

Abstract
Methyl-CpG-binding domain 1 (MBD1) belongs to a family of methyl-CpG-binding proteins that are epigenetic “readers” linking DNA methylation to transcriptional regulation. MBD1 is expressed in neural stem cells residing in the dentate gyrus of the adult hippocampus (aNSCs) and MBD1 deficiency leads to reduced neuronal differentiation, impaired neurogenesis, learning deficits, and autism-like behaviors in mice; however, the precise function of MBD1 in aNSCs remains unexplored. Here, we show that MBD1 is important for maintaining the integrity and stemness of NSCs, which is critical for their ability to generate neurons. MBD1 deficiency leads to the accumulation of undifferentiated NSCs and impaired transition into the neuronal lineage. Transcriptome analysis of neural stem and progenitor cells isolated directly from the dentate gyrus of MBD1 mutant (KO) and WT mice showed that gene sets related to cell differentiation, particularly astrocyte lineage genes, were upregulated in KO cells. We further demonstrated that, in NSCs, MBD1 binds and represses directly specific genes associated with differentiation. Our results suggest that MBD1 maintains the multipotency of NSCs by restraining the onset of differentiation genes and that untimely expression of these genes in MBD1-deficient stem cells may interfere with normal cell lineage commitment and cause the accumulation of undifferentiated cells. Our data reveal a novel role for MBD1 in stem cell maintenance and provide insight into how epigenetic regulation contributes to adult neurogenesis and the potential impact of its dysregulation.
SIGNIFICANCE STATEMENT Adult neural stem cells (aNSCs) in the hippocampus self-renew and generate neurons throughout life. We show that methyl-CpG-binding domain 1 (MBD1), a DNA methylation “reader,” is important for maintaining the integrity of NSCs, which is critical for their neurogenic potency. Our data reveal a novel role for MBD1 in stem cell maintenance and provide insight into how epigenetic regulation preserves the multipotency of stem cells for subsequent differentiation.
Keywords: epigenetics, FACS-seq, lineage restriction, MBD1, neural stem cells, neurogenesis
Introduction
Adult hippocampal neurogenesis contributes to information processing that is critical for cognition, adaptation, learning, and memory (Kempermann et al., 2015). Deficits in adult hippocampal neurogenesis may contribute to shared facets of many neurodevelopmental, psychiatric, and neurodegenerative disorders (Hsieh and Eisch, 2010; Christian et al., 2014). Neural stem cells (NSCs) in the dentate gyrus (DG) of the adult hippocampus self-renew, generating neurons and astrocytes throughout life, and are highly regulated by both intrinsic programs and extrinsic niche signals to strike a balance between stem cell maintenance and lineage differentiation (Jobe et al., 2012). A wealth of research has shown that epigenetic regulation via DNA methylation, histone modifications, and noncoding RNAs play important roles in adult neurogenesis (Jobe and Zhao, 2016); however, the precise epigenetic mechanisms required for maintaining adult NSC identity and multipotency are not fully clear.
Methyl-CpG-binding domain 1 (MBD1) and other members of the methyl-CpG-binding protein (MBP) family, including MeCP2 and MBD1–6, are central players in epigenetic regulation (Shin et al., 2014). MBD1 can bind methylated DNA and interact with a number of chromatin-modifying proteins to mediate gene repression, but the extent to which MBD1 contributes to the maintenance of the epigenome in vivo remains unexplored (Fournier et al., 2012). In humans, mutations or polymorphisms in MBD1 have been identified in sporadic cases of autism spectrum disorder (ASD; Li et al., 2005; Cukier et al., 2010). MBD1 is also contained within the critical region of del(18)(q12.2q21.1) syndrome characterized by developmental delay, hypotonia, obesity, and epilepsy (Imataka et al., 2015). Some cases of atypical Rett syndrome, a severe neurodevelopmental disorder, have also been attributed to del(18)(q12.2q21.1) without the classic MeCP2 mutations (Gustavsson et al., 1999). We have shown that mice with Mbd1 deletion (MBD1-KO) exhibit behavioral deficits associated with ASD, including learning impairment, increased anxiety, reduced social interest, and impaired sensorimotor gating (Zhao et al., 2003; Allan et al., 2008). Unlike deletion of MeCP2 and MBD5, which produce severe defects (Chen et al., 2001; Du et al., 2012), deletion of MBD1 results in comparatively mild phenotypes, yet mutations in these MBPs result in overlapping ASD symptoms (Castro et al., 2013). Therefore, studying the role of MBD1 in neurodevelopment will help us to understand how epigenetic maintenance contributes to the spectrum of subtle neurobehavioral phenotypes that occurs in human populations.
In addition to its significant function, adult hippocampal neurogenesis also provides an excellent model for studying developmental regulation. MBD1-KO mice produce significantly fewer new neurons in the DG of the adult hippocampus, which may contribute to their behavioral deficits (Zhao et al., 2003; Allan et al., 2008). Using neural progenitors derived from the entire adult forebrain (fNPCs), we have shown that MBD1 deficiency leads to increased proliferation and reduced differentiation and we identified several transcriptional targets of MBD1 in fNPCs, including the protein-coding gene Fgf-2 and the noncoding miR-184 and miR-195 (Li et al., 2008, 2010, 2013). However, the function of MBD1 in NSCs residing in the adult DG remains unexplored and the mechanism by which MBD1 deficiency impairs adult DG neurogenesis is unclear. In addition, studies suggest that epigenetic regulation is critical in maintaining the stemness and multipotency of adult stem cells (Avgustinova and Benitah, 2016; Jobe and Zhao, 2016) cells. However, whether a loss of MBD1-mediated maintenance of the epigenome affects gene expression and multipotency of adult stem cells has not been investigated.
Here, we focused on the role of MBD1 in maintaining the multipotency of NSCs during adult hippocampal neurogenesis. We found that NSCs in the MBD1-KO adult DG accumulated and failed to transition into immature neurons. Transcriptome analysis of NESTIN-expressing cells isolated directly from the MBD1-KO adult DG revealed an upregulation of astrocyte genes. We further demonstrated that, in neural stem/progenitor cells derived from the adult DG (dgNPCs), MBD1 repressed lineage differentiation genes and its deficiency led to inappropriate expression of differentiation genes, not only in dgNPCs, but also in differentiated cells. These results suggest an important role for MBD1 in maintaining transcriptional integrity in NSCs and supporting the epigenetic mechanisms that fine-tune the fate specification.
Materials and Methods
Animals.
Animals were handled according to protocols approved by the Animal Care and Use Committee of the University of Wisconsin–Madison. Mice were group housed with the same gender, up to five animals per cage, and maintained on a 14 h light/10 h dark cycle with food and water available ad libitum. All experiments were initiated in 8-week-old mice. MBD1-KO mice (Zhao et al., 2003) were bred onto the C57BL6 genetic background for >10 generations (Liu et al., 2010). Nestin promoter-GFP (Nes-GFP) transgenic mice were described previously (Yamaguchi et al., 2000).
Tissue processing.
Tissues were processed according to standard methods (Guo et al., 2012a; Wang et al., 2015). For in vivo cell proliferation analyses using BrdU labeling, mice were given a single intraperitoneal injection of BrdU (200 mg/kg) 24 h before perfusion. Mice were deeply anesthetized with sodium pentobarbital (30 mg) by intraperitoneal injection followed by transcardial perfusion with 4% paraformaldehyde (PFA). Brains were dissected, postfixed overnight, and then equilibrated in 30% sucrose. Half brains were frozen at −80°C until sectioning. Forty-micrometer-thick sections were prepared using a sliding microtome.
Immunohistochemistry (IHC) of brain tissue.
IHC of brain tissue sections was performed using standard procedures (Guo et al., 2012a; Wang et al., 2015). The primary antibodies used include GFP (chicken, 1:1000, Invitrogen, catalog #A10263), GFAP (rabbit, 1:2000, DAKO, catalog #Z0334), BrdU (rat, 1:2000, Abcam, catalog #ab-6326), MCM2 (rabbit, 1:1000, Cell Signaling Technology, catalog #4007), Doublecortin (rabbit, 1:1000, Cell Signaling Technology, catalog #4604S), NESTIN (chicken, 1:500, Aves Labs, catalog #NES0407), TBR2 (chicken, 1:2000, Millipore, catalog #AB15894), β-galactosidase (β-gal, mouse, 2 μg/ml, Developmental Studies Hybridoma Bank, catalog #40–1a), β-gal (chicken, 1:500, Abcam, catalog #ab9361), S100β (rabbit, 1:1000, DAKO, catalog #A5110), SOX2 (goat, 1:500, Santa Cruz Biotechnology, catalog #SC-17320), TBR1 (chicken, 1:500, Millipore, catalog #Ab2216), NeuN (mouse, 1:1000, Millipore, catalog #MAB377), and βIII-tubulin (Tuj1; mouse, 1:1000, Promega, catalog #G712A). Fluorescent secondary antibodies (1:1000 dilution) were from Invitrogen; Alexa Fluor conjugates were as follows: goat anti-chicken-488 (catalog #A11039), goat anti-mouse-488 (catalog #A11029), goat anti-mouse-568 (catalog #A11031), goat anti-rabbit-568 (catalog #A11011), goat anti-rat-568 (catalog #A11077), goat anti-rabbit-647 (catalog #A21245), donkey anti-rabbit-488 (catalog #A21206) donkey anti-goat-568 (catalog #A11057), and donkey anti-rabbit-647 (catalog #A31573).
Stereology and cell counts.
Total cell MCM2+ or GFP+ numbers in the DG were determined by unbiased stereology methods using StereoInvestigator software (MBF Biosciences) as described previously (Guo et al., 2012a; Wang et al., 2015). z-stack images (2 μm interval) were acquired using an AxioImagerZ2 ApoTome confocal microscope (Plan-APOCHOROMAT, 20×, numerical aperture = 0.8; Zeiss). For GFP, MCM2, and GFAP, total cell numbers in the DG were determined by counting one in six coronal serial sections containing the hippocampus. The experimenter was blinded to the identity of the samples. To determine the percentages of marker (GFP or DCX)-positive cells among BrdU+ cells, 40–80 BrdU+ cells were analyzed in three to four sections per animal as described previously (Wang et al., 2015). The percentages of S100β and SOX2 among GFP+ cells were determined by analyzing ∼150 GFP+ cells in randomly selected sites of the DG of each animal. Images were processed for publication using SteroInvestigator and Adobe Photoshop CS5 software.
FACS.
