

Abstract
Recently, we identified a novel phosphodiesterase 10A (PDE10A) inhibitor, PDM‐042 ((E)‐4‐(2‐(2‐(5,8‐dimethyl‐[1,2,4]triazolo[1,5‐a]pyrazin‐2‐yl)vinyl)‐6‐(pyrrolidin‐1‐yl)pyrimidin‐4‐yl)morpholine). PDM‐042 showed potent inhibitory activities for human and rat PDE10A with IC 50 values of less than 1 nmol/L and more than 1000‐fold selectivity against other phosphodiesterases. Tritiated PDM‐042, [3H]PDM‐042, had high affinity for membranes prepared from rat striatum with a K d value of 8.5 nmol/L. The specific binding of [3H]PDM‐042 was displaced in a concentration‐dependent manner by PDM‐042 and another structurally unrelated PDE10A inhibitor, MP‐10. In rat studies, PDM‐042 showed excellent brain penetration (striatum/plasma ratio = 6.3), occupancy rate (86.6% at a dose of 3 mg/kg), and good oral bioavailability (33%). These data indicate that PDM‐042 is a potent, selective, orally active, and brain‐penetrable PDE10A inhibitor. In behavioral studies using rat models relevant to schizophrenia, PDM‐042 significantly antagonized MK‐801‐induced hyperlocomotion (0.1–0.3 mg/kg) without affecting spontaneous locomotor activity and attenuated the conditioned avoidance response (CAR) (0.3–1 mg/kg). In tests for adverse effects, PDM‐042 had a minimal effect on catalepsy, even at a much higher dose (10 mg/kg) than the minimal effective dose (0.3 mg/kg) in the CAR. Furthermore, PDM‐042 had no effect on prolactin release or glucose elevation up to 3 mg/kg, while risperidone increased prolactin release and olanzapine enhanced glucose levels at doses near their efficacious ones in the CAR. Our results suggest that PDM‐042 is a good pharmacological tool that can be used to investigate the role of PDE10A and may have therapeutic potential for the treatment of schizophrenia.
Keywords: Antipsychotics, catalepsy, occupancy, PDE10A, PDM‐042, schizophrenia
Abbreviations
- AMG‐7980
5‐(6,7‐Dimethoxycinnolin‐4‐yl)‐N‐isopropyl‐3‐methylpyridin‐2‐amine
- AUC
area under the curve
- CAR
conditioned avoidance response
- CS
conditioned stimulus
- DPM
disintegrations per minute
- EPS
extrapyramidal symptoms
- i.v.
intravenously
- Kd
dissociation constant
- Ki
inhibition constant
- LC‐MS/MS
liquid chromatography with tandem mass spectrometry
- MK‐801
(5R,10S)‐(+)‐5‐Methyl‐10,11‐dihydro‐5H‐dibenzo[a,d]cyclohepten‐5,10‐imine
- MP‐10
2‐[4‐(1‐methyl‐4‐pyridin‐4‐yl‐1H‐pyrazol‐3‐yl)‐phenoxymethy]‐quinoline
- NMDA
N‐methyl‐d‐aspartate
- PDE
phosphodiesterase
- PDM‐042
(E)‐4‐(2‐(2‐(5,8‐dimethyl‐[1,2,4]triazolo[1,5‐a]pyrazin‐2‐yl)vinyl)‐6‐(pyrrolidin‐1‐yl)pyrimidin‐4‐yl)morpholine
- SKF82598
(±)‐6‐Chloro‐7,8‐dihydroxy‐3‐allyl‐1‐phenyl‐2,3,4,5‐tetrahydro‐1H‐3‐benzazepine hydrobromide
- T‐773
1‐[2‐fluoro‐4‐(tetrahydro‐2H‐pyran‐4‐yl)phenyl]‐5‐methoxy‐3‐(1‐phenyl‐1H‐pyrazol‐5‐yl)pyridazin‐4(1H)‐one
- TAK‐063
1‐[2‐fluoro‐4‐(1H‐pyrazol‐1‐yl)phenyl]‐5‐methoxy‐3‐(1‐phenyl‐1H‐pyrazol‐5‐yl)pyridazin‐4(1H)‐one
- THPP‐1
2‐(6‐chloropyridin‐3‐yl)‐4‐(2‐methoxyethoxy)‐7,8‐dihydropyrido[4,3‐d]pyrimidin‐6(5H)‐yl](imidazo[1,5‐a]pyridin‐1‐yl)methanone
- TP‐10
2‐{4‐[‐pyridin‐4‐yl‐1‐(2,2,2‐trifluoroethyl)‐1H‐pyrazol‐3‐yl]‐phenoxymethyl}‐quinoline succinic acid
- UCS
unconditioned stimulus
Introduction
Phosphodiesterases (PDEs) are enzymes involved in regulating intracellular signaling by second messengers cAMP and cGMP. PDEs are encoded by 21 genes and subdivided into 11 families, PDE1 through PDE11, according to their structural and functional properties (Bender and Beavo 2006). PDE10A is a dual substrate PDE that hydrolyzes cAMP and cGMP, which is highly expressed in the brain and has limited expression in peripheral tissues (Fujishige et al. 1999; Loughney et al. 1999; Soderling et al. 1999). In the brain, PDE10A is expressed at high levels in medium spiny neurons of the striatum (Seeger et al. 2003; Coskran et al. 2006; Xie et al. 2006) and regulates striatal output by its effects on cAMP and cGMP signaling cascades. Medium spiny neurons are divided into the direct and indirect striatal output pathways expressing dopamine D1 and D2 receptor, respectively (Graybiel 1990, 2000). Direct and indirect pathway neurons induce opposing effects on output neurons in the internal segment of the globus pallidus and the substantia nigra pars reticulate, resulting in disinhibition and proinhibition of output, respectively (Nishi and Snyder 2010). The clinical effects of most currently available antipsychotics are caused mainly by D2 receptor blockade (Kapur and Remington 2001; Kapur and Mamo 2003). However, most antipsychotics also cause the extrapyramidal symptoms (EPS) by D2 receptor blockade (Pierre 2005; Krebs et al. 2006). In addition, some atypical antipsychotics elicit other adverse effects such as hyperprolactinemia and glucose intolerance (Dunlop et al. 2003; Krebs et al. 2006; Peuskens et al. 2014). Because PDE10A is expressed in both direct and indirect pathways (Nishi et al. 2008; Sano et al. 2008; Nishi and Snyder 2010), it has been hypothesized that inhibition of PDE10A in the striatum will result in both D2 antagonism and D1 agonism. Therefore, pharmacological inhibition of PDE10A can represent a unique approach that does not involve direct blockade of the D2 receptor for the treatment of schizophrenia.
