

Abstract
Maintenance of telomeres by telomerase permits continuous proliferation of rapidly dividing cells, including the majority of human cancers. Despite its direct biomedical significance, the architecture of the human telomerase complex remains unknown. Generating homogeneous telomerase samples has presented a significant barrier to developing improved structural models. Here we pair single-molecule Förster resonance energy transfer (smFRET) measurements with Rosetta modeling to map the conformations of the essential telomerase RNA core domain within the active ribonucleoprotein. FRET-guided modeling places the essential pseudoknot fold distal to the active site on a protein surface comprising the C-terminal element, a domain that shares structural homology with canonical polymerase thumb domains. An independently solved medium-resolution structure of Tetrahymena telomerase provides a blind test of our modeling methodology and sheds light on the structural homology of this domain across diverse organisms. Our smFRET-Rosetta models reveal nanometer-scale rearrangements within the RNA core domain during catalysis. Taken together, our FRET data and pseudoatomic molecular models permit us to propose a possible mechanism for how RNA core domain rearrangement is coupled to template hybrid elongation.
Keywords: single-molecule FRET, modeling, Rosetta, telomerase, pseudoknot, polymerase
INTRODUCTION
The telomerase catalytic core consists of the telomerase reverse transcriptase (TERT) protein subunit and an integral RNA component (hTR) that carries the template for telomerase reverse transcription (Zhang et al. 2011). Telomerase differs from other reverse transcriptases in its ability to add multiple telomere repeats to its DNA substrate during a single binding event, an activity termed repeat addition processivity (RAP) (Fig. 1A). Mutations in several conserved telomerase protein and RNA domains abrogate RAP (Lai et al. 2002; Zaug et al. 2008; Robart and Collins 2011; Eckert and Collins 2012), yet the precise molecular mechanism for how these domains contribute to processive telomere repeat addition is not well understood. Structures of several conserved vertebrate hTR fragments have been reported, including two RNA motifs required for catalytic activity: the CR4/5 three-way junction and the pseudoknot fold (Kim et al. 2008, 2014). A recent crystal structure of the CR4/5 domain bound to the telomerase RNA binding domain revealed the details of this protein–RNA interaction (Huang et al. 2014); yet, the precise function of this RNA element is unclear. Similarly, the structure of the native RNA pseudoknot fold is solved, but its location and function within the current low-resolution structure of human telomerase remains unknown (Kim et al. 2008; Sauerwald et al. 2013).
FIGURE 1.

Single-molecule FRET network in the active telomerase RNP. (A) A simplified catalytic mechanism of telomerase is delineated. Telomerase binds to a DNA substrate and reverse transcribes the integral RNA template. The nascent DNA is realigned with the downstream template region, allowing addition of a subsequent telomere repeat to the DNA substrate. (B) Secondary structures of core and CR4/5 domains used in telomerase reconstitution. Five label sites (denoted by black circles) span the two domains and create a network of FRET pairs. Secondary structure information was adapted from the telomerase database (Podlevsky et al. 2008). (C) Dye-labeled RNA fragments are reconstituted with TERT to generate catalytically active RNPs. Each complex is surface immobilized through a 5′ biotinylated DNA primer (TG)6TTAGGG that hybridizes to the template region of telomerase RNA.
Sparse biophysical and biochemical data can be integrated with structural modeling as an effective strategy for generating nanometer resolution pseudoatomic models of complex biological systems (Magnus et al. 2014; Cheng et al. 2015b). Here, we have integrated single-molecule Förster resonance energy transfer (smFRET) measurements with a Rosetta-based modeling approach to establish the architecture of the human telomerase core RNP and reveal novel conformational changes in hTR. FRET is a biophysical structure-probing tool that monitors the distance-dependent energy transfer between a donor dye and an acceptor dye strategically coupled to the molecule of interest. Moreover, FRET at the single-molecule level permits dissection of heterogeneous and dynamic enzyme subpopulations. To date, most FRET studies of the telomerase RNP have focused on gaining qualitative insight about structure and molecular dynamics (Stone et al. 2007; Wu et al. 2010; Berman et al. 2011; Mihalusova et al. 2011; Hengesbach et al. 2012; Parks and Stone 2014; Akiyama et al. 2015). In the cases where FRET was used for molecular distance constraints, modeling uncertainty remained high due to the difficulty of converting FRET to precise distance values (Gavory et al. 2006; Cole et al. 2012). Much of this difficulty results from the errors introduced by the unique dye environments in each FRET sample (Muschielok and Michaelis 2011). However, these challenges can be overcome by several methods, including triangulation of dye positions through multiple FRET measurements (Andrecka et al. 2009; Muschielok and Michaelis 2011). Our study uses the Rosetta modeling algorithm that predicts de novo RNA structure through an energy minimization approach (Cheng et al. 2015a). We expand on this approach by imposing compliant distance ranges for each FRET constraint rather than discrete distances, devaluing precise calibration of individual distances and emphasizing redundancy in the FRET distance network. This modeling decision avoids the need for complex biophysical parameterization that is currently not feasible for telomerase due to its low reconstitution efficiency.
In the present work, we report an interdomain FRET network that maps the conformation of the RNA core domain (hTR 32-195) within the catalytically active RNP. The FRET data alone provide qualitative insights about the RNA conformation within the RNP as well as the RNA dynamics associated with catalytic activity. In order to expand our interpretation of the FRET data, we convert the FRET data into flexible distance constraints for Rosetta modeling to produce pseudoatomic models of the hTR pseudoknot domain architecture in the assembled RNP complex. Interestingly, the modeling reveals a convergent solution to the RNA core domain architecture wherein the catalytically indispensable pseudoknot fold (Fig. 1, red, hTR 93–121, 171–184) contacts a protein surface distant from the RNA template. Upon stimulation of telomerase activity, smFRET-Rosetta modeling provides a means for visualizing the RNA conformational rearrangements observed in the single-molecule data. Our FRET data and models are consistent with a model wherein the RNA core domain rearrangement is coupled to DNA synthesis at the active site.
RESULTS
Mapping telomerase RNA structure within the RNP using a single-molecule FRET network
To investigate the relative positioning of essential RNA structural elements within the assembled telomerase RNP complex we devised a network of smFRET pairs including five distinct labeling sites within the hTR core domain and CR4/5 domains (Fig. 1B). Four label sites in the core domain were strategically chosen to analyze the core domain architecture, as well as the relative orientation of the pseudoknot fold and template region. Knowledge of the high-resolution position of the CR4/5 domain label site (U312) relative to the telomerase protein, assessed by homology modeling with the reported O. latipes structure, provided an important reference point for smFRET measurements to all other label sites in the core domain (Huang et al. 2014). For all measurements used in our study, the core and CR4/5 domains were prepared as two separate RNA fragments and reconstituted in trans into active telomerase RNP complexes (Fig. 1; Supplemental Fig. S1). This approach simplifies preparation of the dye-labeled RNAs and has previously been shown to support reconstitution of catalytically active telomerase (Parks and Stone 2014). We prepared telomerase enzymes harboring one of 10 unique smFRET pairs, across the two RNA domains, using established telomerase reconstitution and immunopurification techniques (Chen and Greider 2003). Each labeled telomerase enzyme was then surface-immobilized for smFRET analysis via binding to a 5′-terminal biotinylated DNA primer with sequence (TG)6TTAGGG (Fig. 1C). This DNA primer possesses native telomere DNA sequence exclusively at the 3′ terminus, ensuring that all complexes are bound through the same 7-nt DNA/RNA hybrid in the telomerase RNP (Berman et al. 2011; Akiyama et al. 2015). We note that the repeating (TG) dinucleotide sequence present in this telomere DNA primer does not alter the telomerase reaction product profile when assayed in vitro (Supplemental Fig. S1, lanes 1,2). Moreover, while unassembled smFRET RNA can, in principle, form the same 7-bp hybrid with the modified primer, control experiments revealed that RNA alone did not form a stable hybrid with the primer, demonstrating the specificity of the primer–telomerase RNP interactions in our experiments (Supplemental Fig. S2). Thus, specific immobilization of telomerase complexes through the high-affinity (TG)6 TTAGGG DNA primer permits analysis of “stalled” telomerase RNP complexes (Wallweber et al. 2003).
Single-molecule FRET measurements were made using a prism-type total internal reflection fluorescence (TIRF) microscope, and each experiment included data collected from at least 50 individual telomerase complexes. The stalled telomerase complexes revealed predominantly unimodal smFRET distributions, consistent with a stably assembled and homogeneous RNP structure. All smFRET values fell within the sensitive range of the FRET response (FRET = 0.22–0.76) (Fig. 2 A–J; Table 1). FRET measurements between U92–U184 yielded a similar distribution to previous work (Hengesbach et al. 2012), suggesting the pseudoknot fold triplex is also formed in the assembled human RNP (Fig. 2H, red). FRET measurements between U312 in CR4/5 and two positions neighboring the template unambiguously place the P6.1 stem of CR4/5 closer to the 3′ template region than the 5′ template region (Fig. 2D vs. G, red). The FRET results within the core domain, both within the RNP and for the free RNA, are generally consistent with a previously proposed model of the protein-free hTR core domain (Supplemental Fig. S3; Zhang et al. 2010). Our smFRET data therefore support the hypothesis of a triangular core domain architecture, which is largely preserved within the assembled telomerase RNP.
FIGURE 2.