FACS was performed as described previously (Gao et al., 2016) based on Beckervordersandforth et al. (2010). All samples were sorted on a FACSAria II (BD Biosciences) at the University of Wisconsin Carbone Cancer Center Flow Cytometry Laboratory. Nestin-GFP;WT and Nestin-GFP;MBD1-KO 8-week-old littermate pairs (n = 3 pairs) were used for all FACS isolations to reduce genetic variation and to control for variation in the isolation and sorting procedures. A negative control was used to draw gates for GFP+ and GFP− cell populations. A total of 10,000–20,000 GFP+ cells per mouse were sorted directly into TRIzol and stored at −80°C.
RNA isolation and RNA sequencing.
RNA was isolated from TRIzol samples using the Direct-zol kit following the manufacturer's instructions (Zymo, catalog #R2050). In-column DNase treatment was included. RNA isolated from sorted cells was analyzed by Agilent 6000 RNA Pico (catalog #5067–1513) before sequencing and only samples with an RIN >9.0 were selected for library preparation. RNA-sequencing and library preparation was performed by the University of Wisconsin Biotech Center. RNA input ranged from 2.5 to 4 ng of total RNA. Ovation Single Cell RNA-Seq System (Nugen, catalog #0342) was used to amplify and generate sequencing libraries. An Illumina HiSeq 2500 generated 100 base pair paired-end reads with an average sequencing depth of 36,865,141 reads per sample (minimum: 20,806,226; maximum: 48,891,436 reads). Accuracy of base calling was assessed using Phred scores. Mean scores for reads ranged from 36.62 to 38.05 and 93.6% of all bases had Phred scores >30.
Bioinformatics analysis.
RSEM was used to align read pairs to the mm9 transcriptome and estimate gene expression levels (Li and Dewey, 2011). The following command was used to execute alignment and quantification: rsem-calculate-expression--paired-end--bowtie2 -p 8 $sample_forward.fastq $sample_reverse.fastq $rsem_index $sample_output. We used a table of RSEM's “expected counts” as expression values in downstream analyses. EBSeq was used to test for differential expression in each set of paired WT and KO samples and genes were considered to be significant if they were changed in at least two pairs (FDR-adjusted p < 0.05). Overrepresentation of gene sets was statistically assessed within identified gene clusters against the transcriptome background in R using the modular single-set enrichment test (Eisinger et al., 2013). For principal component (PC) analysis, DEseq was used to compare the top 500 most variable genes by ANOVA. Gene ontology analysis was performed with the PANTHER functional classification (Thomas et al., 2003). KEGG pathway enrichment analysis was performed using the WebGESTALT (Web-Based Gene Set Analysis Toolkit) with Benjamini and Hochberg multiple test adjustment (Zhang et al., 2005). Proteins with known physical interactions with the products of DE genes were extracted from the BioGRID database (Stark et al., 2006), release 3.4.141, and characterized using DAVID 6.8 with the “high” stringency setting for clustering of related terms, as described previously (Gao et al., 2016).
Real-time PCR.
Quantitative PCR (qPCR) was performed using standard methods as described previously (Guo et al., 2011, 2015). The first-strand cDNA was generated by reverse transcription with random primers using Transcriptor First Strand cDNA Synthesis Kit (Roche, catalog #04896866001). cDNA was subjected to qPCR using a StepOne Real-Time PCR System (Applied Biosystems) and Universal SYBR Green PCR Supermix (Bio-Rad, catalog #172–5124). PCRs contained 20–40 ng of cDNA and 300 nm of forward and reverse primers in a final reaction volume of 20 μl. The ratio of different samples was calculated by the ΔΔCt method using β-actin as a reference gene (Clouaire et al., 2010; Voronova et al., 2011). The sequences of primers are available upon request.
Adult dgNPCs and in vitro analyses.
Adult dgNPCs were isolated from 8-week-old MBD1-KO mice and WT littermates as described previously (Guo et al., 2012b). Immunocytochemistry was performed as described previously (Liu et al., 2013; Guo et al., 2015). Three coverslips of cells were analyzed per cell isolation and three independently isolated cell cultures were used for analysis. The percentage of differentiated cells was calculated as the number of βIII-tubulin or GFAP-labeled cells divided by the total number of DAPI+ cells. For statistical analysis, cell counts were normalized to the average of all WT replicates. The size and shape of βIII-tubulin cells were analyzed by FIJI (ImageJ; Schindelin et al., 2012). Images were collected using the same exposure settings on a Zeiss AxioImagerZ2. βIII-tubulin cells were chosen using the region of interest (ROI) selection tool in FIJI using equivalent threshold settings for all images. ROIs that overlapped with the boundary of the image and those that contained multiple cells were discarded. Cell area and intensity per cell area/ROI was calculated in FIJI. At least 50 cells were counted per genotype and data were analyzed using two-tailed unpaired t tests.
Lentiviral and retroviral production.
Lentivirus production was performed as described previously (Barkho et al., 2008; Li et al., 2008). Briefly, lentiviral vectors or retroviral vectors expressing sh-Mbd1 or sh-NC (Li et al., 2008) were cotransfected with packaging plasmids (pMDL, REV, and pCMV-Vsvg for lentiviral production; pCMV-gp and pCMV-Vsvg for retroviral production) into HEK293Ta cells using calcium phosphate method. The medium containing virus was filtered through a 0.2 μm filter and concentrated using an ultracentrifuge at 19,000 rpm for 2 h at 4°C using a SW32Ti rotor (Beckman). The pellet was washed once and then resuspended in 80 μl of PBS. dgNPCs were plated into coated 6-well plates at 0.5 × 105 cells/cm2, given 4 h to recover, and then 1 × 106 viral particles were added to the medium. At 2 d after infection, cells were washed once and collected in TRIzol for RNA isolation.
In vivo retroviral injection and differentiation analysis.
In vivo virus grating was performed as described previously (Guo et al., 2015; Wang et al., 2015). Briefly, 7- to 8-week-old C57B/L6 male mice were anesthetized with isofluorane and virus (1 μl with titer >5 × 108/ml for retrovirus and 4 × 106/ml for rabies virus) was injected stereotaxically into the DG using the following coordinates relative to bregma, caudal: −2.0 mm; lateral: ±1.6 mm; ventral: −1.9 mm. Seven days after viral grafting, mice were perfused for neuronal differentiation analysis, deeply anesthetized with pentobarbital, and perfused with saline followed by 4% PFA. To analyze the phenotypes of GFP+ cells, we used 1 in 6 series of 40 μm brain sections starting at beginning of hippocampus (relative to bregma, −1.5 mm) to the end of hippocampus (relative to bregma, −3.5 mm). The immature neurons were detected by an antibody against DCX. The Type 2a/b and Type 3 DCX+ was distinguished by the orientation of their processes as described previously (Kempermann et al., 2015). The data were calculated as percentage of total GFP+ cells that are either GFP+DCX+ vertical (Type 3) cells or GFP+DCX+ parallel cells (Type 2ab).
Chromatin immune precipitation (ChIP).
ChIP was performed according to previously published (Wang et al., 2015). Adult dgNPCs grown to 80–90% confluency in 15 cm plates were fixed by adding 1% formaldehyde (Sigma-Aldrich, catalog #S33102) to the culture medium for 20 min at room temperature. The reaction was stopped by the addition of glycine buffer (190 mg of glycine in 1 ml of H2O). After washing with cold PBS, cells were collected with cold PBS, washed, suspended in 1 ml of cold cell lysis buffer (5 mm PIPES, pH 8.0, 85 mm KCl, 0.5% NP40, and 1× complete proteinase inhibitor), and incubated on ice for 5 min. Cell lysates were pelleted by centrifugation at 3000 rpm for 5 min, resuspended in 1 ml of cold cell lysis buffer 5 min on ice, and then repelleted to collect nuclei. Nuclei were lysed at room temperature with 500 μl of nuclei lysis buffer (50 mm Tris, pH 8.1, 10 mm EDTA, 1% SDS, and 1× complete protease inhibitor). Nuclear lysates were sonicated using a Bioruptor UCD-200 (Life Technologies) with five cycles; each cycle included five 30 s high-power pulses >10 min. Input samples were removed and stored until DNA extraction. Dynabeads Protein G (Thermo Fisher Scientific, catalog # 10003D) were washed and then incubated with 10 μg of mouse anti-FLAG (Sigma-Aldrich, catalog #F1804). The antibody/bead complex was then incubated with sonicated chromatin from MBD1-FLAG and WT cells overnight at 4°C. A magnetic stand was used to precipitate beads and was washed 4× with RIPA buffer (50 mm HEPES, 1 mm EDTA, 0.7% DOC, 1% IPGEL, 0.5 m LiCl), followed by a single TE wash. Protein–DNA complexes were eluted from the Protein G agarose beads with 200 μl of freshly prepared immunoprecipitation elution buffer (50 mm NaHCO3, 1% SDS) for 15 min at 65°C with periodic brief vortexing. Formaldehyde induced protein–DNA crosslinking was heat reversed by incubating the protein–DNA complex and input fractions at 65°C for 4 h. DNA was purified by phenol:chloroform:isoamyl alcohol (25:24:1; Life Technologies, catalog #15593–031) isolation and precipitated with two volumes of 100% ethanol and 10 μg of linear acrylamide at −20°C overnight. Immunoprecipitated and purified DNA fragments were resuspended in 50 μl of nuclease-free water; 1 μl of DNA was used in 20 μl of SYBR Green real-time PCRs. Primers sequences were spaced at ∼1 kb intervals spanning 5 kb upstream of the TTS. FLAG immunoprecipitations of WT and MBD1-FLAG cells were normalized to input samples. DNA from four independent chromatin preparations was used and all qPCRs were performed in duplicate for each sample on each amplicon.