Preclinical studies of PDE10A inhibition as a treatment for schizophrenia have been conducted using PDE10A inhibitors. Several PDE10A inhibitors, including TP‐10, MP‐10, THPP‐1, and TAK‐063, have been subjected to behavioral and pharmacological analysis (Schmidt et al. 2008; Grauer et al. 2009; Smith et al. 2013; Suzuki et al. 2015). In addition, various radioligands for PDE10A have been developed as translational tools for investigating target engagement. Tritiated PDE10A inhibitors, [3H]AMG‐7980 and [3H]T‐773, showed high affinity and high specificity for PDE10A in striatal homogenates and brain sagittal sections in rats, suggesting that these radioligands for PDE10A could be utilized as positron emission tomography imaging tracers (Hu et al. 2012; Harada et al. 2015).
Recently, we identified a novel PDE10A inhibitor, PDM‐042 ((E)‐4‐(2‐(2‐(5,8‐dimethyl‐[1,2,4]triazolo[1,5‐a]pyrazin‐2‐yl)vinyl)‐6‐(pyrrolidin‐1‐yl)pyrimidin‐4‐yl)morpholine), in the course of extensive chemical optimization. The purpose of this study was to characterize in vitro and in vivo pharmacological profiles of PDM‐042. PDM‐042 was a highly potent and selective PDE10A inhibitor with good oral bioavailability and brain penetration. The in vivo effects of PDM‐042 in rats were tested in MK‐801‐induced hyperlocomotion and conditioned avoidance response (CAR), which have been established as effective models for examining antipsychotic‐like activity (Gattaz et al. 1994; O'Neill and Shaw 1999; Wadenberg and Hicks 1999). In addition, we examined potential liability of PDM‐042 in comparison with those of representative atypical antipsychotics in catalepsy, prolactin release, and glucose elevation in rats. To examine the target engagement of PDM‐042 in rat striatum, in vitro binding and in vivo occupancy were tested using tritiated PDM‐042 ([3H]PDM‐042).
Materials and Methods
Animals
Male Sprague–Dawley rats (6‒7 weeks old, Charles River Laboratories Japan, Inc., Kanagawa, Japan or 6 weeks old, Japan SLC, Inc., Shizuoka, Japan) and F344 rats (4 or 7 weeks old, Charles River Laboratories Japan, Inc.) were purchased. Male beagle dogs (6 months old, Marshall Bioresources Inc., Beijing, China) were purchased. All rats or dogs were group‐housed or individually housed in an air‐conditioned room (room temperature; 23 ± 3°C, humidity; 55 ± 15%) with a 12‐h light‐dark cycle (lights on: 07:30‒19:30). Each rat had free access to standard chow (CE‐2, CLEA Japan, Inc., Tokyo, Japan) and tap water. At least 7 days were allowed for acclimatization to the facility before starting the experiments. Each dog was offered 250 g per day of standard diet (DS‐A, Oriental Yeast Co., Ltd., Tokyo, Japan) and had free access to tap water.
All experimental procedures were approved by the Institutional Animal Care and Use Committee complied with the Japanese law “Act on Welfare and Management of Animals” and the guidelines from the Ministry of Health, Labor and Welfare of Japan.
Drugs
The structures of PDM‐042 and [3H]PDM‐042 are shown in Figure 1A and B, respectively. PDM‐042 was synthesized in‐house and used as a free base in all experiments. [3H]PDM‐042 (662 GBq/mmol) was synthesized by Sekisui Medical Co., Ltd. (Tokyo, Japan). PDM‐042 was dissolved in dimethylsulfoxide and stored at −20°C to perform in vitro assays. PDM‐042 was suspended in 0.5% methylcellulose solution (Wako Pure Chemical Industries, Ltd., Osaka, Japan) and orally administered at a volume of 5 mL/kg in rats and 2 mL/kg in dogs. When PDM‐042 was intravenously (i.v.) administered at a volume of 1 mL/kg in the pharmacokinetic study, it was dissolved in 50% (v/v) N, N‐dimethylacetamide (Wako Pure Chemical Industries) and 50% (v/v) polyethylene glycol 400 (Wako Pure Chemical Industries). Risperidone (Toronto Research Chemicals Inc., Toronto, Canada) and olanzapine (U.S. Pharmacopeial Convention, Rockville, MD) were dissolved in minimal amounts of aqueous hydrochloric acid solution, diluted to the required concentrations with physiological saline, and subcutaneously administered at a volume of 1 mL/kg. MK‐801 ((5R,10S)‐(+)‐5‐methyl‐10,11‐dihydro‐5H‐dibenzo[a,d]cyclohepten‐5,10‐imine hydrogen maleate, Sigma‐Aldrich Co. LLC, St. Louis, MO) was dissolved in physiological saline and subcutaneously administered at a volume of 1 mL/kg.
Figure 1.
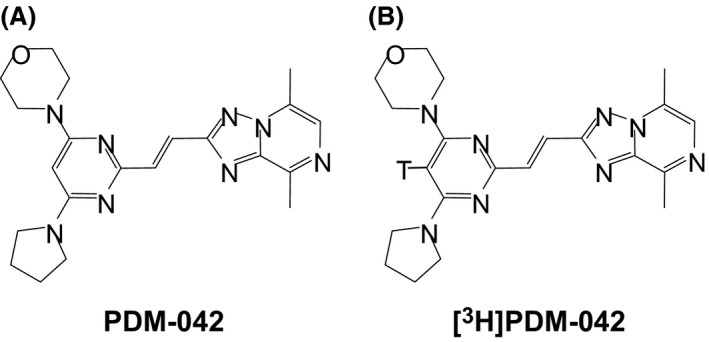
Chemical structures of PDM‐042 (A) and [3H]PDM‐042 (B).