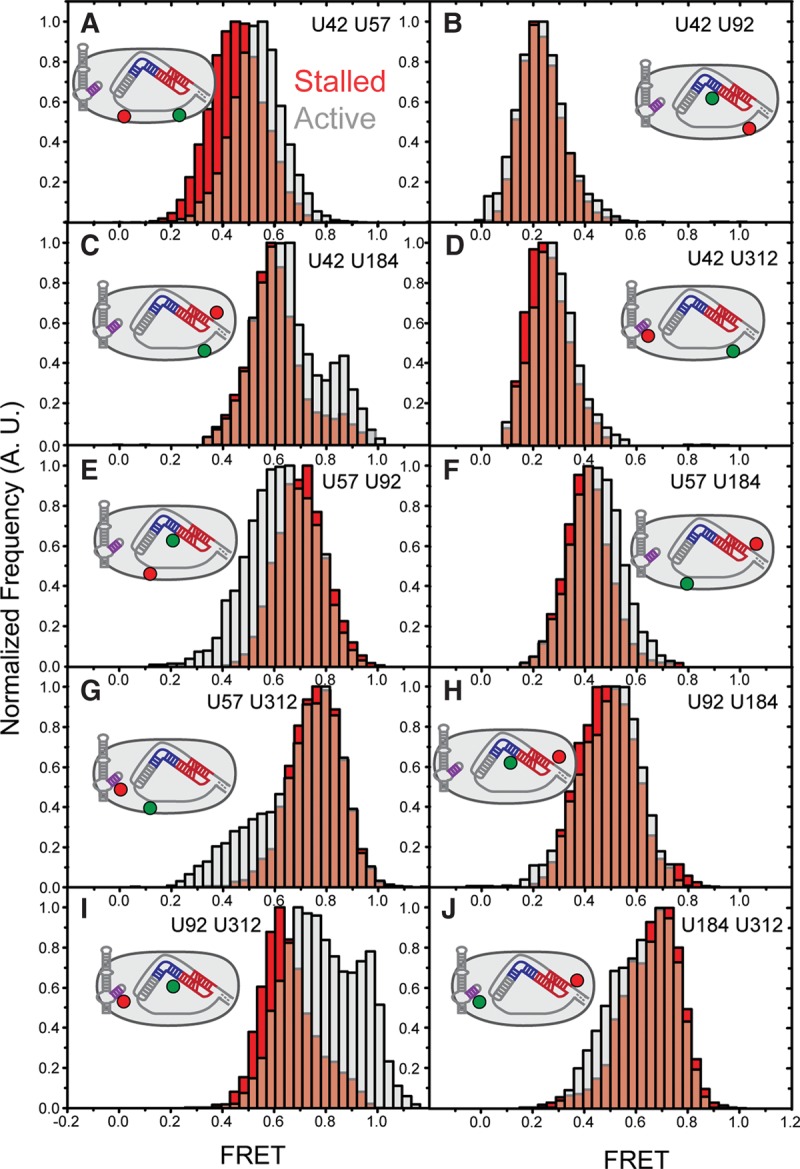
Telomerase activity modulates RNA conformation inducing multiple FRET changes. (A−J) Each panel represents reconstructed FRET histograms generated by compiling single-molecule traces. The FRET label sites are listed in plain text in the panel and depicted in the 2D schematic of telomerase. Red distributions were obtained by binding individual telomerase complexes to 5′-Bio-(TG)6TTAGGG-3′, capturing time resolved traces and then binning the corrected data into a histogram. Gray histograms represent exposure of the telomerase complexes to telomerase activity buffer, followed by similar data collection and analysis. All histograms were generated from at least 50 molecules that exhibited the proper photo-bleaching events for photo-physical correction.
TABLE 1.
FRET distance constraints used for Rosetta modeling
To specifically test whether the core domain architecture is modified during telomerase RAP activity, we initiated telomerase catalysis by introducing activity buffer containing dGTP, dTTP, and dATP to the surface-immobilized complexes. Previous smFRET studies employing this same approach revealed the majority (>90%) of the surface-immobilized telomerase complexes are catalytically active under our experimental conditions (Parks and Stone 2014). Addition of the telomerase activity buffer specifically promotes rapid extension of the DNA substrate to the template boundary, followed by the relatively slow translocation substep (Fig. 1A; Latrick and Cech 2010; Parks and Stone 2014).
Analysis of the active telomerase enzymes revealed significant changes in five of the 10 smFRET distributions, which collectively indicate a conformational rearrangement of the RNA core domain during telomerase catalysis (Fig. 2, compare red and gray histograms). The shift in the distribution measuring template hybrid conformation indicates the template RNA undergoes a conformational rearrangement during telomere repeat synthesis (Fig. 2A, red vs. gray). FRET dyes probing directly 3′ of the RNA template, revealed shifted distributions supporting the notion of dynamics in this region of hTR during telomerase catalysis (Fig. 2E and G, red vs. gray; Berman et al. 2011). Measurements at the 5′ end of the template (Fig. 2C, red vs. gray) revealed a new peak at ∼0.85 FRET suggesting that the P1 stem may exist in multiple distinct conformations during the repeat addition cycle (Chen and Greider 2003). Surprisingly, the most pronounced FRET change was observed in the distribution of the U92-U312 smFRET pair, which probed the relative positions of the CR4/5 domain and the RNA pseudoknot fold (Fig. 2I, red vs. gray). As expected, the CR4/5 domain did not show activity-dependent structural changes with relation to the U42 position, which was shown in previous work to be static during telomerase catalysis (Fig. 2D, red vs. gray; Parks and Stone 2014). The notion of a static CR4/5 domain is also consistent with the extensive contacts made between the CR4/5 RNA and the RNA binding domain of TERT in the O. latipes structure (Huang et al. 2014). Thus, the prominent shift in the U92–U312 FRET distribution (Fig. 2I) likely represents motion of the RNA pseudoknot fold during catalysis. Finally, we did not detect any significant FRET change across the RNA pseudoknot fold, indicating that the human pseudoknot fold is stably folded throughout the catalytic cycle at the time resolution of our experiments (Fig. 2H, red vs. gray; Hengesbach et al. 2012). Moreover, the observed FRET value of the RNA pseudoknot fold within the RNP context is in excellent agreement with expectation based upon the reported NMR structure (Kim et al. 2008), providing a useful internal distance control for our smFRET measurements. These qualitative interpretations of the smFRET data provide useful information about RNA domain structure and dynamics within the catalytically active telomerase complex. However, we sought a more detailed perspective of the RNP structure and dynamics by using automated molecular modeling paired with the experimentally derived structural constraints provided by our smFRET approach.
Modeling telomerase RNP organization with smFRET-Rosetta
Prior to modeling the hTR core domain within the telomerase RNP, we first combined known telomerase RNA and protein substructures to create a modeling scaffold. The smFRET results were consistent with a homogeneous domain architecture and a native conformation of the pseudoknot fold; therefore, we integrated known structures of hTR core domain motifs into our scaffold. Solution structures of the J2a/b and pseudoknot fold were treated as rigid bodies during modeling (Fig. 3). The template–primer hybrid was modeled as an ideal A-form helix, based on our smFRET binding data and the crystal structure of the T. castaneum TERT bound to a model DNA/RNA hybrid (Mitchell et al. 2010). The P2a.1 region was also modeled as an A-form helix based on previous chemical mapping experiments (Ly et al. 2003). Lastly, while smFRET data conclusively demonstrated stable P1 stem formation in RNA alone (Supplemental Fig. S4), data taken within the assembled RNP could be consistent with either formation or remodeling of the P1 helix. Therefore, to minimize the number of assumptions made in our modeling we did not enforce formation of the P1 stem within the RNP.
FIGURE 3.

A FRET constrained Rosetta modeling workflow. Known telomerase substructures were integrated to generate a modeling scaffold. Along with RNA conformation, a Scoreclash term is generated in order to prevent steric clashing of the RNA with the protein homology model. Lastly, the FRET distance constraints are integrated through a ScoreFRET term that is defined by a smooth square well potential that penalizes relatively large differences between modeled distances and FRET-defined distances.
Next we assembled a static structure of the telomerase reverse transcriptase protein component (hTERT). A homology model of hTERT was built by modifying a previously published homology model with an updated homology model of the TRBD (Fig. 3; Steczkiewicz et al. 2011). We note that the position of the hTERT essential N-terminal (TEN) domain in the protein scaffold is speculative; therefore, control modeling runs were also performed in the absence of this domain (see below). To complete our modeling scaffold, we used two published RNP structures of telomerase subcomplexes to fix known RNA/protein interaction sites (Fig. 3). The template hybrid was positioned by aligning the 3′ end of the DNA in the protein active site based on the T. castaneum TERT structure bound to an A-form helix (Mitchell et al. 2010). A recent structure of the O. latipes TRBD in complex with CR4/5 allowed us to position the P6.1 helix directly on the homology model TRBD through alignment (Huang et al. 2014). Combining all of this prior knowledge, the initial modeling point can be envisioned as a telomerase RNP complex where the P6.1 stem and template hybrid were fixed with respect to the hTERT homology model. The P2a.1, J2a/b, and the pseudoknot fold were free to move relative to TERT, and the remaining RNA (40 nucleotides) was structurally unconstrained.