Results
MBD1 is expressed in neural stem cells and neurons in the adult DG
To investigate the role of MBD1 in adult hippocampal neurogenesis, we examined MBD1 expression in the DG using cell-lineage-specific markers. Because MBD1 lacks a specific antibody suitable for histology, we used MBD1-KO mice in which LacZ replaces the first nine coding exons in the targeted allele (Zhao et al., 2003) so that β-gal expression can be used to visualize where MBD1 would be expressed (Fig. 1A). β-gal staining is present in MBD1-KO animals, but not in WT animals (Fig. 1B). To investigate the expression patterns of MBD1 during DG neurogenesis, we used heterozygous (het) MBD1-KO mice that also carried a Nes-GFP transgene (Fig. 1A; Yamaguchi et al., 2000). GFP expression in this Nes-GFP line is commonly used to identify stem/progenitor cells (Type 1 and Type 2a+b; Filippov et al., 2003; Gao et al., 2009; Knobloch et al., 2013) and we confirmed that GFP fluorescence is consistent with NESTIN staining (Fig. 1C). The β-gal staining in Nes-GFP mice shows that MBD1 colocalized with the 52.2% of GFP+ stem/progenitor cells, including GFAP+ Type 1 cells (Fig. 1D,E, arrows), but not with GFAP+ (Fig. 1D) or S100β+ (Fig. 1F,G) astrocytes. We also assessed the expression of SOX2, a marker that is expressed in both Type 1 and Type 2a cells and in some astrocytes throughout the brain (Suh et al., 2007). β-gal/MBD1 was colocalized with SOX2+/S100β− cells, but not SOX2+/S100β+ cells (Fig. 1G), further confirming the lack of expression of MBD1 in astrocytes. β-gal did not colocalize with the vast majority of TBR2+ cells (Fig. 1H,J), a marker for Type 2b cells (Hodge et al., 2008). It colocalized with only 17.9% of DCX+ cells, a marker of Type 2b and Type 3 cells (Fig. 1I). Therefore MBD1 expression is downregulated during early phase of differentiation. However, β-gal is present in 59.7% TBR1 (T-box brain 1; Fig. 1K,L)-positive cells, which marks maturing neurons in late neurogenesis (Hsieh, 2012), as well as 61.1% of NeuN-positive mature neurons (Fig. 1M,N), indicating that after initial differentiation, MBD1 levels increase as neurons become more mature. These results are summarized in Figure 1O, which presents the progression of adult neurogenesis along with MBD1expression and markers used to identify each cell type. GFP expression identifies Type 1 and Type 2a/b cells; Type 1 cells can be distinguished by morphology and GFAP expression while Type 2b cells can be distinguished by DCX expression. In summary, MBD1 is expressed in Type 1 and Type 2a but, but low levels in Type 2b or Type 3 cells, but likely not in astrocytes. This pattern of expression is similar to NRSF/REST, which, in addition to its role in transcriptional regulation in neurons, also restrains the neurogenic program in stem cells (Gao et al., 2011). Our finding that MBD1 was expressed in stem/progenitor cells but was undetectable in immature neurons or astrocytes suggested that MBD1 may be involved in repressing differentiated lineage-specific genes in stem cells.
Figure 1.
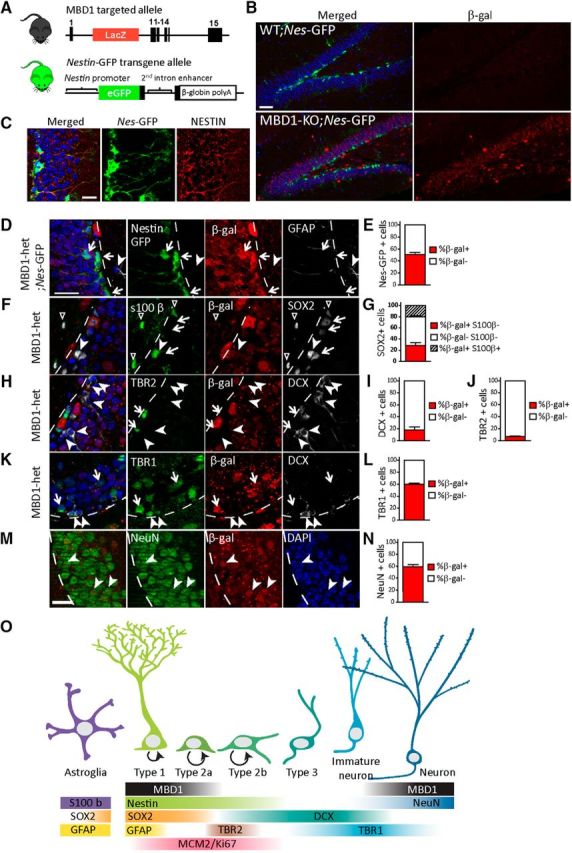
MBD1 is expressed in NSCs, immediate progenitors, and neurons, but is undetectable in immature neurons in the adult DG. A, Mouse lines used in the experiments. For the MBD1-KO allele, the lacZ gene was knocked in to replace the first nine coding exons; therefore, the expression of β-gal represents the expression patterns of endogenous MBD1. Enhanced GFP is driven by the endogenous Nestin promoter in transgenic Nes-GFP mice. B, β-gal (red) and Nes-GFP (green) expression in MBD1-KO and WT animals. Scale bar, 20 μm. C, NESTIN staining (red) and Nes-GFP expression (green). Scale bar, 20 μm. D, Expression of β-gal (red) in MBD1-het;Nes-GFP mice. MBD1 colocalizes with Nes-GFP+ stem cells (green, arrows), but not with GFAP+ astrocytes in the hilus (white, arrowhead). Scale bar, 25 μm. For all images, the SGZ boundary is marked by the dashed line, DAPI (blue). E, Proportion of Nes-GFP+ cells that are β-gal+. F, β-gal (red) colocalizes with SOX2+ (white) stem cells (arrows) and with some SOX2+ S100β astrocyte progenitors (arrowhead), but not with S100β (green) astrocytes in the (arrowhead outline). G, Proportion of SOX2+ cells that are β-gal+ S100β−, β-gal+ S100β− or β-gal+ S100β+. H, β-gal (red) does not colocalize with TBR2+ neural progenitors (green, arrows) or with DCX+ immature neurons (white, arrowheads). I, Proportion of DCX+ cells that are β-gal+. J, Proportion of TBR2+ cells that are β-gal+. K, β-gal (red) colocalizes with some TBR1+ maturing neurons (green, arrows), but does not colocalize with the majority (18%) of DCX+ immature neurons (white, arrowheads). L, Proportion of TBR1+ cells that are β-gal+. M, β-gal (red) colocalizes with many NeuN+ maturing neurons (green, arrowheads). N, Proportion of NeuN++ cells that are β-gal+. O, Depiction of MBD1 expression (black bar) during the stages of adult neurogenesis. MBD1 is expressed in NESTIN+SOX2+Type 1/Type 2a stem and progenitor cells and maturing TBR1+NEUN+ neurons, but undetectable in GFAP+S100β+SOX2+astrocytes or TBR2+DCX+ Type 2b/Type 3 neuronal progenitors and immature neurons.
MBD1 deletion leads to increased Type 2a/b cells but reduced differentiation into Type 3 cells
Since MBD1 is expressed in most Type 1 and Type 2a cells, it likely plays a regulatory role in early neurogenesis. Previous studies have shown that neural progenitor cells isolated from entire forebrain of adult MBD1-KO mice exhibit increased proliferation compared with control WT cells (Liu et al., 2010), but the impact of MBD1 deletion on NSCs in the adult DG in vivo has not been analyzed. To determine the proportion of GFP-expressing stem cells that are in a proliferative state, we used the marker MCM2 (Fig. 2A). MCM2 is used to indicate activated stem/progenitor cells because it is expressed in all active stages of the cell cycle as well as in cells in G0 that are “licensed” to proliferate (Blow and Hodgson, 2002; Maslov et al., 2004; Bonaguidi et al., 2011). Surprisingly, the proportion of MCM2–positive cells in the GFP+ population was not significantly different between WT and MBD1-KO for either Type 1 or Type 2a/b cells (Fig. 2B). However, there were more Type 2a/b cells, but not Type 1 cells, in MBD1-KO animals compared with WT mice (Fig. 2C). Therefore, MBD1 deficiency leads to increased number of Type 2a/b cells without a change in proliferation.
Figure 2.

MBD1-KO mice have more immature cells in the adult DG. A, Sample confocal images of the DG in WT and MBD1-KO animals, Scale bar, 10 μm. Type 1 cells (arrows) are positive for both Nes-GFP (green) and GFAP (white, in the radial process). Type 2(a/b) cells (arrowheads) are Nes-GFP positive and also frequently MCM2 (red) positive (asterisks). B, Proportion of MCM2+ Type 1 and Type 2(a/b) cells is not significantly different between genotypes. C, Number of Type 2(a/b) cells per cubic millimeter is significantly greater in MBD1-KO compared with WT mice, but the number of Type 1 cells is not significantly different. Data are presented as mean ± SEM (n = 7/genotype) 2-way ANOVA, post hoc Bonferroni t test, ****p < 0.001.
Given that long-term neuronal differentiation is reduced in MBD1-KO mice (Zhao et al., 2003), and that there was no change in proliferation of GFP+ cells, we reasoned that the increase in these cells in MBD1-KO animals may result from reduced neuronal differentiation. To test this hypothesis, we quantified the number of cells expressing only Nes-GFP (Type 1 and 2a), both Nes-GFP and DCX (Type 2b), or only DCX (Type 3 and immature neurons; Fig. 3A,B; Kempermann et al., 2015). In agreement with our previous findings, MBD1-KO brains had more Nes-GFP-positive cells (Fig. 3C) and many more cells expressed both GFP and DCX (Fig. 3D) compared with WT mice. However, there were fewer DCX+ cells in MBD1-KO compared with WT mice (Fig. 3E). Even though Mbd1 KO mice exhibit no obvious deficits during embryonic development (Zhao et al., 2003), MBD1 deficiency may nonetheless have a developmental impact on adult NPCs. We therefore used a retrovirus that only infected dividing cells (Guo et al., 2015) to deliver a small inhibitory RNA against MBD1 to NPCs in the adult DG (Fig. 3F). Viral infected NPCs either continue to divide or differentiate. By 1 week after viral targeting, ∼55% GFP+ cells have differentiated into DCX+ neurons. The stage of the differentiation can be determined by the shape and orientation of their processes (Fig. 3F,G). We found that acute knock-down (KD) of MBD1 in adult NPCs led to reduced DCX+ cells with vertical processes (Type 3 cells; Fig. 3F,K) without affecting DCX+ cells with parallel processes (Type 2a/b cells; Fig. 3F,I), consistent with the phenotype of Mbd1 KO mice. In summary, MBD1-KO mice have more GFP+ cells (GFP+ and GFP+DCX+) but fewer immature neurons (GFP-DCX+), supporting our hypothesis that Nes-GFP+ cells accumulate in Type 2a and Type 2b stages in MBD1-KO mice.
Figure 3.

