (Thermo) was used for RNA isolation. Cell lysates were then incubated with anti-HA-agarose beads (Sigma) and washed five times with lysis buffer; and the pellets were resuspended in 50 µL of 1× SDS sample buffer, which was loaded either directly to a SDS-PAGE gel for Western blotting analysis using anti-Flag (M2, Sigma) and anti-HA (12CA5, Roche) monoclonal antibodies or together with 12.5 ng of in vitro transcribed human GAPDH RNA (Thermo) for total RNA isolation and RT-qPCR. After DNase I digestion, reverse transcription was performed by Revert Aid first strand cDNA synthesis kit (Thermo) with random primers, and the obtained cDNAs were diluted and amplified in triplicate by a CFX Connect real-time PCR machine (Bio-Rad) using SYBR Green real-time PCR master mix (Toyobo) according to the manufacturer's protocol. Primers for quantitative PCR are listed in Supplemental Table S3.
High-salt disruption
Yeast cell lysates were incubated with 1 mM glucose for 10 min at 25°C to deplete the endogenous ATP, and then the salt concentration was adjusted to 1 M NaCl followed by incubation for 1 h at 4°C. The lysates were diluted to return salt concentration to 150 mM for 3 h at 4°C followed by centrifugation to remove any aggregates. Coimmunoprecipitation and Western blotting were then carried out as described above.
Supplementary Material
Acknowledgments
We are grateful to T. Bowman and members of the Query and Xu laboratories for helpful discussions and critical readings of the manuscript. This work was supported by grants from the National Natural Science Foundation of China (NSFC; 31525022, 91440109, and 31472045) and Chinese Academy of Sciences (KJZD-EW-L12) to Y.-Z.X, National Institutes of Health grant GM57829 to C.C.Q., and a Cancer Center Support (core) grant from the National Cancer Institute to the Albert Einstein College of Medicine. C.C.Q. is a scholar of the Irma T. Hirschl Trust.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.291872.116.
References
- Abdel-Wahab O, Levine R. 2011. The spliceosome as an indicted conspirator in myeloid malignancies. Cancer Cell 20: 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. 2016. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 7: 10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade MA, Petosa C, O'Donoghue SI, Muller CW, Bork P. 2001. Comparison of ARM and HEAT protein repeats. J Mol Biol 309: 1–18. [DOI] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. 2012. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491: 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SM, Guthrie C. 1993. Beat the clock: paradigms for NTPases in the maintenance of biological fidelity. Trends Biochem Sci 18: 381–384. [DOI] [PubMed] [Google Scholar]
- Burgess S, Couto JR, Guthrie C. 1990. A putative ATP binding protein influences the fidelity of branchpoint recognition in yeast splicing. Cell 60: 705–717. [DOI] [PubMed] [Google Scholar]
- Cingolani G, Petosa C, Weis K, Muller CW. 1999. Structure of importin-β bound to the IBB domain of importin-α. Nature 399: 221–229. [DOI] [PubMed] [Google Scholar]
- Cretu C, Schmitzova J, Ponce-Salvatierra A, Dybkov O, De Laurentiis EI, Sharma K, Will CL, Urlaub H, Luhrmann R, Pena V. 2016. Molecular architecture of SF3b and structural consequences of its cancer-related mutations. Mol Cell 64: 307–319. [DOI] [PubMed] [Google Scholar]
- Cvitkovic I, Jurica MS. 2013. Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res 41: D132–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. 2015. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep 13: 1033–1045. [DOI] [PubMed] [Google Scholar]
- DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. 2015. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol 11: e1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al. 2012. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486: 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, et al. 2013. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov 3: 1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galej WP, Wilkinson ME, Fica SM, Oubridge C, Newman AJ, Nagai K. 2016. Cryo-EM structure of the spliceosome immediately after branching. Nature 537: 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golas MM, Sander B, Will CL, Luhrmann R, Stark H. 2003. Molecular architecture of the multiprotein splicing factor SF3b. Science 300: 980–984. [DOI] [PubMed] [Google Scholar]
- Golas MM, Sander B, Will CL, Luhrmann R, Stark H. 2005. Major conformational change in the complex SF3b upon integration into the spliceosomal U11/U12 di-snRNP as revealed by electron cryomicroscopy. Mol Cell 17: 869–883. [DOI] [PubMed] [Google Scholar]