Models of the entire core domain RNA within the RNP were generated using a version of the previously reported fragment assembly approach (Cheng et al. 2015a). This method was modified to incorporate smFRET distance constraints and steric clashes between the RNA and the protein (Fig. 3). Two separate modeling runs were performed, using either the FRET data collected on stalled or active enzymes. Modeling runs consisted of approximately 2500 independent trials, each resulting in a final model (Supplemental Fig. S5A,B). To utilize the FRET constraints in the modeling procedure, it was necessary to convert our measured FRET efficiencies to physical distances using the standard FRET expression:
| (1) |
Previous work has highlighted the importance of obtaining appropriate photo-physical correction terms to reliably convert FRET efficiency measurements into accurate distances (Muschielok et al. 2008). While our study permitted us to make several of the necessary corrections by analyzing and adjusting individual FRET trajectories (see Materials and Methods), accurate experimental determination of the Förster radius (Ro) for each FRET pair was not feasible due to difficulties generating large quantities of each smFRET-labeled telomerase. However, several observations suggest that the approximation Ro = 5.6 nm for the Cy3/Cy5 pair used in our modeling is reasonable (Andrecka et al. 2009). First, previous work characterizing the observed experimental variation of Ro has shown that, on average, Ro deviates from the standard values by approximately ±0.2 nm (Andrecka et al. 2009). Second, we conservatively set the ScoreFRET term to only introduce a penalty if the modeled distance deviates more than approximately ±0.5 nm from the calculated distance, which encompasses even the worst reported errors in Ro (Fig. 3; Table 1; Andrecka et al. 2009). Such a high level of compliance in the distance constraints might be expected to prohibit convergence during modeling; however, the intrinsic redundancy within the FRET network helped localize each dye position within the network.
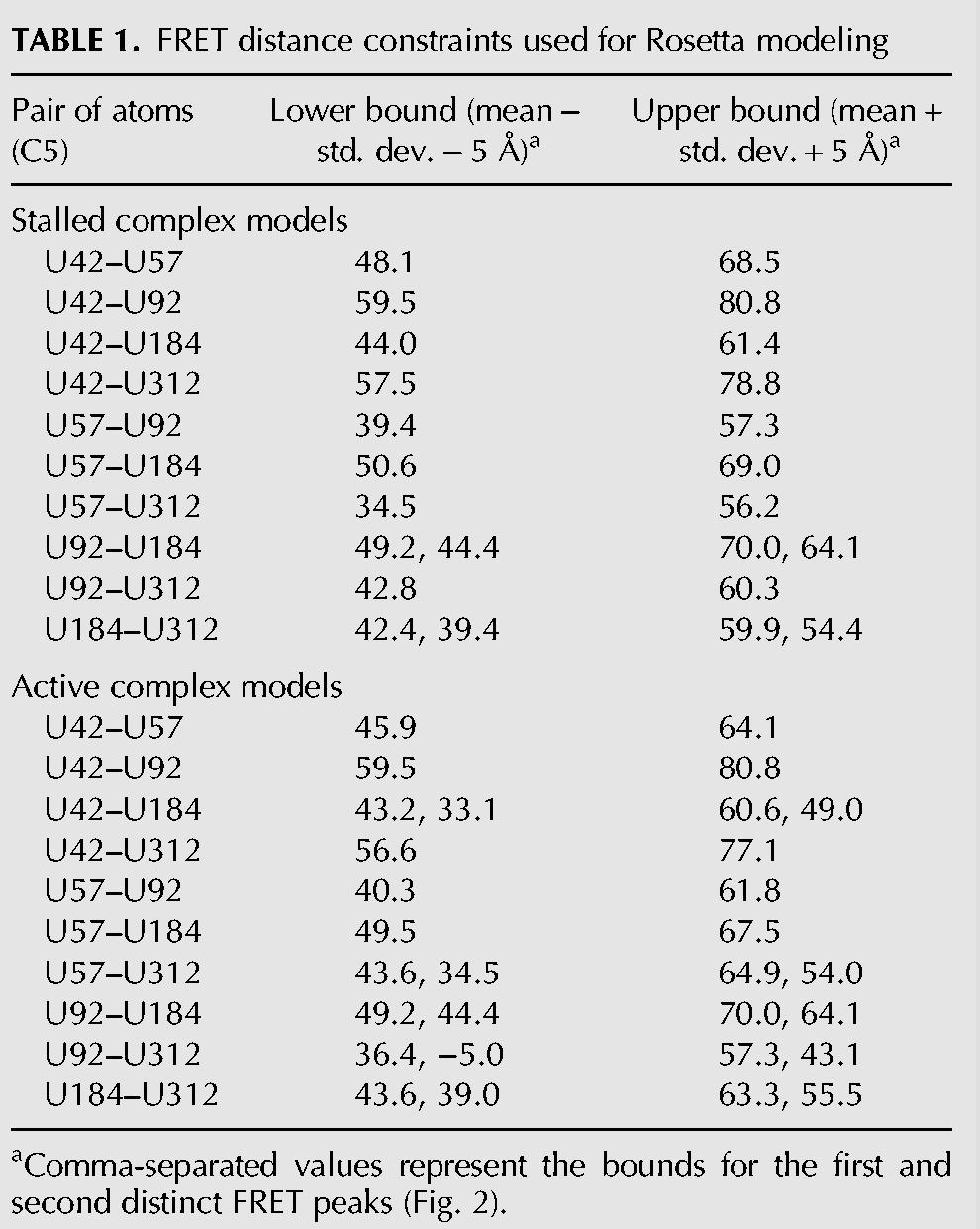
Modeling of the RNA core domain around the RNP scaffold yielded convergent models for the stalled complexes (Fig. 4A; Supplemental Fig. S5A,C). The best scoring models were clustered as described previously (Cheng et al. 2015a). Each cluster represented a single model class for the RNA core domain, supporting the notion that our network of compliant distance constraints was capable of producing a specific RNP architecture (Supplemental Fig. S5C). Modeling of the stalled complex yielded two low-scoring clusters: one of these clusters was disregarded due to steric clashes with the proposed position of the CR4/5 domain (Supplemental Fig. S5A; Huang et al. 2014). Comparison of predicted FRET values (extracted from the converged models of the sterically viable cluster) versus the experimental FRET values revealed a high degree of correlation, confirming that the experimental FRET distance constraints were satisfied during the modeling process (Fig. 4B). In contrast, modeling runs in which FRET constraints were not enforced failed to achieve comparable convergence to a single model class and gave predicted FRET values that did not correlate well with experimental values (Fig. 4C,D). Surprisingly, in the FRET-guided models, the catalytically essential pseudoknot fold was placed on a protein face opposite the active site, contacting the TERT C-terminal extension (CTE) domain (Fig. 4A; Supplemental Fig. S5C). The intrinsic bend of the J2a/b bulge permitted the P2 stem structure to wrap around the CTE, guiding the free template strand toward the active site. Notably, the general architecture of the RNA core domain in the models was not impacted by removing the TEN domain from the modeling scaffold, or by enforcing formation of the P1 stem and docking this stem onto the TERT scaffold based upon a homologous structure from Tetrahymena telomerase (Supplemental Fig. S6A,B; Jansson et al. 2015). As an additional test of this architecture, new models including P4 and P5 stems confirmed that CR4/5 and core domains could be bridged in stereochemically viable conformations (Supplemental Fig. S7).
FIGURE 4.
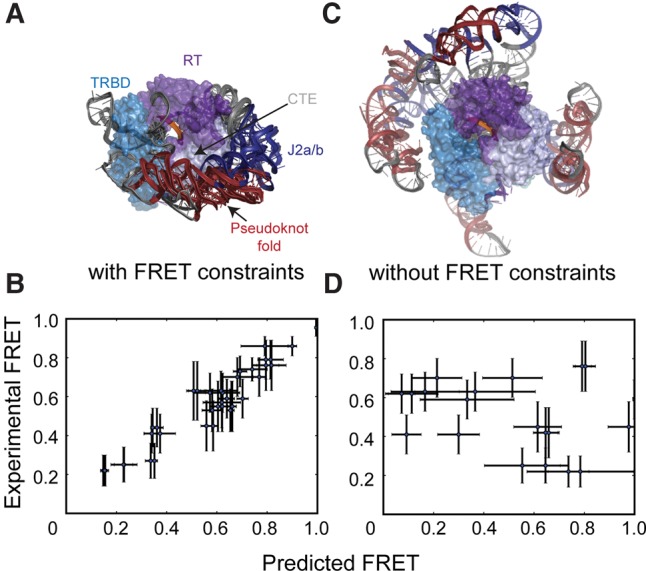
FRET-Rosetta modeling produces convergent solutions to the core domain architecture. (A) Superposition of top scoring RNP models generated with an unformed P1 stem for stalled telomerase complexes. (B) Distances from the Rosetta models utilizing the FRET distance constraints are converted to predicted FRETs and plotted versus the corresponding experimental FRET values. The correlation confirms that the modeling process is satisfying all distance constraints during the modeling process. (C) Lowest energy models for RNP architecture when the ScoreFRET term is removed from the scoring function. (D) Removal of the FRET distance constraints during Rosetta modeling results in predicted FRETs that are uncorrelated from the experimental FRET values, suggesting the ScoreRNA and Scoreclash are insufficient for producing an accurate representation of the experimental FRET values. Error bars (B,D) in the x-direction are computed by taking the standard deviation of the predicted FRET values for the 10 lowest scoring models in the largest cluster. Error bars in the y-direction are the standard deviations of the peaks from the experimental FRET histograms.