In vitro assays
Recombinant PDE10A (human and rat) was purchased from BPS Bioscience, Inc. (San Diego, CA). The inhibitory activity and inhibition constant (K i) of PDM‐042 for each recombinant PDE10A were determined using IMAP TR‐FRET Screening Express (Molecular Devices LLC., Sunnyvale, CA). PDM‐042 was incubated with each recombinant PDE10A for 5 min. After a subsequent incubation with fluorescein‐labeled cAMP for 60 min, followed by incubation with the IMAP binding solution for more than 3 h, the TR‐FRET signal was measured with ARVO‐Sx (Perkin Elmer Inc., Waltham, MA). For IC50 determination, maximal inhibition (100% activity) and 0% activity in the assay were defined as the no enzyme control and no compound control, respectively. Each assay was performed in quadruplicate at 8 PDM‐042 concentrations (0.003–10 nmol/L). Inhibition (%) and IC50 values were calculated using Assay Explorer (Accelrys, Inc., San Diego, CA). For K i determination, substrate‐velocity curves were calculated in the presence of several concentrations of PDM‐042 and fluorescein‐labeled cAMP. Each assay was performed in duplicate at 7 PDM‐042 concentrations (0.03–30 nmol/L) with 7 fluorescein‐labeled cAMP concentrations (3–1000 nmol/L for human PDE10A and 3–3000 nmol/L for rat PDE10A). K i values were calculated using GraphPad Prism Software, version 5 (GraphPad Software Inc., San Diego, CA).
PDE selectivity and off‐target activity
The effects of PDM‐042 on the activities of 13 PDE isozymes were tested at a concentration of 1 μmol/L using the PDE Selectivity Screen from Ricerca Biosciences (Concord, OH), consisting of human PDE1A, human PDE2A, human PDE3A, human PDE4A1A, human PDE4B1, human PDE5A, bovine PDE6, human PDE7A, human PDE7B, human PDE8A1, human PDE9A2, human PDE10A2, and human PDE11A4. The selectivity of PDM‐042 was further assessed against 137 molecular targets, including neurotransmitter receptors, enzymes, ion channels, and transporters at a concentration of 10 μmol/L, using the Cerep BioPrint In Vitro Pharmacology Profiling from Cerep SA (l'Evescault, France).
Pharmacokinetic study
The pharmacokinetic profile of PDM‐042 was examined using male Sprague–Dawley rats and male beagle dogs. Male F344 rats were used to examine the brain penetration of PDM‐042. PDM‐042 was intravenously administered at a dose of 0.1 mg/kg to rats or orally administered at a dose of 0.3 mg/kg to rats and dogs. Blood samples were collected at appropriate time intervals after the administration of PDM‐042 and centrifuged at 2150g for 15 min at 4°C to obtain plasma. Each plasma sample was extracted by ethyl acetate using 96 well Isolute SLE+ plate (Biotage AB, Uppsala, Sweden). The extracts were evaporated to dryness, reconstituted in 45% (v/v) methanol and 55% (v/v) 10 mmol/L ammonium acetate, and analyzed by liquid chromatography with tandem mass spectrometry (LC‐MS/MS). In the study of brain penetration, PDM‐042 was orally administered at a dose of 0.3 mg/kg to rats. Blood samples were collected 60 min after the administration of PDM‐042, after which the striata were immediately collected from both sides of the brain. The collected striata were homogenized in five times its volume of H2O using a Micro Smash TM‐100 (Tomy Seiko Co., Ltd., Tokyo, Japan), the internal standard was added, and the homogenates were centrifuged at 17,800g for 15 min at 4°C to yield supernatants, and analyzed by LC‐MS/MS. Pharmacokinetic parameters were calculated via noncompartmental analysis using WinNolin version 6.2 (Pharsight, Palo Alto, CA).
In vitro binding assay
Membrane fractions from rat striatum were prepared by the protocol described by Kotera et al. (2004) with a minor modification. In brief, male Sprague–Dawley rats were decapitated and brain tissues were rapidly dissected on an ice‐cold dish. The striata from both sides of the brain were immediately weighed and homogenized in ice‐cold HB‐A buffer (20 mmol/L Tris‐HCl, pH 7.5, 2 mmol/L Mg(CH3COO)2, 0.3 mmol/L CaCl2, 1 mmol/L dithiothreitol, supplemented with protease inhibitor cocktail (Complete, EDTA‐free in easy pack, Roche Diagnostics Ltd., Indianapolis, IN)) using a teflon homogenizer. The homogenates were centrifuged at 100,000g for 60 min at 4°C, after which the resulting pellets were homogenized in ice‐cold HBT‐A buffer (HB‐A buffer containing 0.5% Triton X‐100) using a teflon homogenizer. The homogenates were incubated for 30 min at 4°C and centrifuged at 100,000g for 60 min at 4°C. The supernatant of each sample was collected as the membrane fraction, aliquoted, and stored at −80°C. The protein concentration of each membrane fraction was measured using the Bio‐Rad DC Protein Assay Reagents Package Kit from Bio‐Rad Laboratories, Inc. (Hercules, CA) with bovine serum albumin as a standard.
[3H]PDM‐042 was diluted with reaction buffer (20 mmol/L Tris‐HCl, pH 7.5, 2 mmol/L Mg(CH3COO)2, 0.3 mmol/L CaCl2) in binding assay. The membrane fractions (50 μg/well) were incubated with various concentrations (0.78–50 nmol/L) of [3H]PDM‐042 for 60 min at room temperature in a final volume of 200 μL/well. Nonspecific binding was determined by measuring binding in the presence of unlabeled PDM‐042 at 10 μmol/L. The displacement study was conducted by incubating membrane fractions with [3H]PDM‐042 at 15 nmol/L in the presence of increasing concentrations of PDM‐042 or MP‐10 ranging from 0.1 pmol/L to 1 μmol/L. Bound and free radioligand were separated by rapid filtration using MultiScreen FB GF/B filter plates (Millipore Corp, Billerica, MA) presoaked in 0.5% polyethyleneimine and a MultiScreen Vacuum Manifold (Millipore Corp). Filter‐bound radioactivity was counted using a Liquid Scintillation Analyzer Tri‐Carb 2910 TR (Perkin Elmer) after the addition of Ecoscint XR (National Diagnostics, Atlanta, GA). The specific binding of [3H]PDM‐042 was calculated by subtracting nonspecific binding from total binding. The dissociation constant (K d) and maximal number of binding sites (B max) of [3H]PDM‐042 in rat striatal membrane were determined by nonlinear regression and fitting to a one‐site binding model using GraphPad Prism Software, version 5 (GraphPad Software).