- Gozani O, Potashkin J, Reed R. 1998. A potential role for U2AF–SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol Cell Biol 18: 4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. 1999. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell 96: 99–110. [DOI] [PubMed] [Google Scholar]
- Hahn CN, Scott HS. 2012. Spliceosome mutations in hematopoietic malignancies. Nat Genet 44: 9–10. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. 2013. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 45: 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Moore MJ. 2012. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem Sci 37: 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvorecny KL, Prelich G. 2010. A systematic CEN library of the Saccharomyces cerevisiae genome. Yeast 27: 861–865. [DOI] [PubMed] [Google Scholar]
- Kesarwani AK, Ramirez O, Gupta AK, Yang X, Murthy T, Minella AC, Pillai MM. 2016. Cancer-associated SF3B1 mutants recognize otherwise inaccessible cryptic 3′ splice sites within RNA secondary structures. Oncogene. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarska MM, Query CC. 2005. Insights into the mechanisms of splicing: more lessons from the ribosome. Genes Dev 19: 2255–2260. [DOI] [PubMed] [Google Scholar]
- Kong Y, Krauthammer M, Halaban R. 2014. Rare SF3B1 R625 mutations in cutaneous melanoma. Melanoma Res 24: 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koodathingal P, Staley JP. 2013. Splicing fidelity: DEAD/H-box ATPases as molecular clocks. RNA Biol 10: 1073–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, et al. 2013. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152: 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardelli RM, Thompson JX, Yates JR III, Stevens SW. 2010. Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA 16: 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser CF, Guthrie C. 1993. Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics 133: 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WW, Cheng SC. 2015. A novel mechanism for Prp5 function in prespliceosome formation and proofreading the branch site sequence. Genes Dev 29: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, Travaglino E, Groves MJ, Godfrey AL, Ambaglio I, et al. 2011. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 118: 6239–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayas RM, Maita H, Staley JP. 2006. Exon ligation is proofread by the DExD/H-box ATPase Prp22p. Nat Struct Mol Biol 13: 482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPheeters DS, Muhlenkamp P. 2003. Spatial organization of protein–RNA interactions in the branch site-3′ splice site region during pre-mRNA splicing in yeast. Mol Cell Biol 23: 4174–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald AF, Hirano T. 2000. HEAT repeats associated with condensins, cohesins, and other complexes involved in chromosome-related functions. Genome Res 10: 1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Day CL, Dalbadie-McFarland G, Abelson J. 1996. The Saccharomyces cerevisiae Prp5 protein has RNA-dependent ATPase activity with specificity for U2 small nuclear RNA. J Biol Chem 271: 33261–33267. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. 2011. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365: 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Query CC, Konarska MM. 2004. Suppression of multiple substrate mutations by spliceosomal prp8 alleles suggests functional correlations with ribosomal ambiguity mutants. Mol Cell 14: 343–354. [DOI] [PubMed] [Google Scholar]
- Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez-Trillos A, et al. 2012. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44: 47–52. [DOI] [PubMed] [Google Scholar]
- Rauhut R, Fabrizio P, Dybkov O, Hartmuth K, Pena V, Chari A, Kumar V, Lee CT, Urlaub H, Kastner B, et al. 2016. Molecular architecture of the Saccharomyces cerevisiae activated spliceosome. Science 353: 1399–1405. [DOI] [PubMed] [Google Scholar]
- Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, Fangazio M, Vaisitti T, Monti S, Chiaretti S, et al. 2011. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood 118: 6904–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby SW, Chang TH, Abelson J. 1993. Four yeast spliceosomal proteins (PRP5, PRP9, PRP11, and PRP21) interact to promote U2 snRNP binding to pre-mRNA. Genes Dev 7: 1909–1925. [DOI] [PubMed] [Google Scholar]