Modeling the RNA core domain with distance constraints generated from active telomerase complexes returned two classes of sterically viable models (Fig. 5A; Supplemental Fig. S5B,C). The two distinct model classes are a result of the two peaks in the U92–U312 activity histogram (Fig. 2I, gray). Comparison of these activity-dependent models (state II and state III) with the stalled model (state I) reveals a repositioning of the RNA pseudoknot fold around the CTE, consistent with the increase in FRET associated with telomerase activity (Figs. 2I, 5A). As before, the extracted FRET values from the converged models exhibited a high degree of correlation with the experimental data (Fig. 5B). Notably, under the conditions of our experiments, initiation of telomerase activity results in rapid extension of the DNA:RNA hybrid, followed by a subsequent pause at the template boundary. In previous work, this catalytic substep was termed the “pre-translocated” state to contrast with the “post-translocated” state of the enzyme in which the realigned DNA:RNA hybrid is stably engaged in the TERT active site and poised for the subsequent round of telomere repeat synthesis (Parks and Stone 2014). Thus, the activity-dependent conformations of the RNA pseudoknot fold (states II and III) revealed in the current study correlate with the previously described “pre-translocated” catalytic substep.
FIGURE 5.
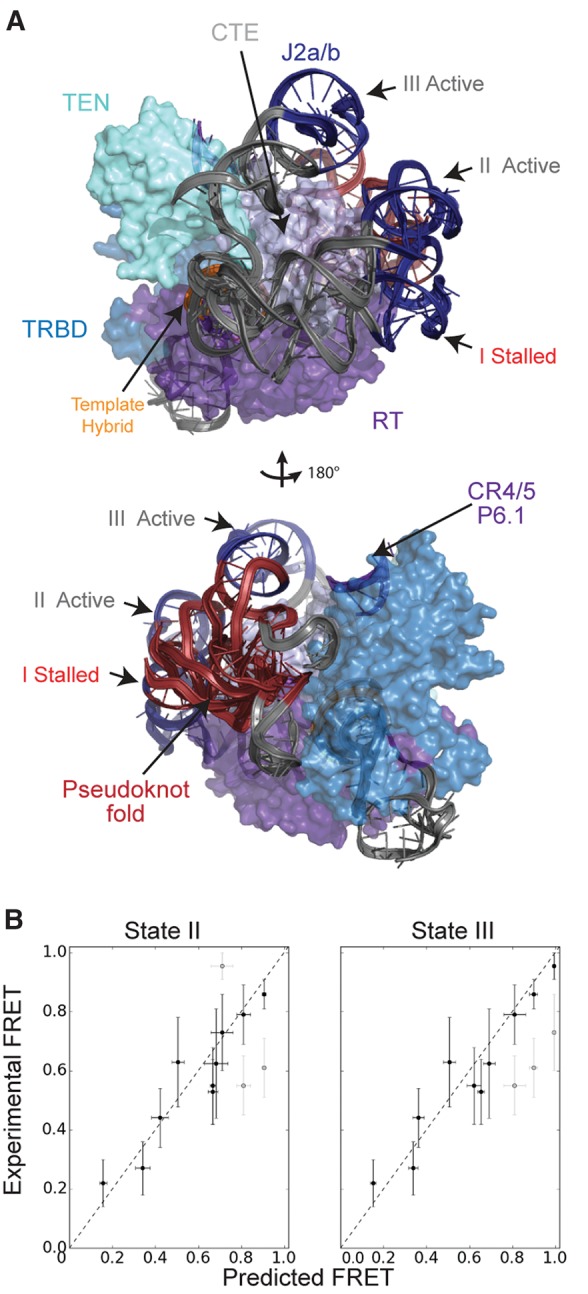
Single-molecule FRET-Rosetta predicts activity-induced motion of the core domain around the C-terminal element. (A) Three distinct RNA states emerged from the modeling when using both the stalled and active data. State I is produced when telomerase is modeled with the stalled data (red histograms, Fig. 2). States II and III are a result of modeling the RNP with the active data (gray histograms, Fig. 2). Both states II and III show a rotation of the pseudoknot fold around the complex away from its original stalled position of state I. (B) Distances were measured in top scoring models from clusters representing each of the states, then converted to FRET values and averaged. These averages are plotted with the standard deviations shown as the error bars in the x-direction. Error bars in the y-direction are the standard deviations of the peaks from the experimental FRET histograms. Separate points are shown for each of the U92–U312, U57–U312, and U42–U184 peaks in the states II and III plots. The unsatisfied peaks in each of these plots are shown in light gray. The difference between states II and III is due to the two U92–U312 FRET peaks in the activity data. Strong correlation between the experimental FRET and predicted FRET confirms that the models satisfy all the distance constraints.
Comparison of our human RNP models with the recently reported 9 Å cryoEM structure of Tetrahymena telomerase provided an excellent blind test for the accuracy of our modeling (Jiang et al. 2015). Excitingly, the human telomerase models place the pseudoknot fold on the same protein face and in a similar position as in the 9 Å Tetrahymena telomerase structure (Fig. 6). Out of these models, the active complex (state III) is most similar to the Tetrahymena structure, which is consistent with the notion that the recent structure represents the pre-translocated state (Fig. 6C). This comparison represents an ideal test case, shedding light on the resolution and accuracy of our modeling methodology, as well as the shared domain organization of telomerase enzymes from diverse species.
FIGURE 6.
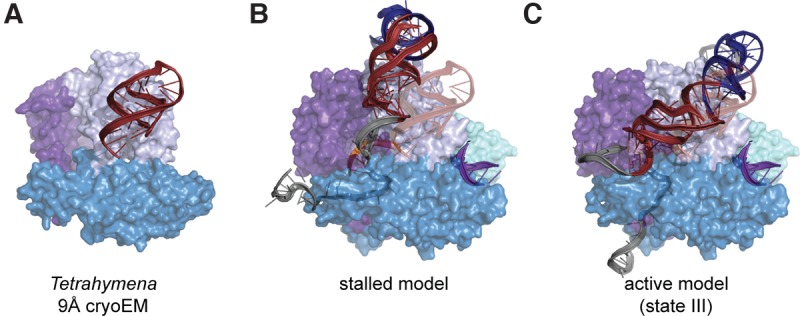
Human RNP models reveal structural homology between human and Tetrahymena RNA architectures. (A) The Tetrahymena cryoEM structure is reduced to depiction of the TERT protein component and the pseudoknot fold. (B) Comparison of P1 not formed core domain (red) model versus the Tetrahymena pseudoknot fold (opaque red overlay; distance between pseudoknot fold centers-of-mass: 1.9nm). (C) Comparison of state iii activity-based RNA architecture versus the Tetrahymena pseudoknot fold (opaque red overlay; distance between pseudoknot fold centers-of-mass: 1.5 nm).
Pseudoknot fold motion tracks telomerase catalysis through the TRE
When considering possible sources of the observed RNA core domain conformational change, we recognized that each round of telomere DNA synthesis may induce template hybrid rotation and extrusion out of the active site. Interestingly, during this putative hybrid rotation, the 3′ end of the RNA template would track the proposed pseudoknot fold motion. This correlation raised the possibility that telomere DNA repeat synthesis at the telomerase active site may be physically coupled to pseudoknot fold motion through the intervening single-stranded RNA (Fig 1, nucleotides 57–63). Similarly, this hypothesis suggested that physical connectivity between the template region and the pseudoknot fold may be essential for telomerase processivity.
We set out to directly test this notion by physically decoupling the template hybrid from the pseudoknot fold using a circular permutation of the core domain (cp-hTR) that introduces a physical break between hTR nucleotides 62–63. We reconstituted telomerase enzymes with the cp-hTR core domain, and elected to use the FRET pair (U92–U312) that was most sensitive to conformational rearrangements induced by telomerase catalysis. When stalled cp-hTR telomerase enzymes were analyzed at the single-molecule level, we observed a very similar FRET distribution in comparison to wild-type telomerase (Fig. 7A,B). Strikingly, addition of activity buffer to the cp-hTR telomerase completely failed to shift the major FRET peak to the higher value that gave rise to the “state II” conformation of the RNA core domain in our modeling (compare Fig. 7C,D). In contrast, the active cp-hTR telomerase was competent to sample the minor high FRET population that coincided with the “state III” conformation of the core domain. Taken together, these results demonstrate that physical connectivity between the template and the pseudoknot fold is strictly required for the activity-dependent conformational change into the “state II” conformation of the core domain. Next, we performed bulk telomerase activity assays comparing wild-type and cp-hTR telomerase complexes. Both enzymes were competent to bind telomere DNA primer and extend to the end of the first repeat, but the cp-hTR telomerase was unable to perform the translocation step that is required for processive addition of multiple telomere DNA repeats (Fig. 7E). This result is consistent with a recent report analyzing activity defects of circularly permuted full-length hTR constructs (Mefford and Zappulla 2015), and reveals that physical connectivity between the template hybrid and pseudoknot fold couples telomere DNA synthesis to pseudoknot fold motion during telomerase processivity (Fig. 7E).
FIGURE 7.