In vivo occupancy test
[3H]PDM‐042 diluted with physiological saline was intravenously administered to male F344 rats (3 μCi/0.2 mL/rat) 50 min after the administration of PDM‐042. The rats were decapitated 10 min after the administration of [3H]PDM‐042 and brain tissues were rapidly dissected on an ice‐cold dish. The striata from both sides of the brain were immediately weighed and homogenized in 10 mL ice‐cold buffer (25 mmol/L sodium phosphate, pH 7.4) using a teflon homogenizer. The homogenates (5 mL) were filtered through Whatman GF/B filters presoaked in 0.3% polyethyleneimine and washed twice with 5 mL of ice‐cold buffer. The filters were immersed in 3 mL of scintillation fluid and radioactivity was counted with a Tri‐Carb 2910TR (Perkin Elmer). Nonspecific binding was defined as the radioactivity after the administration of unlabeled PDM‐042 at a dose of 30 mg/kg. The specific binding for each group was calculated by subtracting nonspecific binding from the binding observed in the vehicle and drug‐treated groups. Data were obtained as disintegrations per minute (DPM)/mg wet tissue. Enzyme occupancy was calculated as follows:
The occupancy curve was fitted with GraphPad Prism Software, version 5 (GraphPad Software).
Locomotor activity
Locomotor activity was measured using a digital counter system with an infrared sensor (SUPERMEX, Muromachi Kikai Co., Ltd., Tokyo, Japan). Male Sprague–Dawley rats were habituated to the test room for at least 60 min before testing. Each rat was treated with PDM‐042 or olanzapine and immediately placed into a novel plastic cage (width, 270 mm × depth, 440 mm × height, 187 mm) with clean paper chips. Spontaneous locomotor activity was measured for 60 min. In the antagonism study, PDM‐042 or olanzapine was administered to each rat 60 min before administration of MK‐801 (0.2 mg/kg). Immediately after the administration of MK‐801, each rat was placed into a novel plastic cage with clean paper chips and locomotor activity was measured for 60 min.
CAR
The CAR was assessed in male F344 rats using Plexiglas shuttle boxes that were divided by a guillotine door into two compartments and enclosed in sound‐attenuating chambers (MED Associates Inc., St. Albans, VT). Each of the two floors of the shuttle box comprised a series of metal grid rods equipped with scrambled electric shockers. Each side of the shuttle box was equipped with a stimulus light, tone, and multiple infrared beam detectors to locate the animal within the box.
Each trial consisted of a 10 sec stimulus light and tone (CS, conditioned stimulus) followed by a 0.8 mA, 10 sec electric shock (UCS, unconditioned stimulus), which was presented through the grid floor on the side where the animal was located in the presence of the light and tone. Crossing to the opposite compartment during the 10 sec CS was recorded as an avoidance response, crossing during the UCS was recorded as an escape, and a failure to cross was recorded as an escape failure. Each session consisted of 30 CS‐UCS trials that were separated by intertrial intervals of a randomized duration (between 7.5 and 22.5 sec) and started immediately after the animal entered the left side of the box and the guillotine door opened. The number of trials in which the animals avoided shock, escaped shock, and failed to respond was recorded by the computer program.
Regular training was continued until the rats successfully avoided the UCS. Only rats displaying stable performance (more than 80% avoidance response on the three consecutive days before the test day) were considered as trained rats and investigated on the test day. To examine the effects of the test drugs, the selected rats were assigned to experimental groups based on the baseline avoidance response. PDM‐042 was administered 60 min before the test session, while risperidone or olanzapine was administered 30 min before the test session. The numbers of avoidances, escapes, and escape failures were recorded over 30 trials, after which the rate of each response was calculated for each treatment.
Catalepsy
Male Sprague–Dawley rats were subjected to catalepsy testing. PDM‐042 and olanzapine were administered 60 and 30 min, respectively, before the test. Catalepsy was assessed by placing both forepaws of the rat on a horizontal bar raised approximately 10 cm above the floor. The latencies required for the rat to remove its forepaws, move a hindlimb, and climb down from the bars into a normal posture were recorded with a cut‐off time of 90 sec.
Prolactin release
Male Sprague–Dawley rats were decapitated 60 and 30 min after the administration of PDM‐042 and risperidone, respectively. Blood was collected from the sacrificed rats into tubes containing heparin. All blood samples were collected during the daytime and centrifuged at 1600g for 20 min at 4°C to obtain plasma samples, which were transferred into new tubes and stored at −20°C. Plasma prolactin concentrations were measured with the RAT PROLACTIN ENZYME IMMUNOASSAY KIT from SPI‐BIO (Montigny le Bretonneux, France).
Glucose elevation
One day before the test, male Sprague‐Dawley rats were fasted overnight. On the test day, the rats were divided into each group based on body weight to minimize the difference of average weight of rats among all treatment groups. PDM‐042 or olanzapine was administered 60 min before intraperitoneal glucose challenge at a dose of 2 g/kg. Blood samples were collected before compound administration, 5 min before glucose challenge, and 15, 30, 60, and 120 min after glucose challenge. Blood samples were centrifuged at 2150g for 15 min at 4°C to obtain plasma samples. Plasma glucose levels were measured with the Glucose CII‐Test Wako from Wako Pure Chemical Industries and calculated as the area under the curve (AUC) from 0 to 120 min.
Statistical analysis
Student's t‐test was used to analyze the effects of MK‐801 and risperidone in the locomotor activity test and prolactin release test, respectively. The effects of the test compounds were analyzed by one‐way analysis of variance and post hoc comparisons were performed by Dunnett's test (locomotor activity, prolactin release, and glucose elevation) and Steel's test (CAR and catalepsy). The ED50 values in the CAR test were calculated by nonlinear regression analysis using GraphPad Prism Software, version 5 (GraphPad Software). A probability level of <0.05 was considered statistically significant.
Results
In vitro and pharmacokinetic profiles of PDM‐042
The inhibitory activity of PDM‐042 was examined using recombinant human and rat PDE10A. PDM‐042 inhibited recombinant human and rat PDE10A with IC50 values of 0.83 and 0.82 nmol/L, respectively. Inhibition constants were examined using fluorescein‐labeled cAMP and recombinant human and rat PDE10A. PDM‐042 inhibited human and rat PDE10A with K i values of 0.36 and 0.59 nmol/L, respectively. The selectivity of PDM‐042 was examined against other PDEs. PDM‐042 inhibited all tested PDE isozymes by less than 50% at a concentration of 1 μmol/L (Table 1), suggesting that PDM‐042 has more than 1000‐fold selectivity against other PDEs. The selectivity of PDM‐042 was also examined against 137 other molecular targets (Table S1). PDM‐042 exhibited weak inhibitory effects of greater than 50% in only 2 receptor binding assays (muscarinic acetylcholine receptor M2 and neurokinin 1 receptor) and 1 enzyme assay (PDE6) at a concentration of 10 μmol/L. However, PDM‐042 showed neither agonistic nor antagonistic activity against muscarinic acetylcholine receptor M2 and neurokinin 1 receptor at a concentration of 10 μmol/L.
Table 1.
