

Abstract
PVR, the Drosophila homolog of the PDGF/VEGF receptor, has been implicated in border cell migration during oogenesis and hemocyte migration during embryogenesis. It was earlier shown that Mbc, a CDM family protein, and its effector, Rac, transduced the guidance signal from PVR during border cell migration. Here we demonstrate that PVR is also required for the morphogenetic process, thorax closure, during metamorphosis. The results of genetic and biochemical experiments indicate that PVR activates the JNK pathway. We present evidence showing Crk (an adaptor molecule), Mbc, ELMO (a homolog of Caenorhabditis elegans CED-12 and mammalian ELMO), and Rac to be mediators of JNK activation by PVR. In addition, we suppose that not only Rac but also Cdc42 is activated and involved in JNK activation downstream of PVR.
Keywords: Crk–Mbc–ELMO complex, JNK activation, PVR, Rac, thorax closure
Introduction
Cell migration is important not only for development but also for wound healing, and it is likely that the genes involved in these processes have been evolutionally conserved. In mammals, platelet-derived growth factors (PDGF-A and -B) and their receptors (PDGFRs) have been shown to direct cell migration, to cause changes in cell shapes, and to play a role in wound healing and development (Hoch and Soriano, 2003). A large number of Src homology (SH) 2 domain proteins have been shown to bind to autophosphorylated PDGFRs. Although phospholipase C-γ (PLC-γ) and phosphatidylinositol 3-kinase (PI3K) appear to be involved in PDGF-stimulated cell migration and chemotaxis of cultured mammalian cells, the precise roles of these SH2 proteins in cell migration during development remain to be elucidated (Heldin and Westermark, 1999).
The Drosophila Pvr gene (also called Vegfr/stasis) encodes the Drosophila homolog of the PDGF/vascular-endothelial-cell growth factor (VEGF) receptor (Duchek et al, 2001; Cho et al, 2002). PVR and its ligands, PVFs, are involved in blood cell (hemocyte) migration during embryogenesis and in border cell migration during oogenesis (Duchek et al, 2001; Cho et al, 2002). Curiously, Drosophila PLC-γ and PI3K homologs have no effects on border cell migration (Duchek and Rørth, 2001). The only molecules that have been shown to interact genetically with Pvr are Myoblast city (Mbc) and the Rho family GTPase Rac (Duchek et al, 2001). Although Rac is a key regulator of the actin cytoskeleton, it is still unclear whether PVR is involved in other developmental processes requiring Rac besides border cell migration. Mbc is also involved in the epidermal cell migration during embryogenesis called dorsal closure (abbreviated here as DC), in addition to border cell migration.
The CDM family proteins (i.e., Caenorhabditis elegans CED-5, mammalian Dock180, and Mbc) are important for Rac activation during cell migration and engulfment of dying cells (Hasegawa et al, 1996; Erickson et al, 1997; Wu and Horvitz, 1998; Albert et al, 2000; Grimsley et al, 2004). Recently, mammalian Dock180 was shown to be a guanine nucleotide exchange factor (GEF) that specifically activates Rac through the newly recognized GEF motif named Docker (Brugnera et al, 2002). Dock180 biochemically interacts with the small adaptor protein Crk (Hasegawa et al, 1996) as well as with the recently described CED-12/ELMO, an upstream regulator of Rac that directly binds to CED-5/Dock180 (Gumienny et al, 2001; Zhou et al, 2001). Although Drosophila Crk has been identified as an Mbc-binding molecule by two-hybrid screening (Galletta et al, 1999), little is known of Crk function during Drosophila development because no Crk mutants have yet been described.