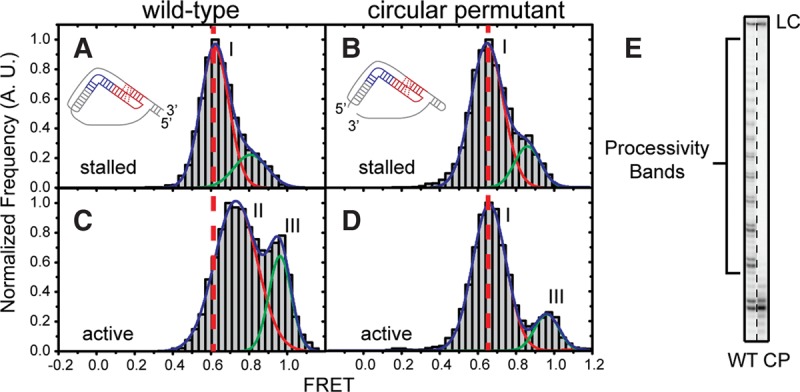
Template-pseudoknot fold connectivity is crucial for motion of the pseudoknot fold and telomerase processivity. (A,C) Single-molecule FRET histograms generated by a WT RNP containing a U92-U312 FRET pair before (A) and after (C) addition of activity buffer. The RNA cartoon depicts the 5′ and 3′ ends of the core domain used for telomerase reconstitution. The vertical dotted line delineates the observed change in FRET upon addition of activity buffer. (B,D) Single-molecule FRET histograms of RNPs containing a circular permutant RNA at hTR 62–63 RNP and a U92–U312 FRET pair, before (B) and after (D) addition of activity buffer. The vertical dotted line delineates the lack of FRET change observed upon addition of activity buffer. Roman numeral peak labels (i,ii, and iii) reference the different FRET peaks that give rise to the separate models in Figure 3. (E) Activity assay of WT and circular permutant enzymes with a (TG)6TTAGGG primer substrate. WT products show binding, extension, and translocation, resulting in processivity bands. Circular permutant complexes exhibit binding and extension, but failed translocation and therefore a lack of processivity bands.
DISCUSSION
Structural studies of human telomerase remain difficult due to the low efficiency of reconstitution and purification of homogeneous telomerase ribonucleoprotein (RNP) complexes. In the present study, we address this challenge using a single-molecule Förster resonance energy transfer (smFRET) assay that permits direct structural characterization of individual, catalytically active telomerase enzymes. Data derived from a series of independent smFRET measurements provided distance constraints that together established a FRET network for the human telomerase RNA (hTR) within either stalled or catalytically active RNP complexes. This distance constraint network was then utilized to guide automated computational modeling of hTR structure within the functional RNP context using a modified Rosetta energy function. Specifically, the smFRET-Rosetta approach generated pseudoatomic structural models of the hTR core domain in several distinct functional substates. Our results suggest that the evolutionarily conserved RNA pseudoknot fold remains stably folded during telomerase catalysis, but exhibits nanometer-scale motion with respect to the rest of the RNP complex during telomerase catalysis. Coupling of pseudoknot fold motion to telomere DNA synthesis in the active site requires physical connectivity between the RNA template and pseudoknot fold. Introduction of a discontinuity in the intervening single-stranded RNA region within hTR using a circularly permuted RNA construct abolished the ability of telomerase to undergo processive telomere repeat addition, and significantly altered the structural properties of the RNA core domain during catalysis when assayed by smFRET. Interestingly, our smFRET-guided molecular modeling unambiguously places the RNA pseudoknot fold on a surface of the TERT protein that is distant from the active site, where it is poised to interact with the TERT C-terminal extension (CTE) domain.
The functional significance of the RNA pseudoknot fold within hTR has been reported; however, the mechanism by which this conserved RNA motif contributes to telomerase catalysis is not well understood. Genetically inherited mutations within the RNA pseudoknot fold have been linked to several human diseases that are characterized by defects in highly proliferative cell types (Vulliamy et al. 2001). Structural characterization of these pathogenic mutations revealed the pseudoknot fold is in a dynamic equilibrium between multiple conformations, leading to the suggestion that a conformational switch within the RNA pseudoknot fold may be important for catalytic function (Theimer et al. 2003). In the present work, the pseudoknot fold appeared to remain stably folded during telomerase catalysis and did not exhibit any detectable transition to an alternative conformation. As previously suggested, this result implies that internal structural dynamics within the RNA pseudoknot fold may play an important role during RNP assembly rather than during telomerase catalysis (Jiang et al. 2015). In a separate study, investigation of pseudoknot fold function in both yeast and human telomerases suggested that the hydroxyl groups of several ribose moieties within the RNA pseudoknot fold may directly participate in telomerase catalysis in a manner that is analogous to the mechanism of self-splicing RNAs (Qiao and Cech 2008). However, our modeling results place the RNA pseudoknot fold at a large distance from the active site, where it is proximal to the TERT CTE, and would be unable to directly participate in the chemistry of telomerase catalysis. Consistent with our human telomerase model, the recently reported medium resolution EM reconstruction of the Tetrahymena telomerase holoenzyme also indicated that the RNA pseudoknot fold is located far from the template in the active site in the vicinity of the TERT CTE (Jiang et al. 2015). Thus, the deleterious impact of removing the 2′-OH groups on several critical ribose sugars may be due to disruption of critical protein–RNA contacts rather than a direct catalytic defect.
In our study, activation of telomerase catalysis appeared to induce motion of the RNA pseudoknot fold into at least two alternative conformations, which we term a “pseudoknot tracking” model (Fig. 5A, state II and state III). Previously reported studies have demonstrated that the rate-limiting step during processive telomere repeat synthesis is realignment of the nascent DNA with the downstream hTR template region and the repositioning of this newly formed hybrid into the TERT active site (Qi et al. 2012; Parks and Stone 2014). Thus, under the conditions of our assay, it is expected that the active telomerase complexes will spend the majority of the time in this so-called “pre-translocation” state. Our biochemical experiments indicate that connectivity between the hTR template and the RNA pseudoknot fold is strictly required for the translocation step during multiple rounds of telomere repeat synthesis. However, the smFRET analysis revealed that while the “state II” conformation is completely abolished when there is a nick between the hTR template and RNA pseudoknot fold, the “state III” conformation can still be sampled. This result suggests that direct coupling between the hTR template and the RNA pseudoknot fold is required for sampling the “state II” conformation, which is likely necessary for completing the translocation step during processive telomerase action. Further studies will be needed to determine whether the additional “state III” conformation of the core domain represents a functionally critical state of the telomerase RNP. In addition, more detailed analysis is required to determine whether flexibility within the J2a/b bulge of the core domain contributes to the observed conformational changes (Zhang et al. 2010).
How might motion of the pseudoknot fold contribute to the processive action of the telomerase RNP when it is located so far from the enzyme active site? One interesting possibility integrates a recent proposal that suggests the CTE domain is mobile during the processive action of telomerase (Yang and Lee 2015). This hypothesis arises from the structural homology between the TERT CTE and the thumb domain of other polymerases. In this putative mechanism, the synthesis of a complete telomere DNA repeat triggers a conformational change within the TERT CTE, which in turn destabilizes DNA–RNA hybrid binding in the active site and promotes telomerase translocation. In the present work, the conformation of the telomerase RNA core domain was monitored by smFRET, while the TERT protein was treated as a rigid body during computational modeling for simplicity. As a result, completion of the telomere repeat synthesis reaction appeared to induce a “sliding” motion of the pseudoknot fold across the surface of the TERT CTE (Fig. 8). However, a more likely scenario that is in line with the model put forth by Yang and Lee (2015) would be that the pseudoknot fold motion would stabilize an alternative conformation of the TERT CTE that exists during the slow translocation step of telomerase activity. Future studies will be required to directly evaluate this hypothesis by using this smFRET-Rosetta technique with FRET labels on distinct TERT protein domains. Similarly, integrating mobile protein domains during the modeling procedure would provide a more detailed view of the RNP architecture during activity. In addition, physical parameters such as electrostatic interactions between RNA and protein surfaces can also be introduced into the modeling procedure.
FIGURE 8.
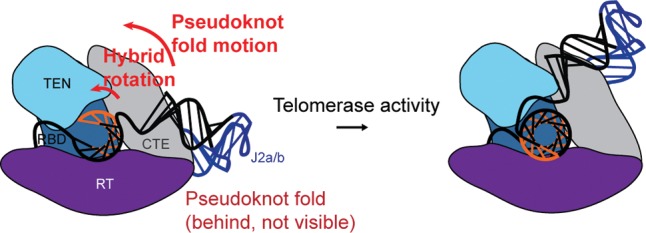
Pseudoknot fold tracking model for RNA conformational changes during telomere DNA repeat synthesis.
Rosetta modeling integrated with smFRET distance constraints is a “molecular motion capture” technique, wherein sparse experimental data can be used to guide pseudoatomic modeling of molecular complexes in order to refine structure–function relationships. The approach described here for studying the telomerase RNP should be generally applicable to diverse biological assemblies that are refractory to traditional structural analyses. For example, the recent structural characterization of the spliceosome by electron microscopy could be integrated with smFRET-Rosetta to help dissect conformational changes associated with the multitude of spliceosome substates. Moreover, while the modeled telomerase assembly in the present work integrated known high-resolution RNA structures, our general modeling method can also be applied to completely de novo RNA structure prediction. Indeed, a standardized algorithm for de novo modeling of RNA structure will be particularly useful in combination with low-mid resolution density maps generated by cryoEM (Sauerwald et al. 2013).