MBD1-KO NSCs are impaired in transition to neuronal fate. A, Sample confocal images of Nes-GFP (green), DCX (red), and DAPI (blue) staining of brain sections from WT;Nes-GFP and MBD1-KO Nes-GFP mice. Scale bar, 20 μm B, Summary of quantification of Nes-GFP+ (Type 1 and Type 2a), Nes-GFP+DCX+ (Type 2b), and DCX+ (Type 3/immature neurons) in WT and KO mice. C–E, Quantification of individual cell types in the adult DG of WT and KO mice: C, GFP+DCX− cells, D, GFP+DCX+ and E, GFP−DCX+ (n = 7 per genotype), 2-way ANOVA, post hoc Bonferroni t test *p < 0.05, **p < 0.01. F, Schematic illustrations of retroviral expressing shMbd1 as well as GFP (Retro-shMbd1) injected into the adult DG. A timeline of the in vivo labeling of newborn neurons in the DG experiment and illustrations of vertical and parallel neurons is shown. G, Confocal images showing examples of retroviral-labeled (GFP+) and DCX+ (white) vertical and parallel neurons. Scale bar, 20 μm. H, I, Quantitative analysis showing that Retro-shMbd1-infection resulted in reduced differentiation into vertical neurons (H), but not parallel neurons (I). J, Sample confocal images of BrdU+ (red) cells in the SGZ identified with cell-type-specific markers: Nes-GFP+ (green) Type 2a cells (arrowhead, top), DCX and Nes-GFP double-positive Type 2b cells (asterisks, middle), DCX+ (white) Type 3 cells (arrows, bottom). Scale bar, 10 μm. K, Quantitative data showing the percentage of each cell type among total BrdU+ cells. After 24 h, MBD1-KO mice had significantly more BrdU+ cells that were Nes-GFP+ and significantly fewer were DCX+ compared with WT mice. L, Hypothetical model based on these results showing reduced transition of BrdU-labeled cells to Type 3 (DCX+) in MBD1-KO mice at 24 h after BrdU labeling. Data are presented as mean ± SEM, WT (n = 7), KO (n = 6), 2-way ANOVA, post hoc Bonferroni's t test, *p < 0.05, **p < 0.01.
To further test the hypothesis that Type 1 and Type 2a/b cells had deficits transitioning into Type 3 (DCX+) cells MBD1-KO mice, we used a single injection of BrdU to label the dividing stem/progenitors (Type 1, Type 2a, and Type 2b). At 24 h after injection, when a portion of the BrdU+ cells should have differentiated into DCX+ immature neurons, we analyzed the composition of BrdU-labeled cells (Fig. 3J). We found that, in MBD1-KO animals, significantly more BrdU+ cells were GFP+ compared with those in WT mice (2-way ANOVA, p < 0.05; Fig. 3K, green bar), which is consistent with the long-term NSC accumulation phenotype of MBD1-KO animals (Fig. 2). We then analyzed DCX+ cells. Although BrdU injection can label DCX+ transit-amplifying progenitors (Type 2b) cells, <20% of DCX+ cells are mitotically active (Jessberger et al., 2005; Kempermann et al., 2015); therefore, most BrdU+DCX+ cells that we analyzed at 24 h after BrdU injection were differentiated from Type 2a/b progenitors. In WT, 13% of BrdU+ cells colocalized solely with DCX (BrdU+GFP−DCX+; Fig. 3K, gray bar), whereas none of these cells was found in MBD1-KO animals, suggesting that almost no BrdU-labeled cells had fully transitioned to Type 3 cells in MBD1-KO animals. These data support the idea that Type 2a/b cells accumulate in MBD1-KO animals because they fail to differentiate to immature neurons efficiently (Fig. 3L). However, because MBD1 was undetectable in Type 2b or Type 3 cells (Fig. 1), we reasoned that this outcome must be a downstream effect of the loss of MBD1 in Type 1 and Type 2a cells.
RNA sequencing reveals upregulation of astrocyte transcripts in MBD1-KO aNSCs
To investigate the molecular mechanism underlying MBD1-regulated fate commitment, we isolated Nes-GFP cells from the DG of 8-week-old WT and KO littermates using FACS (Fig. 4A,B). Using qPCR, we confirmed that sorted GFP+ cells were highly enriched with Nestin and eGFP, but not with NeuroD1 or NeuN, relative to total (input) cells (Fig. 4C). MBD1-KO animals had a higher percentage of GFP+ cells relative to the total cell gate compared with WT animals (Fig. 4D), which is consistent with the results determined by stereological estimates (Fig. 3C). RNA isolated from 10,000–20,000 GFP+ cells per animal was subjected to RNA sequencing. To confirm the identity of our sorted cell population, we performed a PC analysis on our data, along with Nes-GFP and Dcx-DsRed single-cell RNA-sequencing datasets (Shin et al., 2015; Gao et al., 2016; Fig. 4E). This analysis showed that our Nes-GFP population is similar to Nes-GFP single cells and relatively dissimilar to Dcx-DsRed single cells.
Figure 4.
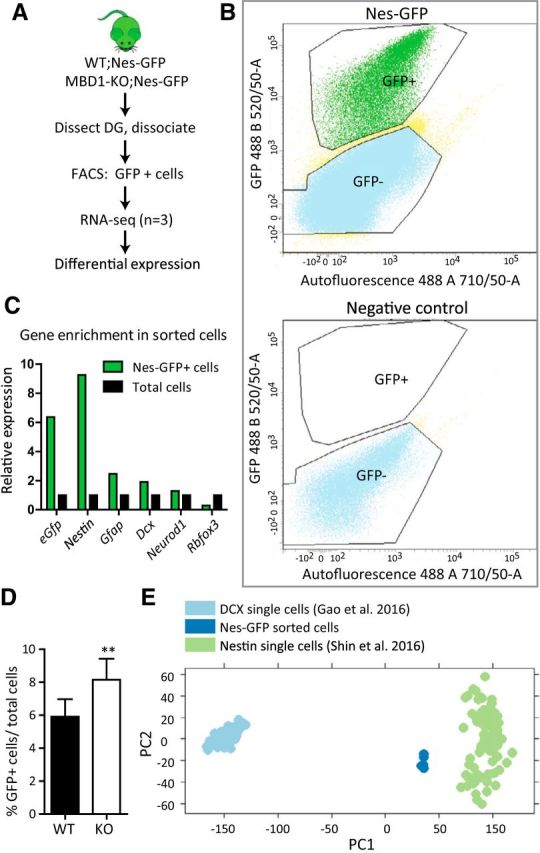
The DG of adult MBD1-KO mice yielded more Nes-GFP+ cells compared with WT mice. A, Experimental workflow of FACS-seq showing dissection of adult DG, direct cell isolation using FACS, RNA-seq, and bioinformatics analysis. B, Example of sorting gates used to separate for GFP+ and GFP− single cells dissociated from DG tissue. For each sorting, gates were drawn based on the profile of a WT mouse that did not express GFP (right). C, Relative enrichment of each gene was determined by qPCR, with equal numbers of sorted cells used to generate cDNA. Cells from the GFP+ gate (green) relative to the total cell gate (black) shows a pronounced enrichment of stem cell markers (eGFP, Nestin, and Gfap), mild enrichment of early neuronal markers (Dcx and NeuroD1), and depletion of mature neuronal marker (NeuN) in the GFP+ cell population (n = 1 with triplicates of qPCR). D, Proportion of GFP+ cells among total (input) cells is significantly greater in MBD1-KO compared with WT samples as assessed by cell counts. Cells were isolated from littermate pairs (n = 10 pairs); data are presented as mean ± SEM, paired t test, p = 0.003. E, Principal component analysis comparing Nes-GFP sorted cells (dark blue) with single Nes-GFP cell (green; Shin et al., 2015) or single Dcx-DsRed cell (light blue; Gao et al., 2016).
To minimize batch effects resulting from different litters and gating procedures between the three FACS isolations, differential expression was analyzed pairwise by comparing three sets of WT–KO littermates. The intersection of these three datasets was taken and any genes that were not differentially expressed (DE) in the same direction (FDR-adjusted p < 0.05) in at least two pairs were discarded. This yielded 124 genes that are upregulated and 146 that are downregulated in MBD1-KO cells. MeCP2 KO models have revealed that changes in gene expression levels related to DNA-binding proteins are often subtle and spread across a large number of genes (Lyst and Bird, 2015); therefore, it is expected that a relatively small number of genes achieved statistical significance in these cells.
We used PANTHER [35] to classify DE genes by biological processes (Fig. 5A) and WebGestalt (Zhang et al., 2005) to identify pathways affected by the loss of MBD1 (Table 1). Upregulated genes were significantly enriched for metabolic pathways (adjusted p = 4.86E-09). Genes implicated in Alzheimer's and Huntington's diseases, two neurodegenerative disorders with altered metabolic function (Cai et al., 2012), were also enriched within upregulated MBD1-KO genes (adjusted p = 1.07E-02, adjusted p = 1.09E-02, respectively). Downregulated genes were enriched for MAPK signaling (adjusted p = 1.8E-03), focal adhesion (adjusted p = 6.4E-03), and cancer pathways (adjusted p = 6.3E-03). Because Type 2a/b cells accumulated in MBD1-KO mice and displayed deficits in lineage commitment, we wondered whether the DE genes corresponded to cell-lineage-specific gene expression data for cortical neurons, astrocytes, microglia, endothelial, and oligodendrocytes (Zhang et al., 2014). We compared the DE genes in MBD1-KO with the top 500 lineage-enriched genes for each cell type in the Zhang et al. (2014) dataset using an overrepresentation analysis (Eisinger et al., 2013). We found that astrocyte-associated genes were significantly overrepresented in the set of upregulated genes (p = 0.0001; Fig. 5B, Table 2). Conversely, neuronal and myelinating oligodendrocytes genes were enriched among the set of downregulated genes (p = 0.016, p = 0.0002, respectively; Fig. 5B). This suggests that MBD1 may maintain the neurogenic potency of NSCs by suppressing astrocyte lineage commitment genes and permitting the expression of genes related to neuronal fate. To explore potential downstream effects of these gene changes, we curated sets of proteins known to interact with the products of upregulated and downregulated genes, reasoning that their activity would likely be influenced by altered levels of their interacting partners. Proteins that interact with both upregulated and downregulated gene products were strongly associated with transcriptional regulation, whereas proteins that interact with downregulated gene products were specifically related to the structural maintenance of chromosomes and cell division, suggesting that the presence of MBD1 supports protein networks that preserve the proliferative capacity of aNSCs through an epigenetic mechanism (Fig. 5C). To validate our sequencing results, we acutely knocked down MBD1 in primary dgNPCs with lentiviral shRNA and confirmed expression changes of Mbd1 (Fig. 5D) and four upregulated genes (Fig. 5E). We confirmed that two of the genes, Grin 2C and Cd38, were also upregulated in KD cells compared with WT cells. Therefore, MBD1-deficient Type 1/2 cells exhibit altered expression of genes related to diverse cellular functions and protein pathways that are particularly associated with cell identity and lineage specification.