PDE selectivity of PDM‐042 at 1 μmol/L
| PDE | Species | Inhibition (%) |
|---|---|---|
| 1A | Human | 12 |
| 2A | Human | 23 |
| 3A | Human | −4 |
| 4A1A | Human | 12 |
| 4B1 | Human | 9 |
| 5A | Human | 13 |
| 6 | Bovine | 28 |
| 7A | Human | 38 |
| 7B | Human | 10 |
| 8A1 | Human | 0 |
| 9A2 | Human | −1 |
| 10A2 | Human | 101 |
| 11A4 | Human | −4 |
PDM‐042 showed good oral bioavailability of 33% in rats and 42% in dogs at a dose of 0.3 mg/kg (Table S2). PDM‐042 also exhibited excellent brain penetration 60 min after the administration of 0.3 mg/kg to rats (Table S2).
In vitro binding of [3H]PDM‐042 to PDE10A in rat striatal homogenates
An in vitro binding study of the radiotracer, [3H]PDM‐042, was performed using rat striatal homogenates. The specific binding of [3H]PDM‐042 to PDE10A in rat striatal membrane fractions was saturable (Fig. 2A). [3H]PDM‐042 had high affinity for membrane fractions prepared from rat striatum and showed a B max value of 7.2 pmol/mg protein and a K d value of 8.5 nmol/L. To determine whether the radiotracer could be displaced, the binding of [3H]PDM‐042 to rat striatal membrane fractions was tested with unlabeled PDM‐042 and the structurally unrelated PDE10A inhibitor, MP‐10. PDM‐042 and MP‐10 displaced [3H]PDM‐042 binding to PDE10A in a concentration‐dependent manner (Fig. 2B and C).
Figure 2.

In vitro binding of [3H]PDM‐042 to membranes prepared from rat striatum. Saturation binding was determined by incubating [3H]PDM‐042 (0.78–50 nmol/L) with rat striatal membranes for 60 min (A). Specific binding of [3H]PDM‐042 was displaced by PDM‐042 (filled circles) (B) and structurally unrelated PDE10A inhibitor, MP‐10 (filled triangles) (C). Data are expressed as the mean of specific binding in the experiment performed in duplicate.
In vivo occupancy in rat striatum
An in vivo occupancy study was conducted using [3H]PDM‐042 in rats. [3H]PDM‐042 increased striatal radioactivity in a concentration‐dependent manner. The PDM‐042 occupancy rate in the striatum increased in a dose‐dependent manner and was 86.6% following administration of the highest tested dose of 3 mg/kg (Fig. 3).
Figure 3.

In vivo occupancy of PDM‐042 in rat striatum. PDM‐042 (0.1‒3 mg/kg, p.o.) was administered 60 min before decapitation. [3H]PDM‐042 was administered 10 min before decapitation. Data are expressed as the mean of 3 animals.
Locomotor activity
The effects of PDM‐042 and olanzapine on spontaneous locomotor activity, as well as the antagonistic effects of these compounds against MK‐801‐induced hyperlocomotion, were tested in rats. PDM‐042 significantly decreased spontaneous locomotor activity at doses of 1 and 3 mg/kg (Fig. 4A). PDM‐042 significantly antagonized MK‐801‐induced hyperlocomotion at doses from 0.1 to 0.3 mg/kg without affecting spontaneous locomotor activity (Fig. 4A and 5A). Olanzapine significantly decreased spontaneous locomotor activity at a dose of 3 mg/kg (Fig. 4B), while olanzapine significantly antagonized the MK‐801‐induced hyperlocomotion at doses of 1 and 3 mg/kg (Fig. 5B).
Figure 4.

Effects of PDM‐042 (A) and olanzapine (B) on spontaneous locomotor activity in rats. Spontaneous locomotor activity was measured during a 60 min observation period immediately after the administration of PDM‐042 (0.1–3 mg/kg, p.o.) or olanzapine (0.1–3 mg/kg, s.c.). Data are expressed as mean ± S.E.M. of 6 animals. Asterisks represent a significant difference from the vehicle‐treated group, *P < 0.05, **P < 0.01 (Dunnett's test).
Figure 5.

Effects of PDM‐042 (A) and olanzapine (B) on MK‐801‐induced hyperlocomotion in rats. PDM‐042 (0.03–0.3 mg/kg, p.o.) or olanzapine (0.3–3 mg/kg, s.c.) was administered 60 min before the administration of MK‐801 (0.2 mg/kg, s.c.). MK‐801‐induced hyperlocomotion was measured during a 60 min observation period immediately after the administration of MK‐801. Data are expressed as mean ± S.E.M. of 6–10 animals. Sharps represent a significant difference from the vehicle + vehicle‐treated group, # # P < 0.01 (Student's t‐test). Asterisks represent a significant difference from the vehicle + MK‐801‐treated group, *P < 0.05, **P < 0.01 (Dunnett's test).
CAR
We examined the effects of PDM‐042, risperidone, and olanzapine on the CAR in rats. PDM‐042 attenuated the CAR in a dose‐dependent manner with an ED50 of 0.44 mg/kg (Fig. 6A). Risperidone and olanzapine also attenuated the CAR in a dose‐dependent manner with ED50 of 0.088 and 1.4 mg/kg, respectively (Fig. 6B and C). None of the tested drugs produced significant escape failure response at any tested doses.
Figure 6.

Effects of PDM‐042 (A), risperidone (B), and olanzapine (C) on the CAR in rats. PDM‐042 (0.1–1 mg/kg, p.o.) was administered 60 min before the test session. Risperidone (0.01–0.2 mg/kg, s.c.) or olanzapine (0.3–3 mg/kg, s.c.) was administered 30 min before the test session. Data are expressed as mean ± S.E.M. of 5–7 animals. Asterisks represent a significant difference from the vehicle‐treated group, *P < 0.05, **P < 0.01 (Steel's test).
Catalepsy
The cataleptic effect of PDM‐042 was tested at doses from 0.3 to 10 mg/kg in rats. PDM‐042 significantly induced catalepsy at doses from 1 to 10 mg/kg in a manner that was not dose‐dependent (Table 2). In contrast, olanzapine significantly induced catalepsy at doses from 1 to 10 mg/kg, as shown in Table 2.
Table 2.