MATERIALS AND METHODS
Telomerase reconstitution and activity
In vitro RNA preparation
In vitro reconstitution of telomerase enzyme was performed using the established two RNA fragment system in which the essential RNA core domain and the CR4/CR5 domain are added in trans. Unlabeled CR4/CR5 RNA (hTR 239–328) was generated by in vitro transcription using T7 RNAP and polyacrylamide gel electrophoresis (PAGE) purified. Site-specific RNA labeling within the core domain and CR4/5 domains was achieved using DNA-splinted RNA ligation techniques as described previously (Parks and Stone 2014). Unlabeled fragments of the RNA core domain (hTR 63–195, hTR 120–195, and cphTR 120–195 + 32–62) were prepared by in vitro transcription using T7 RNAP and purified using a 6% denaturing PAGE gel. Subsequently, the RNA was phosphatase (CIP, NEB) treated to remove the 5′ triphosphate, phenol/chloroform extracted to remove CIP, T4 PNK (NEB) treated to add a 5′ monophosphate, phenol/chloroform extracted to remove PNK and PAGE purified to remove excess nucleotide. Both enzymatic reactions were carried out using NEB specifications.
Dye labeling of synthetic RNA
Synthetic RNA fragments were purchased from Dharmacon containing internal aminoallyl uridines used for dye labeling reactions. Specific RNA sequences are listed in Supplemental Table S1. RNAs were generally processed as previously described (Hengesbach et al. 2012). RNAs from Dharmacon were directly dissolved in 50 μL 0.1 M NaHCO3 and then used to solubilize a single mono-reactive dye pack (GE). The labeling reaction was held at 37°C for 2 h. The RNA was precipitated and deprotected according to the manufacturer's protocol (Dharmacon). RNA was again precipitated and then dissolved in 60 μL of 0.1 M TEAA pH 7.5. An analytical C-8 reverse column was used to purify the labeled from unlabeled RNA fractions as previously described (Akiyama and Stone 2009). After purification the RNAs were precipitated, pooled, and stored at −70°C until use in ligation.
Ligation of RNA fragments
Modifications of the previously reported procedure included addition of 1 U µL−1 RNasin and carrying out the ligation overnight at 30°C (Akiyama and Stone 2009; Hengesbach et al. 2012). The DNA splints for each RNA ligation are listed in Supplemental Table S1. Note for the doubly labeled core domain RNA constructs, the 32–62 fragment was first ligated to the 63–120 fragment, denaturing PAGE purified, then ligated to the 120–195 fragment, and finally purified again by denaturing PAGE gel. During the PK ligations, when the required RNA fragment did not contain a label, the unlabeled fraction of the synthetic oligo was used.
Telomerase reconstitution and purification
Telomerase reconstitution was performed using the TnT Quick Coupled Transcription/Translation system (Promega) (Hengesbach et al. 2012). For every 100 µL of TnT Quick mix, 2 µg of pNFLAG-hTERT was added. RNAs were then added to the reaction mixture where dye-labeled RNAs were added to a final concentration of 0.05 µM, whereas all unlabeled RNAs were added to 1 µM. As an exception, the circular permutant dye-labeled RNA was reconstituted at 1 µM. The reconstitution mixture was held at 30°C for 3 h.
Directly after reconstitution, telomerase enzymes were purified using an N-terminal Flag tag on hTERT. To pull down the enzyme, Sigma Anti-FLAG M2-agarose bead slurry was used at 50 µL bead slurry per 200 µL lysate. The beads were first washed four times with wash buffer 1 (50 mM Tris HCl, pH 8.3, 3 mM MgCl2, 2 mM DTT, 1 mM spermidine, 100 mM KCl), spinning at 2350 r.c.f., 4°C, for 1 min between washes. The beads were blocked while being agitated in 750 µL blocking buffer (50 mM Tris HCl, pH 8.3, 3 mM MgCl2, 2 mM DTT, 1 mM spermidine, 0.5 mg mL−1 BSA, 0.05 mg mL−1 glycogen, 0.1 mg mL−1 yeast tRNA) for 15 min at 4°C. After spinning the beads at 2350 r.c.f., 4°C, for 1 min the supernatant was discarded. The blocking step was repeated (without discarding supernatant) and then the beads were added to the lysate. The bead–lysate mixtures were agitated at 4°C for 2 h allowing the prepared enzyme to associate with the anti-Flag beads. The mixture was spun at 2350 r.c.f., 4°C, for 1 min and the supernatant was flash frozen and stored for analytical purposes. The beads were then washed with 750 µL of wash buffer 2 (50 mM Tris HCl, pH 8.3, 3 mM MgCl2, 2 mM DTT, 1 mM spermidine, 300 mM KCl) and spun at 2350 r.c.f., 4°C, for 1 min. This process was repeated three times to remove residual lysate. The beads were washed three more times with 750 µL of wash buffer 1. To elute the enzyme, 60 µL of elution buffer (50 mM Tris HCl, pH 8.3, 3 mM MgCl2, 1 mM spermidine, 0.5 mg mL−1 BSA, 20% glycerol, 0.75 mg mL−1 3 × Flag peptide) was added for every 200 µL of lysate and the slurry was incubated at 4°C for 1 h. Beads were removed by centrifugation filtration using a Nanosep MF 0.45 µm filter. From this solution, 10-μL aliquots were prepared, flash frozen in liquid nitrogen and stored at −70°C until use.
Telomerase activity assay
Activity assays were performed using 5 µL of purified enzyme diluted to a final volume of 10µL in 1 × activity buffer. Each reaction maintained a final concentration of 1 µM primer substrate, 250 µM dATP, 250 µM dTTP, 1 µM α32P-dGTP (PerkinElmer BLU514Z500UC), and 1 × activity buffer (50 mM Tris HCl, pH 8.3, 3 mM MgCl2, 2 mM DTT, 1 mM spermidine). The reactions were initiated by addition of dNTPs and incubated at 30°C for 90 min. Reactions were terminated by addition of 190 µL stopping buffer containing the loading control (10 mM Tris HCl, pH 7.5, 1 mM EDTA, 0.1% w/v SDS). The reactions were mixed vigorously with 100 µL of phenol to remove protein components followed by a 10 min spin at 15,000 r.c.f. The aqueous layer was further extracted with 100 µL of chloroform and spun at 15,000 r.c.f. for 2 min. After isolation of the aqueous phase, the radiolabeled DNA was precipitated and separated on a 12% denaturing PAGE gel (19:1 crosslinking ratio). The gel was dried, exposed on a phosphorimager screen overnight and scanned using a Typhoon scanner. Quantification of the bands was accomplished by using SAFA v11b (Das et al. 2005).
Single-molecule FRET experiments
Slide preparation
Quartz slides (Finkenbeiner Inc.) were cleaned and pegylated using the following protocol (Roy et al. 2008). The slides were boiled in water to remove parafilm and coverslips from previous experiments. The slides were then scrubbed with alconox (Alconox Inc.), rinsed and placed into a solution containing 10% alconox. After sonication for 20 min, the slides were rinsed and placed in water and sonicated for 5 min. Slides were then sonicated in acetone for 15 min. The slides were directly transferred into 1 M KOH and sonicated for 20 min. After thorough rinsing, the slides surfaces were flame dried and cleaned using a butane torch (BernzOmatic). While the slides were cooling, a silanizing solution containing 100 mL of methanol, 5 mL of glacial acetic acid, and 1 mL of N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (UCT) was prepared. The cooled slides were placed in the solution, sonicated for 1 min and allowed to stand in solution at room temperature for at least 20 min. While incubating slides, 200 mg of mPEG-Succinimidyl Valerate MW 5000 (Laysan Bio, Inc.), was resuspended in 400 µL of 0.1 M NaHCO3. In addition, 2 mg of Biotin-PEG-Succinimidyl Valerate MW 5000 (Laysan Bio, Inc.) was resuspended in 200 µL of 0.1 M NaHCO3. After rinsing the slides with dH2O and drying with nitrogen, the solutions were mixed and applied to each slide surface (150 µL per slide). A coverslip was used to cover the solution and incubated overnight at room temperature in a humidor box. The following day, channels were assembled using Paramfilm as a spacer on the pegylated quartz slides and plasma-cleaned coverslips were used as the second chamber face.
Single-molecule experimental procedure
To prepare the slides for molecule deposition, the channels were incubated with 60 µL of 10 mg mL−1 BSA (NEB) for 20 min. The slide was then washed with 100 µL of T50 (10 mM Tris HCl, pH8, 50 mM NaCl) and then incubated with 60 µL of 0.2 mg mL−1 streptavidin in T50 for 5 min. After coating the slide with streptavidin, the excess protein was washed away with 200 µL of T50. The slide surface was coated with immobilization primer by washing 100 nM primer over the surface in T50. Excess primer was then rinsed away with 200 µL of T50. A total of 10 µL telomerase solution from the immunoprecipitation was diluted with 20 µL of imaging buffer (50 mM Tris HCl, pH 8.3, 3 mM MgCl2, 1 mM spermidine, 0.5 mg mL−1 BSA, 4% glucose) and flowed over the slide surface. For labeled RNAs, each core domain construct was diluted to 1 nM and applied to the slide surface. The number of molecules on the surface was observed in real time to reach the desired density for smFRET measurements. Once telomerase molecules/RNAs were deposited on the surface, the stable complexes were retained and imaged after multiple buffer exchanges. Imaging of the FRET-labeled enzymes required an oxygen-scavenging system and a triplet state quencher to increase dye lifetime. Imaging buffer does not substantially impact telomerase activity (Parks and Stone 2014; Akiyama et al. 2015). The buffer was saturated with Trolox (triplet state quencher), passed through a 0.22-µm filtration unit and brought to pH 8.3. Once ready for imaging, the solution was brought to 2 µg mL−1 catalase and 1 mg mL−1 glucose oxidase. The pH was found to be stable for more than 30 min with these conditions (data not shown). In the case of telomerase FRET activity, dATP, dTTP, and dGTP were added as described in the text at a concentration of 200 µM in imaging buffer.