Figure 5.
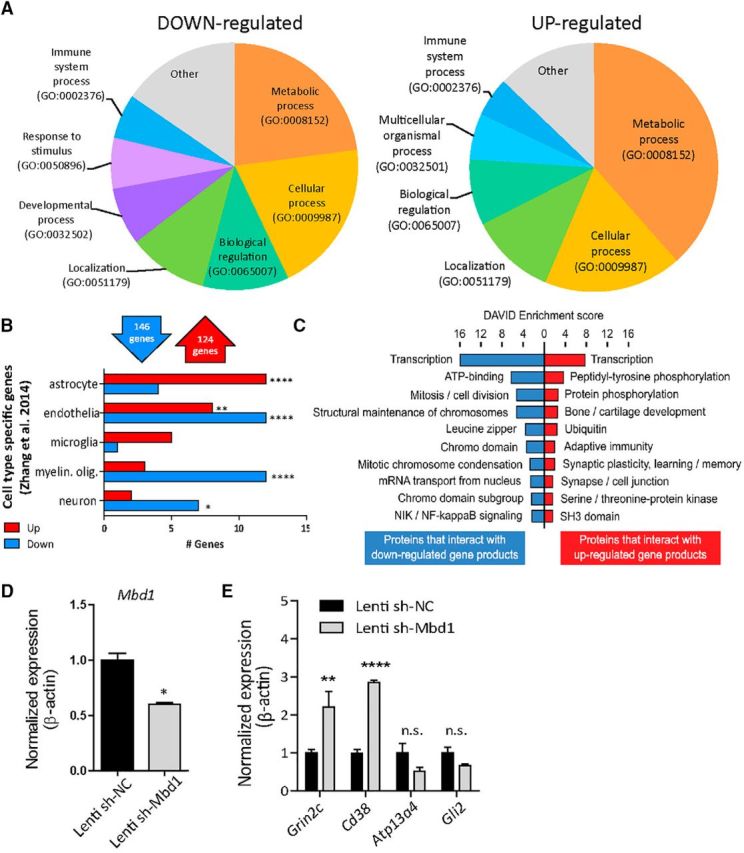
Nes-GFP+ cells isolated from MBD1-KO mice have elevated astrocyte lineage genes. A, Overrepresentation analysis of upregulated and downregulated genes in MBD1-KO cells compared with cell-type-specific genes (Zhang et al., 2014). B, Differential expression analysis of RNA-seq data using pairwise comparison identified 124 upregulated genes and 151 downregulated genes in MBD1-KO-sorted cells. PANTHER was used to categorize up and down differentially expressed genes in MBD1-KO GFP+ NSCs using a gene ontology (GO) biological process. C, DAVID enrichment analysis of proteins known to interact physically with protein products of upregulated and downregulated genes. D, Mbd1 mRNA expression was reduced in lenti-shMbd1-infected WT dgNPCs compared with lenti-shNC-infected WT dgNPCs, when analyzed at 48 h after initial viral infection. E, Expression levels of selected transcripts identified by RNA-seq were assessed in dgNPCs with acute KD of Mbd1 (lentivirus expressing shMbd1) using qPCR. The levels of Grin2C and Cd38 are significantly upregulated in MBD1 acute KD compared with control (lentivirus expressing control sh-NC), n = 3 per condition, t test, Bonferroni correction for multiple comparisons, *p < 0.05, **p < 0.01.
Table 1.
KEGG pathway analysis of up and down-regulated genes in MBD1-KO cells
| Adjusted p-value | Genes | |
|---|---|---|
| Upregulated genes | ||
| Metabolic pathways | 4.86E-09 | Tk1, Gmppa, Naglu, Mvd, Dct, Mri1, Hexb, Hsd11b1, Polr1e, Pi4k2b, Alg6, Cbr3, Ndufs4, Cd38, Khk, Scly, Polr2d |
| Pyrimidine metabolism | 0.0051 | Tk1, Polr2d, Polr1e, Ctss |
| Lysosome | 0.0059 | Hexb, Naglu |
| Alzheimer's disease | 0.0107 | Grin2c, Ndufb11, Ndufs4 |
| Huntington's disease | 0.0109 | Ndufb11, Ndufs4, Polr2d |
| Downregulated genes | ||
| MAPK signaling pathway | 0.0018 | Fos, Map3k8, Mapk11, Cacnb1, Rasrgp1, Nfkb2 |
| Focal adhesion | 0.0064 | Col1a2, Col6a3, Pdgfc, Col4a5 |
| Pathways in cancer | 0.0063 | Ctnna3, Fos, Col4a5, Nfkb2, Pias |
Table 2.
Enrichment of cell-specific genes (Zhang et al., 2014) among genes differentially expressed in MBD1-KO cells
| p-value | Genes | |
|---|---|---|
| Upregulated genes | ||
| Astrocyte | 1.00E-04 | AA387883, Atp13a4, Cd38, Chrdl1, Gjb6, Gli2, Gm973, Gpr179, Grin2c, Hsd11b1, Rhcg, Slc39a12 |
| Endothelial | 0.005 | Ankrd37, Arhgef5, Def6, Gbp4, Ifitm3, Ly6a, Pcp4l1, Slc35f2 |
| Microglia | 0.128 | Ctss, Gal3st4, Hexb, Hfe, Mpeg1 |
| Myelinating oligodendrocytes | 0.495 | Carns1, Fam83d, Ppp1r14a |
| Neuron | 0.730 | Fam65b, Vsnl1 |
| Downregulated genes | ||
| Astrocyte | 0.391 | Col4a5, Fos, Pasgrp1, Tsku |
| Endothelial | 0.000 | Abcc9, Art3, Atp13a5, Cysltr2, Efna1, Gbp9, Gjc1, Ly75, Nid1, Prkd2, Rhoj, Sox7 |
| Microglia | 0.959 | Hk2 |
| Myelinating oligodendrocytes | 2.00E-04 | Apa, Ctnna3, Fam57a, Galnt6, Hcn2, Nkx6-2, Scd3, Smtnl2, Spock3, Synj2, Tmem159, |
| Neuron | 0.016 | 1500009L16Rik, Ccbe1, Col24a1, E130003G02Rik, Pde1a, Sdk2, Slc12a5 |
MBD1-deficient NSCs exhibit increased expression of astrocyte markers
Our sequencing results revealed the misregulation of astrocytic genes in MBD1-KO Nes-GFP+ cells. Therefore, we investigated whether astroglial markers were increased in Nes-GFP+ cells in vivo. In MBD1-KO animals, we found that more Nes-GFP cells were positive for S100β, a marker for astrocytes (Fig. 6A,B). To determine whether MBD1 may be involved directly in the regulation of these genes without the confounding variable of developmental effects of cells isolated from MBD1-KO animals, we knocked down Mbd1 by infecting proliferating dgNPCs with lentivirus expressing sh-Mbd1 (Liu et al., 2010). We found that the expression levels of several astrocyte-lineage genes, Aqp4 and s100β, were elevated, whereas the progenitor cell gene Nestin was unchanged (Fig. 6C). In addition, Tubb3, the gene that codes for βIII-tubulin, and NeuroD1 (ND1) genes expressed during neuronal differentiation were also increased in MBD1-KD cells. Our RNA-seq data showed that Tubb3 was upregulated 1.4- to 1.8-fold in all three pairs of sorted cells, although it was not identified as a statistically significant. Using ChIP, we found that MBD1 binds to the promoters of s100β, Aqp4, and NeuroD1 in proliferating dgNPCs (Fig. 6D–F). In summary, we found that MBD1-deficient NSCs acquired increased expression of genes associated with cellular differentiation.
Figure 6.
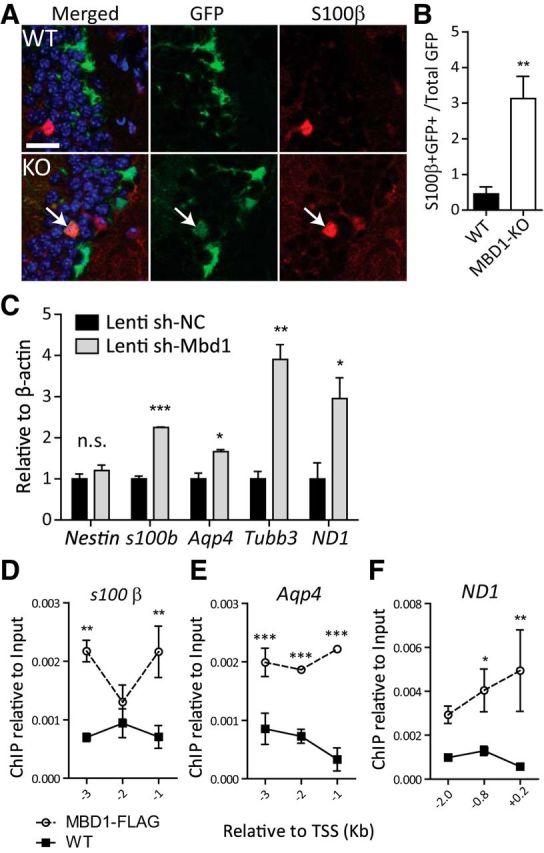
MBD1 represses the expression of lineage differentiation genes in neural stem and progenitor cells derived from adult DG (dgNPCs). A, Sample confocal images of Nes-GFP (green) and S100β (red) staining in WT and MBD1-KO animals. Arrow indicates colocalization. Scale bar, 20 μm. B, Quantification of S100β+ cells among Nes-GFP+ cells, n = 5 per genotype, 2-tailed unpaired t test, p = 0.0034. C, Quantitative analysis of stem cell- and lineage-specific genes using qPCR in proliferating NSCs with acute KD of MBD1 (sh-Mbd1) compared with control (sh-NC), n = 3, 2-tailed unpaired t test, *p < 0.05, **p < 0.01. D–F, ChIP with anti-FLAG antibody in proliferating WT and FLAG-tagged MBD1 dgNPCs followed by qPCR for the genomic sequence of the s100β promoter (D), the Aqp4 promoter (E), and the NeuroD1 promoter (F). Data are presented as mean ± SEM, calculated relative to input sample. The x-axis depicts location of primers relative to the transcriptional start site (TTS) in kilobases (Kb), n = 4, two-way ANOVA, Bonferroni post hoc t test, **p < 0.01.