Effect of PDM‐042 on catalepsy in rats
| Dose | Duration (sec) | |
|---|---|---|
| Vehicle | 0.80 ± 0.05 | |
| PDM‐042 | 0.3 mg/kg, p.o. | 1.5 ± 0.7 |
| PDM‐042 | 1 mg/kg, p.o. | 14 ± 2** |
| PDM‐042 | 3 mg/kg, p.o. | 13 ± 2** |
| PDM‐042 | 10 mg/kg, p.o. | 13 ± 1** |
| Vehicle | 0.49 ± 0.03 | |
| Olanzapine | 1 mg/kg, s.c. | 2.8 ± 1.3* |
| Olanzapine | 3 mg/kg, s.c. | 29 ± 13* |
| Olanzapine | 10 mg/kg, s.c. | 31 ± 8** |
Data are expressed as mean ± S.E.M. of seven animals. Statistical significance between the drug‐treated groups and the vehicle‐treated group was determined by Steel's test. Asterisks represent a significant difference from the vehicle‐treated group, *P < 0.05, **P < 0.01.
Prolactin release and glucose elevation
In prolactin release, risperidone significantly increased the plasma prolactin concentration at a dose of 0.1 mg/kg, while PDM‐042 did not affect plasma prolactin concentration up to 3 mg/kg (data not shown). In the glucose tolerance test, PDM‐042 did not increase plasma glucose levels up to 3 mg/kg (Fig. 7A), while olanzapine significantly increased plasma glucose levels at a dose of 10 mg/kg (Fig. 7B).
Figure 7.

Effects of PDM‐042 (A) and olanzapine (B) on plasma glucose levels during intraperitoneal glucose tolerance tests in rats. PDM‐042 (0.1–3 mg/kg, p.o.) or olanzapine (1–10 mg/kg, s.c.) was administered 60 min before intraperitoneal glucose challenge (2 g/kg, i.p.). Data are expressed as mean ± S.E.M. of 7–8 animals. Asterisks represent a significant difference from the vehicle‐treated group, **P < 0.01 (Dunnett's test).
Discussion
We identified a novel potent, selective, orally active, and brain‐penetrable PDE10A inhibitor, PDM‐042. PDM‐042 showed potent inhibitory activities for human and rat PDE10A with IC50 values of 0.83 and 0.82 nmol/L, respectively, and had no significant off‐target activity against other PDEs or other pharmacological targets, including neurotransmitter receptors, enzymes, ion channels, and transporters. Furthermore, PDM‐042 showed favorable pharmacokinetic profiles with good oral bioavailability and brain penetration in rats and dogs. These results indicate that PDM‐042 is a good pharmacological tool that can be used to investigate the role of PDE10A in vitro and in vivo.
In this study, we compared the effects of PDM‐042 with those of some currently available atypical antipsychotics in rat models relevant to schizophrenia. To examine the antipsychotic potential of PDM‐042, we selected the MK‐801‐induced hyperlocomotion and CAR models. Because atypical antipsychotics attenuate the hyperlocomotion caused by N‐methyl‐d‐aspartate (NMDA) receptor antagonists such as MK‐801 and phencyclidine in rodents, MK‐801‐induced hyperlocomotion is commonly used in preclinical studies to predict the antipsychotic potential of test compounds (Gattaz et al. 1994; O'Neill and Shaw 1999). PDM‐042 significantly antagonized MK‐801‐induced hyperlocomotion at doses from 0.1 to 0.3 mg/kg, but did not decrease spontaneous locomotor activity, indicating that the antagonistic effect of PDM‐042 against MK‐801‐induced hyperlocomotion was not due to behavioral suppression by PDM‐042 itself. Olanzapine significantly antagonized MK‐801‐induced hyperlocomotion at doses of 1 and 3 mg/kg, however, olanzapine significantly decreased spontaneous locomotor activity at a dose of 3 mg/kg, while its antagonistic effect at the same dose was only partial. In this study, PDM‐042 attenuated the hyperlocomotion caused by NMDA receptor blockade with efficacy similar to that of olanzapine, while the minimum effective dose of PDM‐042 was 10‐fold lower than that which decreased spontaneous locomotor activity. These findings suggest that the functional antagonism with PDM‐042 against MK‐801 may be due to the inhibition of PDE10A modulating glutamatergic system.
CAR is considered to possess a high degree of predictive validity (Wadenberg and Hicks 1999). PDM‐042 attenuated the CAR in a dose‐dependent manner and significantly reduced avoidance rates at doses from 0.3 to 1 mg/kg. PDM‐042 did not cause significant escape failure responses at any tested doses, indicating that the effect of PDM‐042 on the CAR was mediated by specific inhibition of PDE10A. In addition, risperidone and olanzapine also significantly attenuated the CAR at doses of 0.1 and 0.2 mg/kg and 1 and 3 mg/kg, respectively, without affecting escape failure responses. These results suggest that PDM‐042 may have antipsychotic potential similar to that of atypical antipsychotics such as risperidone and olanzapine.
To examine the potential liability of PDM‐042 that could be produced along with its antipsychotic‐like effects, we investigated the effects of PDM‐042 in catalepsy, prolactin release, and glucose elevation.
Most antipsychotics are known to cause EPS in clinic, and catalepsy is considered to be a suitable rodent model for detecting EPS liability in humans (Hoffman and Donovan 1995). PDM‐042 did not induce catalepsy at a dose of 0.3 mg/kg, which was a dose sufficient to antagonize MK‐801‐induced hyperlocomotion and the minimum effective dose in the CAR. PDM‐042 significantly induced catalepsy at doses from 1 to 10 mg/kg. However, this cataleptic response did not intensify as the dose was increased to 10 mg/kg. These results suggest that PDM‐042 had a minimal cataleptic effect even at a dose 30‐fold higher than that which antagonized MK‐801‐induced hyperlocomotion and attenuated the CAR. Olanzapine significantly induced catalepsy at doses from 1 to 10 mg/kg, which were near to the dose which antagonized MK‐801‐induced hyperlocomotion and attenuated the CAR. Taken together, our findings suggest that PDM‐042 has a limited cataleptic effect even at doses much higher than its efficacious dose, unlike olanzapine. Because antipsychotics such as olanzapine and risperidone cause EPS by D2 receptor blockade (Pierre 2005; Krebs et al. 2006), the cataleptic effect induced by PDM‐042 may be at least partially mediated by D2 antagonism. However, the cataleptic effect induced by PDM‐042 was minimal, even at a dose of 10 mg/kg (much higher than the dose that antagonized MK‐801‐induced hyperlocomotion and attenuated the CAR), indicating that activation of D1 signaling is likely involved in the limited cataleptic effect of PDM‐042. It was reported that the cataleptic response induced by haloperidol as D2 antagonist was attenuated by D1 agonist, SKF82598 (Megens et al. 2014; Suzuki et al. 2015). In addition, it was recently demonstrated that PDE10A inhibitor, MP‐10 reversed the cataleptic effect induced by haloperidol in a dose‐dependent manner (Megens et al. 2014). These findings indicate that activation of D1 signaling via PDE10A inhibition can minimize the cataleptic effect induced by PDE10A inhibition. Therefore, further studies with D1 agonists/antagonists will be necessary to clarify the contribution of D1 agonism to the cataleptic effect induced by PDM‐042.