- Schellenberg MJ, Edwards RA, Ritchie DB, Kent OA, Golas MM, Stark H, Luhrmann R, Glover JN, MacMillan AM. 2006. Crystal structure of a core spliceosomal protein interface. Proc Natl Acad Sci 103: 1266–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semlow DR, Blanco MR, Walter NG, Staley JP. 2016. Spliceosomal DEAH-box ATPases remodel pre-mRNA to activate alternative splice sites. Cell 164: 985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Kim HS, Cao Y, Xu YZ, Query CC. 2012. A U1–U2 snRNP interaction network during intron definition. Mol Cell Biol 32: 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadaccini R, Reidt U, Dybkov O, Will C, Frank R, Stier G, Corsini L, Wahl MC, Luhrmann R, Sattler M. 2006. Biochemical and NMR analyses of an SF3b155–p14–U2AF–RNA interaction network involved in branch point definition during pre-mRNA splicing. RNA 12: 410–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. 1998. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 92: 315–326. [DOI] [PubMed] [Google Scholar]
- Strefford JC, Sutton LA, Baliakas P, Agathangelidis A, Malcikova J, Plevova K, Scarfo L, Davis Z, Stalika E, Cortese D, et al. 2013. Distinct patterns of novel gene mutations in poor-prognostic stereotyped subsets of chronic lymphocytic leukemia: the case of SF3B1 and subset #2. Leukemia 27: 2196–2199. [DOI] [PubMed] [Google Scholar]
- Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI. 2016. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 35: 2413–2427. [DOI] [PubMed] [Google Scholar]
- Tazi J, Bakkour N, Stamm S. 2009. Alternative splicing and disease. Biochim Biophys Acta 1792: 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa T, Guthrie C. 2005. The Isy1p component of the NineTeen complex interacts with the ATPase Prp16p to regulate the fidelity of pre-mRNA splicing. Genes Dev 19: 1894–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconte V, Rogers HJ, Singh J, Barnard J, Bupathi M, Traina F, McMahon J, Makishima H, Szpurka H, Jankowska A, et al. 2012. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood 120: 3173–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Wan Y, Wu CJ. 2013. SF3B1 mutations in chronic lymphocytic leukemia. Blood 121: 4627–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan R, Yan C, Bai R, Huang G, Shi Y. 2016. Structure of a yeast catalytic step I spliceosome at 3.4 A resolution. Science 353: 895–904. [DOI] [PubMed] [Google Scholar]
- Wang C, Chua K, Seghezzi W, Lees E, Gozani O, Reed R. 1998. Phosphorylation of spliceosomal protein SAP 155 coupled with splicing catalysis. Genes Dev 12: 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, et al. 2011. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365: 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warkocki Z, Odenwalder P, Schmitzova J, Platzmann F, Stark H, Urlaub H, Ficner R, Fabrizio P, Luhrmann R. 2009. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat Struct Mol Biol 16: 1237–1243. [DOI] [PubMed] [Google Scholar]
- Will CL, Luhrmann R. 2011. Spliceosome structure and function. Cold Spring Harb Perspect Biol 3: a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Schneider C, MacMillan AM, Katopodis NF, Neubauer G, Wilm M, Luhrmann R, Query CC. 2001. A novel U2 and U11/U12 snRNP protein that associates with the pre-mRNA branch site. EMBO J 20: 4536–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DS, Keefe AD. 2001. Random mutagenesis by PCR. Curr Protoc Mol Biol 51: 8.3.1–8.3.9. [DOI] [PubMed] [Google Scholar]
- Xu YZ, Query CC. 2007. Competition between the ATPase Prp5 and branch region-U2 snRNA pairing modulates the fidelity of spliceosome assembly. Mol Cell 28: 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YZ, Newnham CM, Kameoka S, Huang T, Konarska MM, Query CC. 2004. Prp5 bridges U1 and U2 snRNPs and enables stable U2 snRNP association with intron RNA. EMBO J 23: 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Wan R, Bai R, Huang G, Shi Y. 2016. Structure of a yeast activated spliceosome at 3.5 Å resolution. Science 353: 904–911. [DOI] [PubMed] [Google Scholar]
- Yang F, Wang XY, Zhang ZM, Pu J, Fan YJ, Zhou J, Query CC, Xu YZ. 2013. Splicing proofreading at 5′ splice sites by ATPase Prp28p. Nucleic Acids Res 41: 4660–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69. [DOI] [PubMed] [Google Scholar]
- Zhang J, Manley JL. 2013. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov 3: 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZM, Yang F, Zhang J, Tang Q, Li J, Gu J, Zhou J, Xu YZ. 2013. Crystal structure of Prp5p reveals interdomain interactions that impact spliceosome assembly. Cell Rep 5: 1269–1278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.