aNSC-intrinsic MBD1 deficiency causes aberrant neuronal lineage commitment in vitro
It has been shown that aberrant expression of differentiation genes in stem cells interferes with proper cell differentiation (Amador-Arjona et al., 2015; Knock et al., 2015). Because both astrocyte and early neuronal genes are misregulated in MBD1-KO dgNPCs, we sought to determine whether the neuronal differentiation deficit observed in MBD1-KO mice was caused by compromised neurogenic potency of aNSCs. We therefore assessed the differentiation potential of dgNPCs isolated from the DG of adult MBD1-KO mice and WT control littermates. At 4 d after growth factor withdrawal, WT cells differentiated into βIII-tubulin-positive (Tuj1, green) neurons with defined, narrow processes and a small nucleus (asterisks) or into GFAP+ astrocytes with short, broad processes and a large nucleus (arrow) (Fig. 7A). However, in MBD1-KO cultures, many cells had astrocyte-like broad processes, but were positive, albeit diffusely, for βIII-tubulin (Fig. 7A, bottom, arrowhead). We used ImageJ to analyze βIII-tubulin+ cells (Fig. 7B). Supporting our previous observations, we found that MBD1-KO βIII-tubulin+ cells have a larger cell area (Fig. 7C), but that the mean intensity of βIII-tubulin labeling per cell is lower (Fig. 7D). In addition, there were more total βIII-tubulin positive cells in MBD1-KO cultures (Fig. 7E), which supports the observed increase in Tubb3 expression in sh-Mbd1 (Fig. 6C). However, when we counted cells using morphology (bright βIII-tubulin labeling, narrow processes) as additional criteria, we found that MBD1-KO cultures had five times as many βIII-tubulin+ cells with a non-neuronal morphology (low level and diffused βIII-tubulin labeling, broad processes) compared with WT cultures (Fig. 7F), but one-third as many “true” neurons (Fig. 7G). Although there was no difference in the number of GFAP+ cells (Fig. 7H), there was a trend (variable) toward an increased proportion of GFAP+ βIII-tubulin+ double-positive cells (Fig. 7I). Therefore, these results support the idea that the conflicting transcriptional programs in MBD1-deficient stem cells disrupt their ability to differentiate into true neurons despite elevated levels of certain neuron-specific genes in MBD1-deficient dgNPCs (Fig. 7J). Our data demonstrate the importance of MBD1 in setting the “transcriptional stage” for proper neuronal differentiation in NSCs.
Figure 7.

MBD1 deficiency leads to aberrant differentiation of dgNPCs. A, Sample images of in vitro differentiated WT and MBD1-KO dgNPCs. Scale bar, 50 μm. In WT, βIII-tubulin/Tuj1 (green) marked neurons with small nuclei and narrow and well defined processes (asterisks) and GFAP (red) marks astrocytes with broad processes and a large nucleus (arrowhead). In MBD1-KO, βIII-tubulin staining was abnormal, characterized by cells with wide βIII-tubulin+ processes (arrows) and coexpression of GFAP (red; arrowhead). B, Analysis of βIII-tubulin+ cell shape using FIJI. Cell contours were selected with the ROI tool using standardized thresholds (right). Note: same image shown as in A. C, Box plot of average cell area in WT (n = 53) and MBD1-KO (n = 55) βIII-tubulin+ cells. D, Box plot of mean intensity per cell, t test, ****p < 0.0001. E–I, Cell quantification. Cell number was determined relative to DAPI+ cells/coverslip. For each cell type, results were normalized to the average of the WT samples, n = 3 pairs, paired t test. E, Total βIII-tubulin+ cells, p = 0.035. F, βIII-tubulin+ cells with neuronal morphology, p = 0.032. G, βIII-tubulin+ cells with non-neuronal morphology, p = 0.013. H, GFAP+ cells, p = 0.58 (n.s.). I, βIII-tubulin+ GFAP+ cells, p = 0.091 (n.s.). J, Proposed model depicting a function of MBD1 in adult neural stem cells and neurogenesis. Loss of MBD1-mediated repression increases expression of astrocyte genes (purple), which impedes the progression of neurogenesis, restricting the “passage (funnel) of differentiation” and causing cells to accumulate at earlier stages despite the upregulation of some early neuronal genes (blue).
Discussion
In this study, we demonstrate that MBD1 is critical for fine-tuning the multipotency of adult NSCs by maintaining appropriate transcriptional state. Our results suggest that the altered gene expression profile in stem cells interferes with normal cell lineage commitment, preventing NSCs from differentiating into neurons and causing the accumulation of undifferentiated cells. Our data reveal a novel role for MBD1 in transcriptional control of stem cell potency and provide new insight into how epigenetic regulation contributes to adult neurogenesis.
Epigenetic mechanisms are closely associated with maintaining the multipotency of embryonic and adult neural stem cells (Namihira et al., 2008). During embryogenesis, DNA methylation helps to define both the developmental potential of progenitor cells as well as the lineage restriction of committed cells (Takizawa et al., 2001; Mohn et al., 2008; Cortese et al., 2011). In embryonic neural stem cells, MeCP2 promotes neuronal fates and restricts astrocytic fates (Tsujimura et al., 2009). MBD3, part of the NuRD complex, is important for cortical NSC fate specification and for maintaining the transcriptional program of stem cells (Knock et al., 2015). Studies in neocortical development have shown that DNA methylation contributes to maintaining astrocyte genes in a poised state in NSCs by “suppressing their expression while maintaining their capacity for activation later during development” (Hirabayashi and Gotoh, 2010). It is likely that similar mechanisms are responsible for maintaining transcriptional programs in adult neural stem cells. Indeed, Sox2 was recently found to be required for the induction of neuronal genes marked by bivalent chromatin in adult stem/progenitor cells (Amador-Arjona et al., 2015). Our results show that transcriptional regulation in stem/progenitor cells by MBD1 is important for setting the stage for subsequent differentiation.
Regulation of DNA methylation is important during multiple stages of adult neurogenesis. In addition to its known role in neuronal maturation, MeCP2 is also involved at earlier stages of neurogenesis because the balance between neural stem/progenitor cell proliferation and neuronal differentiation in the DG is regulated by MeCP2 phosphorylation (Li et al., 2014). Deletion of DNMT3A, a de novo DNA methyltransferase, does not alter cell proliferation, but does decrease the number of DCX+ immature neuroblasts (Wu et al., 2010). DNMT3A promotes the transcription of a number of neurogenic genes by antagonizing PRC2-mediated repression as cells differentiate. The DNMT3A-KO phenotype is intriguingly similar to that of MBD1-KO mice. Given that MBD1 has been shown to interact with hPc2 and RING1B, components of the polycomb repressive complex 1 (PRC1; Sakamoto et al., 2007), it is possible that MBD1 represses transcription in stem cells via interactions with PRC complexes. Multiple studies have found that MBD1 interacts with chromatin modifiers or other repressive complexes to mediate gene repression (Sarraf and Stancheva, 2004; Ichimura et al., 2005). If this is the case, then MBD1 could be involved in establishing a permissive transcriptional state in stem cells for genes required later in differentiation. Future studies might reveal whether MBD1 binds to components of the PRC1 or PRC2 complexes in aNSCs and if it is associated with bivalent chromatin.
In cancer cells, normal transcriptional control is often lost or exploited through abnormal methylation and the misregulation of methyl DNA-binding proteins (Parry and Clarke, 2011). Alterations in MBD1 have been identified in multiple cancers, suggesting that MBD1 is important in maintaining transcriptional regulation in many cell types (Li et al., 2015). DNA methylation is strongly associated with gene repression in many different cells (Miranda and Jones, 2007) and DNA methyl-binding proteins such as MeCP2 and MBD1 have long been considered transcriptional repressors (Boyes and Bird, 1991; Fujita et al., 1999; Ng et al., 2000; Jørgensen et al., 2004). Given a role for MBD1 in transcriptional control in general and our findings of the role of MBD1 in neuronal lineage commitment, we propose that MBD1 maintains stem cell neurogenic potency by maintaining transcriptional control of lineage-specific genes. In this model (Fig. 7I), loss of MBD1-mediated repression increases expression of astrocyte genes (purple) which impedes the progression of neurogenesis, restricting the “funnel” and causing cells to accumulate at earlier stages despite the upregulation of some early neuronal genes (blue).
As described above, we found that astrocyte genes and Neurod1 and Tubb3 were upregulated in MBD1 KO stem cells. Our expression studies determined that MBD1 is not expressed in astrocytes or Type 2b/Type 3 neuronal progenitors and, if MBD1 acts as a repressor, it is logically consistent that genes expressed in astrocytes or neural progenitor cells would be upregulated in its absence. However, the majority of genes were downregulated upon loss of MBD1, including an enriched set of neuronal lineage genes (p = 0.016). These may represent secondary perturbations in transcriptional networks caused by unrestrained elevation of direct MBD1 targets or they may be direct targets of MBD1 acting through a novel mechanism either alone or in concert with other transcriptional regulators to promote the expression of genes involved in neuronal maturation. Further exploration of genome-wide MBD1-binding patterns would yield valuable insight into this possibility. Some neuronal genes downregulated in MBD1-KO stem cells have no known function in the nervous system, whereas SDK2 and SLC12A5 are involved in neuronal maturation (Puskarjov et al., 2014; Krishnaswamy et al., 2015). Therefore, these genes may not be immediately involved in fate specification of neural stem/progenitor cells, but rather in maintaining neurogenic potential of these cells for subsequent differentiation. It is also possible that misregulation of other pathways such as MAPK (Collins et al., 2015), which was identified as significantly enriched among the downregulated genes (p = 0.0018), contributes to reduced neuronal differentiation.
Our data suggest that, in the absence of MBD1, loss of transcriptional regulation in stem cells leads to later deficits in neuronal differentiation. Other mouse models also exhibit no change in cell proliferation but decreased neuronal differentiation. These models involve genes that encode well known neuronal regulators such as NeuroD1 (Gao et al., 2009), BDNF (Waterhouse et al., 2012), REST/NRSF (Gao et al., 2011), and retinoic acid (Jacobs et al., 2006). This indicates that induction of neuronal differentiation is separate from stem cell proliferation, although both are critical to the ongoing supply of new neurons. Interestingly, the upregulation of NeuroD1 in the present study corresponds with βIII-tubulin expression in vitro, but in a context of aberrant neuronal differentiation. In the sorted cells, changes in the expression of early neuronal genes might have been too small to detect because the proportion of Nes−GFP+DCX+ cells in relation to the total number Nes−GFP+ cells is fairly small, ∼30%. Our data suggests that, despite the upregulation of Neurod1 and Tubb3 in the absence of MBD1, aberrant regulation of other genetic programs in stem/progenitor cells interferes with neuronal lineage commitment. One future avenue of research could be to investigate the influence of MBD1 on other master neuronal regulators such as REST/NRSF, which has a similar in vivo expression pattern to MBD1 (Gao et al., 2011).