Microscope setup
Data were collected using a custom-built prism-type microscope, allowing hundreds of molecules to be studied in parallel (Akiyama and Stone 2009). Cy3 donor dyes were directly excited with a 532 nm laser (Laserglow). Depending on the proximity of Cy5 acceptor dye to the donor dye, a variable degree of energy transfer will occur from the excited donor dye to the acceptor dye. Due to this FRET process, both dyes are capable of fluorescing and emitting at distinct wavelengths. Emitted fluorescence was split into separate donor and acceptor channels using dichroic mirrors, and imaged on an EM-CCD camera (Andor Ixon).
Data acquisition and analysis
Imaging fields containing 30–250 molecules were imaged using a 100-msec integration time for fast timescale traces and histograms. Individual traces were parsed out using custom written IDL software where they were corrected for background and dye-crosstalk signal. The individual traces were then filtered in MATLAB manually where only traces that showing FRET, acceptor bleaching and then the donor bleaching pathway were accepted. This specific photo-bleaching process was required for correction of the γ factor at the single-molecule level. FRET intensities were then calculated using the equation (IA − βID)/ (IA + γID) where IA is the acceptor intensity, ID is the donor intensity, γ is the gamma correction factor, and β accounts for the leakage of the donor fluorescence into the acceptor channel. The γ correction factor was determined as described for individual molecules (McCann et al. 2010). The first five seconds of the corrected, individual FRET traces were then binned into FRET histograms. The centers of the FRET distributions were determined by a nonlinear Gaussian fitting algorithm.
Determining distance from FRET
FRET peak centers were converted from FRET efficiency to distance using the Equation 1. In this equation, FRET represents the observed peak center of the corrected smFRET data and R0 is the tabulated Förster radius (5.6 nm) specified by the dye manufacturer (Amersham Biosciences). Typically R0 has been calculated using a number of tabulated parameters and an experimentally determined quantum yield of the donor (Andrecka et al. 2008, 2009; Muschielok et al. 2008; Muschielok and Michaelis 2011). In our experiments, it was not possible to experimentally determine the quantum yield of the donor in the assembled complexes due to low sample concentrations. Therefore, we accounted for errors in R0 through compliance in the FRET scoring parameter in the modeling function.
Rosetta modeling
Generation of modeling scaffold
An initial modeling scaffold was generated using known substructures of human telomerase. The previously published homology model of human TERT was used with an updated model of the TRBD, to reflect information from the recently solved structure of the TRBD from the vertebrate Takifugu rubripes (Harkisheimer et al. 2013). The new homology model of the human TRBD was built using HHPRED and Modeller (Soding et al. 2005; Eswar et al. 2007) using 4LMO, 2R4G, and 3KYL as templates (Rouda and Skordalakes 2007; Mitchell et al. 2010; Harkisheimer et al. 2013). This was integrated back into the original homology model by aligning corresponding residues. The template hybrid was modeled as an ideal A-form helix, and because interactions between the template hybrid and the protein were not modeled explicitly (see below), the template hybrid was fixed relative to the protein. In the case of the stalled complexes, the template hybrid was modeled as a 7-nt hybrid, based on the expected hybrid binding register of the DNA primer. When modeling the active complex, an 11-nt hybrid was modeled in the active site in order to depict a fully extended template hybrid. As our previous work suggested multiple hybrid registers upon addition of activity buffer, we also modeled the activity data with a 7-nt hybrid in the active site. This modeling control did not significantly affect the returned models (data not shown). These results suggest that the length of hybrid in the active site plays an insignificant role for our modeling purposes, and rather the FRET distance constraints are the main contributors to modeling convergence. The DNA in the template hybrid was modeled as RNA for simplicity. The template hybrid was positioned in the human homology model by aligning the end of the template hybrid (5′ end of the template and 3′ end of the primer) to the template hybrid in the active site in the homology model, which was built using information from the T. castaneum crystal structure of the TRBD, RT, and CTE domains bound to the DNA/RNA template hybrid (PDB ID 3KYL) (Mitchell et al. 2010). The structure of the P6.1 helix, residues 301–305 and 311–315, was taken from the crystal structure of the O. latipes TRBD bound to CR4/5 RNA (PDB ID 4O26) (Huang et al. 2014). To position it relative to the human TERT model, the O. latipes TRBD and CR4/5 structure was aligned to the homology model of the human TRBD, and the resulting coordinates of residues 199–203 and 209–213 from the O. latipes CR4/5, which correspond to the human P6.1 stem, were used. For models built with an enforced P1 stem, the recently solved structure of the Tetrahymena TRBD bound to the template boundary element (TBE), was aligned to the human TRBD model, then the P1b helix was aligned to the TBE helix (Jansson et al. 2015). The final scaffold contained the TERT homology model bound to the template hybrid, the P6.1 stem, and with/without P1b helix. In addition to the modeling scaffold, other known RNA structures were treated as rigid bodies that were free to move relative to the modeling scaffold. The solution structure of human J2a/b, PDB ID 2L3E, was used for residues 78–93 and 121–131, and the pseudoknot fold, PDB ID 2KID, for residues 96–118 and 170–183. P2a.1 (residues 64–66, 143–145, 69–72, 136–139) was modeled as an ideal A-form helix.
General algorithm and scoring
Models of the telomerase RNA were generated using a modified version of fragment assembly of RNA (FARFAR), a method for building de novo structures of RNA by sampling known fragments of RNA structures from the protein data bank in a Monte Carlo simulation (Cheng et al. 2015a). Structures are scored with a statistically derived low-resolution potential meant to approximate the free energy of the RNA (ScoreRNA). Here, we modified this method to include smFRET data and protein-RNA sterics in two additional score terms, ScoreFRET and Scoreclash, respectively. Now, the total score of an RNA structure is given by the sum of the original ScoreRNA with ScoreFRET and Scoreclash. ScoreFRET takes the form of a smoothed square well potential. First, the peak measured FRET efficiencies ± approximately one standard deviation were converted into distances, as described above. Then, for each pair of residues for which there are smFRET data, a penalty of zero is assigned if they are within this distance range. The potential is then smoothed up to a penalty of 200 Rosetta units over 5 Å above and below this distance range (Fig. 3). When there were multiple peaks in the FRET distributions, each peak was converted into a separate score term. Each of these terms was calculated for a given model, and the lowest of the scores was applied. In this way, if a modeled distance agreed with one of the FRET peaks, it would not get a penalty if it failed to agree with any other FRET peak. Scoreclash was introduced to account for steric clashes between the RNA and TERT protein, because the original fragment assembly method does not model interactions between RNA and protein residues. For computational efficiency, the coordinates of the atoms of the protein structure, taking into account their van der Waals radii, were placed into a three dimensional grid with bin spacing set at 0.1 Å. Here, we accounted only for clashes with the backbone atoms of the protein structure because the TERT structure was a homology model and we do not allow for any conformational changes of the protein. Each time the RNA structure was scored, it was aligned to this grid using the fixed RNA components of the initial modeling scaffold, then Scoreclash was computed by counting the number of clashes and multiplying by a scaling factor, here 0.3. In this way, conformations of the RNA that had many steric clashes with the protein were penalized. This algorithm is available as part of the Rosetta software package (version 3.7). An example command line is provided below:
rna_denovo -nstruct 500 -params_file 7nt_hybrid_P1.params -fasta 7nt_hybrid_P1.fasta -out:file:silent 7nt_hybrid_P1.out -include_neighbor_base_stacks -minimize_rna false -s 7nt_hybrid+CR45+P1b-TBE.pdb P2a.1_1_helix.pdb P2a.1_2_helix.pdb P2ab_2L3E.pdb pseudoknot-WT_2K95.pdb -input_res 1-5 18-24 131-152 32-34 111-113 37-40 104-107 46-61 89-99 64-86 114-127 -cst_file 7nt_hybrid_P1_FRET_constraints.cst -gradual_constraints false -grid_vdw_weight 0.3 -VDW_rep_screen_info TERT_homology_ model.pdb 914-923 136-145 -VDW_rep_screen_include_sidechains false -output_res_num 33-145 170-191 301-305 311-315 452-458
Analysis
For each of the modeling runs, approximately 2500 structures were generated, 10,130 in total. Convergence of the runs was assessed by clustering the 400 best scoring RNA models, after alignment of all the models to the modeling scaffold, with a range of different radii. The smallest radius that placed at least 25% of these models in the largest cluster estimates the extent of convergence and provides an estimate of the precision of the models (Supplemental Fig. S9; Cheng et al. 2015a).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grant GM095850 to M.D.S.); National Science Foundation Graduate Research Fellowship Program (DGE-0809125), Center for Biomolecular Science and Engineering Research Mentoring Institute Fellowship (National Institutes of Health–National Human Genome Research Institute 1R25HG006836-01A1), and the Eugene Cota-Robles Fellowship (to J.W.P.); National Institutes of Health–National Institute of General Medical Sciences R01 (GM102519) and Burroughs Wellcome Foundation (CASI 1007236) (to R.D.); National Science Foundation Graduate Research Fellowship Program (DGE-114747) and Gabilan Stanford Graduate Fellowship (to K.K.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.058743.116.