Hyperprolactinemia and glucose intolerance as a result of administration of atypical antipsychotics are serious adverse effects in the clinic (Krebs et al. 2006). Risperidone and olanzapine were reported to cause hyperprolactinemia and glucose elevation, respectively (Dunlop et al. 2003; Krebs et al. 2006; Peuskens et al. 2014). Therefore, we compared the effects of PDM‐042 on prolactin release and glucose elevation with those of risperidone and olanzapine, respectively, in rats. PDM‐042 increased neither prolactin nor glucose concentrations in plasma up to 3 mg/kg, which was 10‐fold higher than the minimal effective dose in the CAR. This result was nearly consistent with the previous findings that a structurally unrelated PDE10A inhibitor, TAK‐063 under Phase 2 clinical trial had no effects in prolactin release and glucose elevation up to 30‐fold higher dose (3 mg/kg) than the minimum dose (0.1 mg/kg) required to significantly suppress MK‐801‐induced hyperlocomotion in rats (Suzuki et al. 2015). In contrast, risperidone significantly increased prolactin concentrations at a dose of 0.1 mg/kg, while olanzapine increased glucose levels at a dose 10‐fold higher than the minimal effective dose in the CAR. These findings demonstrate that, unlike risperidone and olanzapine, PDM‐042 may not elicit hyperprolactinemia and hyperglycemia and thus may have a greater therapeutic window than currently available atypical antipsychotics.
We showed that PDM‐042 significantly antagonized MK‐801‐induced hyperlocomotion and attenuated the CAR in rats. Regarding adverse effects, PDM‐042 had a minimal cataleptic effect, even at a dose much higher than the efficacious dose in the CAR, and affected neither prolactin release nor glucose elevation. However, the relationship between PDE10A occupancy and the pharmacological effects of PDM‐042 was unclear. Therefore, we examined the relationship between the dose of PDM‐042 and its occupancy of PDE10A using [3H]PDM‐042, which was newly synthesized as a radioligand for this study. To confirm the specific binding of [3H]PDM‐042 to striatal PDE10A in rats, we first conducted an in vitro binding assay using striatal membrane fractions of rats. [3H]PDM‐042 showed high affinity for PDE10A in striatal membrane fractions, with a B max value of 7.2 pmol/mg protein and a K d value of 8.5 nmol/L, while its specific binding was saturable. Both PDM‐042 and another PDE10A inhibitor, MP‐10, displaced [3H]PDM‐042 from PDE10A in a concentration‐dependent manner. These results suggest that [3H]PDM‐042 specifically binds to striatal PDE10A in rats and can be a suitable radioligand for in vivo occupancy studies. The PDE10A occupancy rate of PDM‐042 increased dose‐dependently and was approximately 90% at a dose of 3 mg/kg. We also estimated the PDE10A occupancy by PDM‐042 at its ED50 and at the dose producing a nearly maximal effect in the CAR using the dose‐occupancy curve of PDM‐042. The PDE10A occupancy rates of PDM‐042 at its ED50 (0.44 mg/kg) and nearly maximal effective dose (1 mg/kg) were 39 and 66%, respectively. These results indicate that PDE10A occupancy of at least 40–70% may be necessary for PDM‐042 to exhibit antipsychotic‐like effects. Further studies with several structurally unrelated PDE10A inhibitors will be necessary to clarify the rate of PDE10A occupancy required to exhibit antipsychotic‐like effects.
In this study, PDM‐042 was confirmed as a highly potent, selective, orally active, and brain‐penetrable PDE10A inhibitor. PDM‐042 exhibited antipsychotic‐like activity with less adverse effect profiles than those of currently available atypical antipsychotics in rats. We also demonstrated that radiotracer [3H]PDM‐042 is useful for measuring target occupancy by PDE10A inhibitors in vivo, enabling us to analyze the relationship between pharmacological effects and PDE10A occupancy in rats. These findings suggest that PDM‐042 may have therapeutic potential with a favorable safety profiles. Moreover, these results demonstrate that inhibition of PDE10A may provide an alternative approach for the treatment of schizophrenia.
Disclosure
All authors are employees of Mochida Pharmaceutical Co., Ltd.
Supporting information
Table S1. Inhibitory effect of PDM‐042 among 137 molecular targets listed in Cerep BioPrint In Vitro Pharmacology Profiling at 10 μmol/L.
Table S2. Pharmacokinetic profiles of PDM‐042 in rats and dogs.
Acknowledgements
We thank Keiko Taguchi and Koko Fujimoto for assistance with in vitro experiments. We thank Rie Yanagawa, Yusuke Nakagawa, Toshihiro Sato, and Naoto Tsuda for assistance with in vivo experiments.