When we analyzed the upregulated and downregulated DE genes by KEGG pathway enrichment, metabolic pathways were significantly enriched within upregulated genes (Table 1). Proper regulation of metabolism is increasingly recognized as a critical aspect of neural development (Ito and Suda, 2014). Perturbation of pathways related to metabolism affect adult neurogenesis (Renault et al., 2009; Buchovecky et al., 2013; Knobloch et al., 2013, 2014; Webb et al., 2013) and metabolism-related genes have been shown to be expressed differentially in different stages of neurogenesis (Shin et al., 2015). In addition, many genes that are specific to astrocytes (33%) are involved in metabolism (Lovatt et al., 2007). Growing evidence has shown that the maintenance of stem cells requires different metabolic conditions than do differentiated cells. Our findings support the idea that metabolism and fate specification are closely linked in neural stem cells and that proper transcriptional regulation by MBD1 is required for both aspects. Whether metabolic dysregulation facilitates MBD1-KO phenotypes or is a reflection of the increased astrocytic character of MBD1-KO stem cells remains to be determined.
In conclusion, our present data have established a new mechanism for MBD1 in regulating neurodevelopment, which is maintaining stem cell identity by restricting the expression of lineage-specific genes. The integrity of aNSCs is critical for the early stages of neuronal fate commitment in adult neurogenesis. In the absence of MBD1, upregulation of astrocyte genes is accompanied by an accumulation of undifferentiated neural progenitors and a decreased transition to immature neurons. Our data indicate that regulation of the stem cell transcriptional state is vital for fate specification and subsequent proper neuronal differentiation of neural stem cells.
Footnotes
This work was supported by the National Institutes of Health (Grants RO1MH080434, RO1MH07897, and R21NS095632 to X. Z.; Grants P30HD03352 and U54 HD090256 to the Waisman Center; Molecular Biosciences Training Grant MBTG: T32GM07215 to E.M.J; and Grant F32NS094120 to B.E.E.) and by a University of Wisconsin–Hilldale Undergraduate Research Fellowship to L.E.K. We thank Maggie Caulkins and Yina Xing for technical assistance; Natalie Patzlaff and Matthew Doers for editing; Karen Ersland, Dagna Sheerar, and Faye Bruggink at the University of Wisconsin Carbone Cancer Center Flow Cytometry Laboratory for cell-sorting assistance; Sandra Splinter BonDurant at the University of Wisconsin–Madison Biotechnology Center for library preparation and sequencing advisement; Karla Knobel at the Waisman Cell and Molecular Neuroscience Core; and Jason Pinnow, Dawna Bollig, and Megan Eastwood at the Waisman Rodent Models Core.
The authors declare no competing financial interests.
References
- Allan AM, Liang X, Luo Y, Pak C, Li X, Szulwach KE, Chen D, Jin P, Zhao X. The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits. Hum Mol Genet. 2008;17:2047–2057. doi: 10.1093/hmg/ddn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amador-Arjona A, Cimadamore F, Huang CT, Wright R, Lewis S, Gage FH, Terskikh AV. SOX2 primes the epigenetic landscape in neural precursors enabling proper gene activation during hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2015;112:E1936–E1945. doi: 10.1073/pnas.1421480112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avgustinova A, Benitah SA. Epigenetic control of adult stem cell function. Nat Rev Mol Biol. 2016;17:643–658. doi: 10.1038/nrm.2016.76. [DOI] [PubMed] [Google Scholar]
- Barkho BZ, Munoz AE, Li X, Li L, Cunningham LA, Zhao X. Endogenous matrix metalloproteinase (MMP)-3 and MMP-9 promote the differentiation and migration of adult neural progenitor cells in response to chemokines. Stem Cells. 2008;26:3139–3149. doi: 10.1634/stemcells.2008-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckervordersandforth R, Tripathi P, Ninkovic J, Bayam E, Lepier A, Stempfhuber B, Kirchhoff F, Hirrlinger J, Haslinger A, Lie DC, Beckers J, Yoder B, Irmler M, Götz M. In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell. 2010;7:744–758. doi: 10.1016/j.stem.2010.11.017. [DOI] [PubMed] [Google Scholar]
- Blow JJ, Hodgson B. Replication licensing-defining the proliferative state? Trends Cell Biol. 2002;12:72–78. doi: 10.1016/S0962-8924(01)02203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, Wheeler MA, Shapiro JS, Stadel RP, Sun GJ, Ming GL, Song H. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell. 2011;145:1142–1155. doi: 10.1016/j.cell.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyes J, Bird A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell. 1991;64:1123–1134. doi: 10.1016/0092-8674(91)90267-3. [DOI] [PubMed] [Google Scholar]
- Buchovecky CM, Turley SD, Brown HM, Kyle SM, McDonald JG, Liu B, Pieper AA, Huang W, Katz DM, Russell DW, Shendure J, Justice MJ. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat Genet. 2013;45:1013–1020. doi: 10.1038/ng.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Cong WN, Ji S, Rothman S, Maudsley S, Martin B. Metabolic dysfunction in Alzheimer's disease and related neurodegenerative disorders. Curr Alzheimer Res. 2012;9:5–17. doi: 10.2174/156720512799015064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro J, Mellios N, Sur M. Mechanisms and therapeutic challenges in autism spectrum disorders: insights from Rett syndrome. Curr Opin Neurol. 2013;26:154–159. doi: 10.1097/WCO.0b013e32835f19a7. [DOI] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Christian KM, Song H, Ming GL. Functions and dysfunctions of adult hippocampal neurogenesis. Annu Rev Neurosci. 2014;37:243–262. doi: 10.1146/annurev-neuro-071013-014134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouaire T, de Las Heras JI, Merusi C, Stancheva I. Recruitment of MBD1 to target genes requires sequence-specific interaction of the MBD domain with methylated DNA. Nucleic Acids Res. 2010;38:4620–4634. doi: 10.1093/nar/gkq228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins LM, Downer EJ, Toulouse A, Nolan YM. Mitogen-activated protein kinase phosphatase (MKP)-1 in nervous system development and disease. Mol Neurobiol. 2015;51:1158–1167. doi: 10.1007/s12035-014-8786-6. [DOI] [PubMed] [Google Scholar]
- Cortese R, Lewin J, Bäckdahl L, Krispin M, Wasserkort R, Eckhardt F, Beck S. Genome-wide screen for differential DNA methylation associated with neural cell differentiation in mouse. PLoS One. 2011;6:e26002. doi: 10.1371/journal.pone.0026002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukier HN, Rabionet R, Konidari I, Rayner-Evans MY, Baltos ML, Wright HH, Abramson RK, Martin ER, Cuccaro ML, Pericak-Vance MA, Gilbert JR. Novel variants identified in methyl-CpG-binding domain genes in autistic individuals. Neurogenetics. 2010;11:291–303. doi: 10.1007/s10048-009-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Liu B, Guo F, Xu G, Ding Y, Liu Y, Sun X, Xu G. The essential role of Mbd5 in the regulation of somatic growth and glucose homeostasis in mice. PLoS One. 2012;7:e47358. doi: 10.1371/journal.pone.0047358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisinger BE, Saul MC, Driessen TM, Gammie SC. Development of a versatile enrichment analysis tool reveals associations between the maternal brain and mental health disorders, including autism. BMC Neurosci. 2013;14:147. doi: 10.1186/1471-2202-14-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov V, Kronenberg G, Pivneva T, Reuter K, Steiner B, Wang LP, Yamaguchi M, Kettenmann H, Kempermann G. Subpopulation of nestin-expressing progenitor cells in the adult murine hippocampus shows electrophysiological and morphological characteristics of astrocytes. Mol Cell Neurosci. 2003;23:373–382. doi: 10.1016/S1044-7431(03)00060-5. [DOI] [PubMed] [Google Scholar]
- Fournier A, Sasai N, Nakao M, Defossez PA. The role of methyl-binding proteins in chromatin organization and epigenome maintenance. Brief Funct Genomics. 2012;11:251–264. doi: 10.1093/bfgp/elr040. [DOI] [PubMed] [Google Scholar]
- Fujita N, Takebayashi S, Okumura K, Kudo S, Chiba T, Saya H, Nakao M. Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms. Mol Cell Biol. 1999;19:6415–6426. doi: 10.1128/MCB.19.9.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Wang F, Eisinger BE, Kelnhofer LE, Jobe EM, Zhao X. Integrative single-cell transcriptomics reveals molecular networks defining neuronal maturation during postnatal neurogenesis. Cereb Cortex. 2016 doi: 10.1093/cercor/bhw040. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Ure K, Ables JL, Lagace DC, Nave KA, Goebbels S, Eisch AJ, Hsieh J. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat Neurosci. 2009;12:1090–1092. doi: 10.1038/nn.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Ure K, Ding P, Nashaat M, Yuan L, Ma J, Hammer RE, Hsieh J. The master negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J Neurosci. 2011;31:9772–9786. doi: 10.1523/JNEUROSCI.1604-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Allan AM, Zong R, Zhang L, Johnson EB, Schaller EG, Murthy AC, Goggin SL, Eisch AJ, Oostra BA, Jin P, Zhao X. Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat Med. 2011;17:559–565. doi: 10.1038/nm.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Murthy AC, Zhang L, Johnson EB, Schaller EG, Allan AM, Zhao X. Inhibition of GSK3β improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum Mol Genet. 2012a;21:681–691. doi: 10.1093/hmg/ddr501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Patzlaff NE, Jobe EM, Zhao X. Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat Protoc. 2012b;7:2005–2012. doi: 10.1038/nprot.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Polich ED, Su J, Gao Y, Christopher DM, Allan AM, Wang M, Wang F, Wang G, Zhao X. Fragile X proteins FMRP and FXR2P control synaptic GluA1 expression and neuronal maturation via distinct mechanisms. Cell Rep. 2015;11:1651–1666. doi: 10.1016/j.celrep.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson P, Kimber E, Wahlström J, Annerén G. Monosomy 18q syndrome and atypical Rett syndrome in a girl with an interstitial deletion (18)(q21.1q22.3) Am J Med Genet. 1999;82:348–351. [PubMed] [Google Scholar]
- Hirabayashi Y, Gotoh Y. Epigenetic control of neural precursor cell fate during development. Nat Rev Neurosci. 2010;11:377–388. doi: 10.1038/nrn2810. [DOI] [PubMed] [Google Scholar]