REFERENCES
- Akiyama BM, Stone MD. 2009. Assembly of complex RNAs by splinted ligation. Methods Enzymol 469: 27–46. [DOI] [PubMed] [Google Scholar]
- Akiyama BM, Parks JW, Stone MD. 2015. The telomerase essential N-terminal domain promotes DNA synthesis by stabilizing short RNA-DNA hybrids. Nucleic Acids Res 43: 5537–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrecka J, Lewis R, Bruckner F, Lehmann E, Cramer P, Michaelis J. 2008. Single-molecule tracking of mRNA exiting from RNA polymerase II. Proc Natl Acad Sci 105: 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrecka J, Treutlein B, Arcusa MA, Muschielok A, Lewis R, Cheung AC, Cramer P, Michaelis J. 2009. Nano positioning system reveals the course of upstream and nontemplate DNA within the RNA polymerase II elongation complex. Nucleic Acids Res 37: 5803–5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman AJ, Akiyama BM, Stone MD, Cech TR. 2011. The RNA accordion model for template positioning by telomerase RNA during telomeric DNA synthesis. Nat Struct Mol Biol 18: 1371–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Greider CW. 2003. Template boundary definition in mammalian telomerase. Genes Dev 17: 2747–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Chou FC, Das R. 2015a. Modeling complex RNA tertiary folds with Rosetta. Methods Enzymol 553: 35–64. [DOI] [PubMed] [Google Scholar]
- Cheng CY, Chou FC, Kladwang W, Tian S, Cordero P, Das R. 2015b. Consistent global structures of complex RNA states through multidimensional chemical mapping. Elife 4: e07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DI, Legassie JD, Bonifacio LN, Sekaran VG, Ding F, Dokholyan NV, Jarstfer MB. 2012. New models of Tetrahymena telomerase RNA from experimentally derived constraints and modeling. J Am Chem Soc 134: 20070–20080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Baker D. 2007. Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci 104: 14664–14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB. 2005. SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA 11: 344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Karanicolas J, Baker D. 2010. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat Methods 7: 291–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert B, Collins K. 2012. Roles of telomerase reverse transcriptase N-terminal domain in assembly and activity of Tetrahymena telomerase holoenzyme. J Biol Chem 287: 12805–12814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. 2007. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci 50: 2.9.1–2.9.31. [DOI] [PubMed] [Google Scholar]
- Gavory G, Symmons MF, Krishnan Ghosh Y, Klenerman D, Balasubramanian S. 2006. Structural analysis of the catalytic core of human telomerase RNA by FRET and molecular modeling. Biochemistry 45: 13304–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkisheimer M, Mason M, Shuvaeva E, Skordalakes E. 2013. A motif in the vertebrate telomerase N-terminal linker of TERT contributes to RNA binding and telomerase activity and processivity. Structure 21: 1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengesbach M, Kim NK, Feigon J, Stone MD. 2012. Single-molecule FRET reveals the folding dynamics of the human telomerase RNA pseudoknot domain. Angew Chem Int Ed Engl 51: 5876–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Brown AF, Wu J, Xue J, Bley CJ, Rand DP, Wu L, Zhang R, Chen JJ, Lei M. 2014. Structural basis for protein-RNA recognition in telomerase. Nat Struct Mol Biol 21: 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson LI, Akiyama BM, Ooms A, Lu C, Rubin SM, Stone MD. 2015. Structural basis of template-boundary definition in Tetrahymena telomerase. Nat Struct Mol Biol 22: 883–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Chan H, Cash DD, Miracco EJ, Ogorzalek Loo RR, Upton HE, Cascio D, O'Brien Johnson R, Collins K, Loo JA, et al. 2015. Structure of Tetrahymena telomerase reveals previously unknown subunits, functions, and interactions. Science 350: aab4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NK, Zhang Q, Zhou J, Theimer CA, Peterson RD, Feigon J. 2008. Solution structure and dynamics of the wild-type pseudoknot of human telomerase RNA. J Mol Biol 384: 1249–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NK, Zhang Q, Feigon J. 2014. Structure and sequence elements of the CR4/5 domain of medaka telomerase RNA important for telomerase function. Nucleic Acids Res 42: 3395–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CK, Miller MC, Collins K. 2002. Template boundary definition in Tetrahymena telomerase. Genes Dev 16: 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latrick CM, Cech TR. 2010. POT1-TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. EMBO J 29: 924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly H, Blackburn EH, Parslow TG. 2003. Comprehensive structure-function analysis of the core domain of human telomerase RNA. Mol Cell Biol 23: 6849–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus M, Matelska D, Lach G, Chojnowski G, Boniecki MJ, Purta E, Dawson W, Dunin-Horkawicz S, Bujnicki JM. 2014. Computational modeling of RNA 3D structures, with the aid of experimental restraints. RNA Biol 11: 522–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann JJ, Choi UB, Zheng L, Weninger K, Bowen ME. 2010. Optimizing methods to recover absolute FRET efficiency from immobilized single molecules. Biophys J 99: 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford MA, Zappulla DC. 2015. Physical connectivity mapping by circular permutation of human telomerase RNA reveals new regions critical for activity and processivity. Mol Cell Biol 36: 251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalusova M, Wu JY, Zhuang X. 2011. Functional importance of telomerase pseudoknot revealed by single-molecule analysis. Proc Natl Acad Sci 108: 20339–20344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell M, Gillis A, Futahashi M, Fujiwara H, Skordalakes E. 2010. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat Struct Mol Biol 17: 513–518. [DOI] [PubMed] [Google Scholar]
- Muschielok A, Michaelis J. 2011. Application of the nano-positioning system to the analysis of fluorescence resonance energy transfer networks. J Phys Chem B 115: 11927–11937. [DOI] [PubMed] [Google Scholar]
- Muschielok A, Andrecka J, Jawhari A, Bruckner F, Cramer P, Michaelis J. 2008. A nano-positioning system for macromolecular structural analysis. Nat Methods 5: 965–971. [DOI] [PubMed] [Google Scholar]
- Parks JW, Stone MD. 2014. Coordinated DNA dynamics during the human telomerase catalytic cycle. Nat Commun 5: 4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlevsky JD, Bley CJ, Omana RV, Qi X, Chen JJ. 2008. The telomerase database. Nucleic Acids Res 36: D339–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Xie M, Brown AF, Bley CJ, Podlevsky JD, Chen JJ. 2012. RNA/DNA hybrid binding affinity determines telomerase template-translocation efficiency. EMBO J 31: 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao F, Cech TR. 2008. Triple-helix structure in telomerase RNA contributes to catalysis. Nat Struct Mol Biol 15: 634–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robart AR, Collins K. 2011. Human telomerase domain interactions capture DNA for TEN domain-dependent processive elongation. Mol Cell 42: 308–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouda S, Skordalakes E. 2007. Structure of the RNA-binding domain of telomerase: implications for RNA recognition and binding. Structure 15: 1403–1412. [DOI] [PubMed] [Google Scholar]
- Roy R, Hohng S, Ha T. 2008. A practical guide to single-molecule FRET. Nat Methods 5: 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerwald A, Sandin S, Cristofari G, Scheres SH, Lingner J, Rhodes D. 2013. Structure of active dimeric human telomerase. Nat Struct Mol Biol 20: 454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33: W244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steczkiewicz K, Zimmermann MT, Kurcinski M, Lewis BA, Dobbs D, Kloczkowski A, Jernigan RL, Kolinski A, Ginalski K. 2011. Human telomerase model shows the role of the TEN domain in advancing the double helix for the next polymerization step. Proc Natl Acad Sci 108: 9443–9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MD, Mihalusova M, O'Connor CM, Prathapam R, Collins K, Zhuang X. 2007. Stepwise protein-mediated RNA folding directs assembly of telomerase ribonucleoprotein. Nature 446: 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theimer CA, Finger LD, Trantirek L, Feigon J. 2003. Mutations linked to dyskeratosis congenita cause changes in the structural equilibrium in telomerase RNA. Proc Natl Acad Sci 100: 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I. 2001. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413: 432–435. [DOI] [PubMed] [Google Scholar]
- Wallweber G, Gryaznov S, Pongracz K, Pruzan R. 2003. Interaction of human telomerase with its primer substrate. Biochemistry 42: 589–600. [DOI] [PubMed] [Google Scholar]
- Wu JY, Stone MD, Zhuang X. 2010. A single-molecule assay for telomerase structure-function analysis. Nucleic Acids Res 38: e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Lee YS. 2015. A DNA-hairpin model for repeat-addition processivity in telomere synthesis. Nat Struct Mol Biol 22: 844–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaug AJ, Podell ER, Cech TR. 2008. Mutation in TERT separates processivity from anchor-site function. Nat Struct Mol Biol 15: 870–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Kim NK, Peterson RD, Wang Z, Feigon J. 2010. Structurally conserved five nucleotide bulge determines the overall topology of the core domain of human telomerase RNA. Proc Natl Acad Sci 107: 18761–18768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Kim NK, Feigon J. 2011. Architecture of human telomerase RNA. Proc Natl Acad Sci 108: 20325–20332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.