Arakawa K., Maehara S., Yuge N., Ishikawa M., Miyazaki Y., Naba H., Kato Y., Nakao K., Pharmacological characterization of a novel potent, selective, and orally active phosphodiesterase 10A inhibitor, PDM‐042 [(E)‐4‐(2‐(2‐(5,8‐dimethyl‐[1,2,4]triazolo[1,5‐a]pyrazin‐2‐yl)vinyl)‐6‐(pyrrolidin‐1‐yl)pyrimidin‐4‐yl)morpholine] in rats: potential for the treatment of schizophrenia, Pharma Res Per, 4(4), 2016, e00241, doi: 10.1002/prp2.241
References
- Bender AT, Beavo JA (2006). Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58: 488–520. [DOI] [PubMed] [Google Scholar]
- Coskran TM, Morton D, Menniti FS, Adamowicz WO, Kleiman RJ, Ryan AM, et al. (2006). Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J Histochem Cytochem 54: 1205–1213. [DOI] [PubMed] [Google Scholar]
- Dunlop BW, Sternberg M, Phillips LS, Andersen J, Duncan E (2003). Disturbed glucose metabolism among patients taking olanzapine and typical antipsychotics. Psychopharmacol Bull 37: 99–117. [PubMed] [Google Scholar]
- Fujishige K, Kotera J, Omori K (1999). Striatum‐ and testis‐specific phosphodiesterase PDE10A isolation and characterization of a rat PDE10A. Eur J Biochem 266: 1118–1127. [DOI] [PubMed] [Google Scholar]
- Gattaz WF, Schummer B, Behrens S (1994). Effects of zotepine, haloperidol and clozapine on MK‐801‐induced stereotypy and locomotion in rats. J Neural Transm Gen Sect 96: 227–232. [DOI] [PubMed] [Google Scholar]
- Grauer SM, Pulito VL, Navarra RL, Kelly MP, Kelley C, Graf R, et al. (2009). Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J Pharmacol Exp Ther 331: 574–590. [DOI] [PubMed] [Google Scholar]
- Graybiel AM (1990). Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci 13: 244–254. [DOI] [PubMed] [Google Scholar]
- Graybiel AM (2000). The basal ganglia. Curr Biol 10: R509–R511. [DOI] [PubMed] [Google Scholar]
- Harada A, Suzuki K, Miura S, Hasui T, Kamiguchi N, Ishii T, et al. (2015). Characterization of the binding properties of T‐773 as a PET radioligand for phosphodiesterase 10A. Nucl Med Biol 42: 146–154. [DOI] [PubMed] [Google Scholar]
- Hoffman DC, Donovan H (1995). Catalepsy as a rodent model for detecting antipsychotic drugs with extrapyramidal side effect liability. Psychopharmacology 120: 128–133. [DOI] [PubMed] [Google Scholar]
- Hu E, Ma J, Biorn C, Lester‐Zeiner D, Cho R, Rumfelt S, et al. (2012). Rapid identification of a novel small molecule phosphodiesterase 10A (PDE10A) tracer. J Med Chem 55: 4776–4787. [DOI] [PubMed] [Google Scholar]
- Kapur S, Mamo D (2003). Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry 27: 1081–1090. [DOI] [PubMed] [Google Scholar]
- Kapur S, Remington G (2001). Dopamine D(2) receptors and their role in atypical antipsychotic action: still necessary and may even be sufficient. Biol Psychiatry 50: 873–883. [DOI] [PubMed] [Google Scholar]
- Kotera J, Sasaki T, Kobayashi T, Fujishige K, Yamashita Y, Omori K (2004). Subcellular localization of cyclic nucleotide phosphodiesterase type 10A variants, and alteration of the localization by cAMP‐dependent protein kinase‐dependent phosphorylation. J Biol Chem 279: 4366–4375. [DOI] [PubMed] [Google Scholar]
- Krebs M, Leopold K, Hinzpeter A, Schaefer M (2006). Current schizophrenia drugs: efficacy and side effects. Expert Opin Pharmacother 7: 1005–1016. [DOI] [PubMed] [Google Scholar]
- Loughney K, Snyder PB, Uher L, Rosman GJ, Ferguson K, Florio VA (1999). Isolation and characterization of PDE10A, a novel human 3’, 5’‐cyclic nucleotide phosphodiesterase. Gene 234: 109–117. [DOI] [PubMed] [Google Scholar]
- Megens AA, Hendrickx HM, Mahieu MM, Wellens AL, de Boer P, Vanhoof G (2014). PDE10A inhibitors stimulate or suppress motor behavior dependent on the relative activation state of the direct and indirect striatal output pathways. Pharmacol Res Perspect 2: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Snyder GL (2010). Advanced research on dopamine signaling to develop drugs for the treatment of mental disorders: biochemical and behavioral profiles of phosphodiesterase inhibition in dopaminergic neurotransmission. J Pharmacol Sci 114: 6–16. [DOI] [PubMed] [Google Scholar]
- Nishi A, Kuroiwa M, Miller DB, O'Callaghan JP, Bateup HS, Shuto T, et al. (2008). Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J Neurosci 28: 10460–10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill MF, Shaw G (1999). Comparison of dopamine receptor antagonists on hyperlocomotion induced by cocaine, amphetamine, MK‐801 and the dopamine D1 agonist C‐APB in mice. Psychopharmacology 145: 237–250. [DOI] [PubMed] [Google Scholar]
- Peuskens J, Pani L, Detraux J, De Hert M (2014). The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs 28: 421–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre JM (2005). Extrapyramidal symptoms with atypical antipsychotics: incidence, prevention and management. Drug Saf 28: 191–208. [DOI] [PubMed] [Google Scholar]
- Sano H, Nagai Y, Miyakawa T, Shigemoto R, Yokoi M (2008). Increased social interaction in mice deficient of the striatal medium spiny neuron‐specific phosphodiesterase 10A2. J Neurochem 105: 546–556. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Chapin DS, Cianfrogna J, Corman ML, Hajos M, Harms JF, et al. (2008). Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther 325: 681–690. [DOI] [PubMed] [Google Scholar]
- Seeger TF, Bartlett B, Coskran TM, Culp JS, James LC, Krull DL, et al. (2003). Immunohistochemical localization of PDE10A in the rat brain. Brain Res 985: 113–126. [DOI] [PubMed] [Google Scholar]
- Smith SM, Uslaner JM, Cox CD, Huszar SL, Cannon CE, Vardigan JD, et al. (2013). The novel phosphodiesterase 10A inhibitor THPP‐1 has antipsychotic‐like effects in rat and improves cognition in rat and rhesus monkey. Neuropharmacology 64: 215–223. [DOI] [PubMed] [Google Scholar]
- Soderling SH, Bayuga SJ, Beavo JA (1999). Isolation and characterization of a dual‐substrate phosphodiesterase gene family: PDE10A. Proc Natl Acad Sci U S A 96: 7071–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Harada A, Shiraishi E, Kimura H (2015). In vivo pharmacological characterization of TAK‐063, a potent and selective phosphodiesterase 10A inhibitor with antipsychotic‐like activity in rodents. J Pharmacol Exp Ther 352: 471–479. [DOI] [PubMed] [Google Scholar]
- Wadenberg ML, Hicks PB (1999). The conditioned avoidance response test re‐evaluated: is it a sensitive test for the detection of potentially atypical antipsychotics? Neurosci Biobehav Rev 23: 851–862. [DOI] [PubMed] [Google Scholar]
- Xie Z, Adamowicz WO, Eldred WD, Jakowski AB, Kleiman RJ, Morton DG, et al. (2006). Cellular and subcellular localization of PDE10A, a striatum‐enriched phosphodiesterase. Neuroscience 139: 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Inhibitory effect of PDM‐042 among 137 molecular targets listed in Cerep BioPrint In Vitro Pharmacology Profiling at 10 μmol/L.
Table S2. Pharmacokinetic profiles of PDM‐042 in rats and dogs.