- Hodge RD, Kowalczyk TD, Wolf SA, Encinas JM, Rippey C, Enikolopov G, Kempermann G, Hevner RF. Intermediate progenitors in adult hippocampal neurogenesis: Tbr2 expression and coordinate regulation of neuronal output. J Neurosci. 2008;28:3707–3717. doi: 10.1523/JNEUROSCI.4280-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J. Orchestrating transcriptional control of adult neurogenesis. Genes Dev. 2012;26:1010–1021. doi: 10.1101/gad.187336.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Eisch AJ. Epigenetics, hippocampal neurogenesis, and neuropsychiatric disorders: unraveling the genome to understand the mind. Neurobiol Dis. 2010;39:73–84. doi: 10.1016/j.nbd.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura T, Watanabe S, Sakamoto Y, Aoto T, Fujita N, Nakao M. Transcriptional repression and heterochromatin formation by MBD1 and MCAF/AM family proteins. J Biol Chem. 2005;280:13928–13935. doi: 10.1074/jbc.M413654200. [DOI] [PubMed] [Google Scholar]
- Imataka G, Ohwada Y, Shimura N, Yoshihara S, Arisaka O. Del(18)(q12.2q21.1) syndrome: a case report and clinical review of the literature. Eur Rev Med Pharmacol Sci. 2015;19:3241–3245. [PubMed] [Google Scholar]
- Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15:243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs S, Lie DC, DeCicco KL, Shi Y, DeLuca LM, Gage FH, Evans RM. Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc Natl Acad Sci U S A. 2006;103:3902–3907. doi: 10.1073/pnas.0511294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Römer B, Babu H, Kempermann G. Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp Neurol. 2005;196:342–351. doi: 10.1016/j.expneurol.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Jobe EM, McQuate AL, Zhao X. Crosstalk among epigenetic pathways regulates neurogenesis. Front Neurosci. 2012;6:59. doi: 10.3389/fnins.2012.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobe EM, Zhao X. DNA methylation and adult neurogenesis. Brain Plasticity. 2016 doi: 10.3233/BPL-160034. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen HF, Ben-Porath I, Bird AP. Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Mol Cell Biol. 2004;24:3387–3395. doi: 10.1128/MCB.24.8.3387-3395.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Song H, Gage FH. Neurogenesis in the adult hippocampus. Cold Spring Harb Perspect Biol. 2015;7:a018812. doi: 10.1101/cshperspect.a018812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Araúzo-Bravo MJ, Kovacs WJ, Karalay O, Suter U, Machado RA, Roccio M, Lutolf MP, Semenkovich CF, Jessberger S. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature. 2013;493:226–230. doi: 10.1038/nature11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, von Schoultz C, Zurkirchen L, Braun SM, Vidmar M, Jessberger S. SPOT14-positive neural stem/progenitor cells in the hippocampus respond dynamically to neurogenic regulators. Stem Cell Reports. 2014;3:735–742. doi: 10.1016/j.stemcr.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knock E, Pereira J, Lombard PD, Dimond A, Leaford D, Livesey FJ, Hendrich B. The methyl binding domain 3/nucleosome remodelling and deacetylase complex regulates neural cell fate determination and terminal differentiation in the cerebral cortex. Neural Dev. 2015;10:13. doi: 10.1186/s13064-015-0040-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy A, Yamagata M, Duan X, Hong YK, Sanes JR. Sidekick 2 directs formation of a retinal circuit that detects differential motion. Nature. 2015;524:466–470. doi: 10.1038/nature14682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Yamagata T, Mori M, Yasuhara A, Momoi MY. Mutation analysis of methyl-CpG binding protein family genes in autistic patients. Brain Dev. 2005;27:321–325. doi: 10.1016/j.braindev.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Li H, Zhong X, Chau KF, Santistevan NJ, Guo W, Kong G, Li X, Kadakia M, Masliah J, Chi J, Jin P, Zhang J, Zhao X, Chang Q. Cell cycle-linked MeCP2 phosphorylation modulates adult neurogenesis involving the Notch signalling pathway. Nat Commun. 2014;5:5601. doi: 10.1038/ncomms6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chen BF, Chan WY. An epigenetic regulator: methyl-CpG-binding domain protein 1 (MBD1) Int J Mol Sci. 2015;16:5125–5140. doi: 10.3390/ijms16035125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Barkho BZ, Luo Y, Smrt RD, Santistevan NJ, Liu C, Kuwabara T, Gage FH, Zhao X. Epigenetic regulation of the stem cell mitogen Fgf-2 by Mbd1 in adult neural stem/progenitor cells. J Biol Chem. 2008;283:27644–27652. doi: 10.1074/jbc.M804899200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Teng ZQ, Santistevan NJ, Szulwach KE, Guo W, Jin P, Zhao X. Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell. 2010;6:433–444. doi: 10.1016/j.stem.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Teng ZQ, McQuate AL, Jobe EM, Christ CC, von Hoyningen-Huene SJ, Reyes MD, Polich ED, Xing Y, Li Y, Guo W, Zhao X. An epigenetic feedback regulatory loop involving microRNA-195 and MBD1 governs neural stem cell differentiation. PLoS One. 2013;8:e51436. doi: 10.1371/journal.pone.0051436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, Goldman SA, Nedergaard M. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci. 2007;27:12255–12266. doi: 10.1523/JNEUROSCI.3404-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Bird A. Rett syndrome: a complex disorder with simple roots. Nat Rev Genet. 2015;16:261–275. doi: 10.1038/nrg3897. [DOI] [PubMed] [Google Scholar]
- Maslov AY, Barone TA, Plunkett RJ, Pruitt SC. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J Neurosci. 2004;24:1726–1733. doi: 10.1523/JNEUROSCI.4608-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007;213:384–390. doi: 10.1002/jcp.21224. [DOI] [PubMed] [Google Scholar]
- Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schübeler D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 2008;30:755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Namihira M, Kohyama J, Abematsu M, Nakashima K. Epigenetic mechanisms regulating fate specification of neural stem cells. Philos Trans R Soc Lond B Biol Sci. 2008;363:2099–2109. doi: 10.1098/rstb.2008.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Jeppesen P, Bird A. Active repression of methylated genes by the chromosomal protein MBD1. Mol Cell Biol. 2000;20:1394–1406. doi: 10.1128/MCB.20.4.1394-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry L, Clarke AR. The roles of the methyl-CpG binding proteins in cancer. Genes and Cancer. 2011;2:618–630. doi: 10.1177/1947601911418499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, Oliver KL, Grinton BE, Vutskits L, Scheffer IE, Petrou S, Blaesse P, Dibbens LM, Berkovic SF, Kaila K. A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Rep. 2014;15:723–729. doi: 10.1002/embr.201438749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault VM, Rafalski VA, Morgan AA, Salih DA, Brett JO, Webb AE, Villeda SA, Thekkat PU, Guillerey C, Denko NC, Palmer TD, Butte AJ, Brunet A. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell. 2009;5:527–539. doi: 10.1016/j.stem.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y, Watanabe S, Ichimura T, Kawasuji M, Koseki H, Baba H, Nakao M. Overlapping roles of the methylated DNA-binding protein MBD1 and polycomb group proteins in transcriptional repression of HOXA genes and heterochromatin foci formation. J Biol Chem. 2007;282:16391–16400. doi: 10.1074/jbc.M700011200. [DOI] [PubMed] [Google Scholar]
- Sarraf SA, Stancheva I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell. 2004;15:595–605. doi: 10.1016/j.molcel.2004.06.043. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Ming GL, Song H. DNA modifications in the mammalian brain. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130512. doi: 10.1098/rstb.2013.0512. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Berg DA, Zhu Y, Shin JY, Song J, Bonaguidi MA, Enikolopov G, Nauen DW, Christian KM, Ming GL, Song H. Single-cell RNA-Seq with waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell. 2015;17:360–372. doi: 10.1016/j.stem.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh H, Consiglio A, Ray J, Sawai T, D'Amour KA, Gage FH. In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell. 2007;1:515–528. doi: 10.1016/j.stem.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell. 2001;1:749–758. doi: 10.1016/S1534-5807(01)00101-0. [DOI] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura K, Abematsu M, Kohyama J, Namihira M, Nakashima K. Neuronal differentiation of neural precursor cells is promoted by the methyl-CpG-binding protein MeCP2. Exp Neurol. 2009;219:104–111. doi: 10.1016/j.expneurol.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Voronova A, Fischer A, Ryan T, Al Madhoun A, Skerjanc IS. Ascl1/Mash1 is a novel target of Gli2 during Gli2-induced neurogenesis in P19 EC cells. PLoS One. 2011;6:e19174. doi: 10.1371/journal.pone.0019174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Tidei JJ, Polich ED, Gao Y, Zhao H, Perrone-Bizzozero NI, Guo W, Zhao X. Positive feedback between RNA-binding protein HuD and transcription factor SATB1 promotes neurogenesis. Proc Natl Acad Sci U S A. 2015;112:E4995–E5004. doi: 10.1073/pnas.1513780112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse EG, An JJ, Orefice LL, Baydyuk M, Liao GY, Zheng K, Lu B, Xu B. BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J Neurosci. 2012;32:14318–14330. doi: 10.1523/JNEUROSCI.0709-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AE, Pollina EA, Vierbuchen T, Urbán N, Ucar D, Leeman DS, Martynoga B, Sewak M, Rando TA, Guillemot F, Wernig M, Brunet A. FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Rep. 2013;4:477–491. doi: 10.1016/j.celrep.2013.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y, Sun YE. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–448. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Saito H, Suzuki M, Mori K. Visualization of neurogenesis in the central nervous system using nestin promoter-GFP transgenic mice. Neuroreport. 2000;11:1991–1996. doi: 10.1097/00001756-200006260-00037. [DOI] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Ueba T, Christie BR, Barkho B, McConnell MJ, Nakashima K, Lein ES, Eadie BD, Willhoite AR, Muotri AR, Summers RG, Chun J, Lee KF, Gage FH. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc Natl Acad Sci U S A. 2003;100:6777–6782. doi: 10.1073/pnas.1131928100. [DOI] [PMC free article] [PubMed] [Google Scholar]