During metamorphosis, the adult dorsal thorax (the notum) develops from the dorsal parts of the wing imaginal discs (Zeitlinger and Bohmann, 1999). They approach each other from either side and fuse at the midline in a process called thorax closure (TC), which closely resembles DC during embryogenesis. Genetic studies have revealed a requirement for cytoskeletal components and a number of signal transduction molecules for DC (Noselli, 1998; Stronach and Perrimon, 1999; Harden, 2002). Activity of the AP-1 transcription factors (D-Jun/Jra and D-Fos/Kayak (Kay)) and an upstream kinase cascade homologous to the Jun NH2-terminal kinase (JNK) pathway in mammals are required in the leading edge (LE) cells during DC (Noselli, 1998; Noselli and Agnes, 1999; Stronach and Perrimon, 1999; Harden, 2002). Some of signaling molecules required for TC were shown to be similar to those involved in DC (e.g., Hemipterous (Hep)/JNK-Kinase (JNKK) and Kay; Glise et al, 1995; Riesgo-Escovar and Hafen, 1997; Agnes et al, 1999; Zeitlinger and Bohmann, 1999; Martin-Blanco et al, 2000). The expression of dominant-negative (DN) and constitutively activated (CA) forms of Rac1 and Cdc42 suggested a requirement for these GTPases during DC. By using CA forms of Rac1 and Cdc42, Glise and Noselli (1997) obtained evidence suggesting that both Rac1 and Cdc42 could act as upstream activators for the JNK pathway. Mbc was proposed as one likely candidate for Rac activation, but the role of Mbc in JNK activation during DC was not well understood (Nolan et al, 1998).
To investigate the role of PVR and its downstream signaling during Drosophila development, we examined PVR involvement in TC. We also studied whether the Crk–Dock180–ELMO ternary complex homolog was involved in Rac activation downstream of PVR.
Results
PVR receptor tyrosine kinase is required for thorax closure
To study the involvement of PVR in TC during metamorphosis, we generated a transgenic construct expressing inverted repeats (IRs) of PVR to knock down its function by RNA interference (RNAi; Kennerdell and Carthew, 2000). Endogenous PVR was almost completely depleted from Schneider 2 (S2) cells when double-stranded (ds) RNA matching the 5′ coding region of the gene was added to the medium (Figure 1A and B; Clemens et al, 2000). By use of the GAL4/UAS system (Brand and Perrimon, 1993), the expression of Pvr-IR can knock down gene function at a given stage and a given place, even if this gene has other functions earlier in the life cycle or in other tissues. First, to test the ability of RNAi to disrupt gene function in the future medial notum, we prepared the UAS-hep-IR transgene (hep is known to be required for TC; Glise et al, 1995; Agnes et al, 1999; Zeitlinger and Bohmann, 1999; Martin-Blanco et al, 2000) driven by pannier (pnr)-GAL4 (abbreviated as pnr>hep-IR in the figure), which is expressed in the dorsal part of wing imaginal discs (Figure 1I, Calleja et al, 1996; Zeitlinger and Bohmann, 1999). hep RNAi often gave rise to flies with a split thorax, resembling a hypomorphic hep mutation (Figure 1C and D; Figure 1D shows one of the strongest phenotypes obtained by hep RNAi). Because it has been demonstrated that Hep is required for Basket (Bsk)/JNK activation during DC (Harden, 2002; Stronach and Perrimon, 2002), we expected that the same signaling pathway was involved in TC. In accord with our hypothesis, bsk RNAi interrupted TC when UAS-bsk-IR was driven by pnr-GAL4 (Figure 1E). When UAS-Pvr-IR flies were driven by pnr-GAL4, we found TC defects similar to those of hypomorphic hep mutants and RNAi knock-down for hep and bsk (Figure 1F–H; Agnes et al, 1999; Tateno et al, 2000). We found that UAS-Pvr-IRs driven by pnr-GAL4 moderately reduced level of PVR (Figure 1I). About 66% of the flies having 1 Pvr-IR transgene driven by pnr-GAL4 were nearly normal (Class I; Figure 1F and J). However, the remaining 34% exhibited defects on their nota, with the midline lacking bristles (Class II, 29%) or split thoraxes (Class III, 5%; Figure 1G, H, and J). Increasing the number of the Pvr-IR transgene to two or three copies increased to almost 17 or 42%, respectively, the flies with cleft nota (Class III; Figure 1J). Correlation between severity of phenotypes and the copy number of UAS-Pvr-IR transgenes suggests that Pvr RNAi did not fully knock out Pvr gene function when we used our PVR-IR construct. Expressing one copy of Pvr-IR with pnr-GAL4 on a Pvrc2195/+ heterozygote background (Pvrc2195 is an amorphic allele; Cho et al, 2002) enhanced the TC defect (24% of flies had Class III phenotype; Figure 1J). These data also support the above implication. The similarity between the adult phenotype of Pvr RNAi flies and that of hep and bsk RNAi flies implied that PVR was involved in JNK activation during TC.
Figure 1.
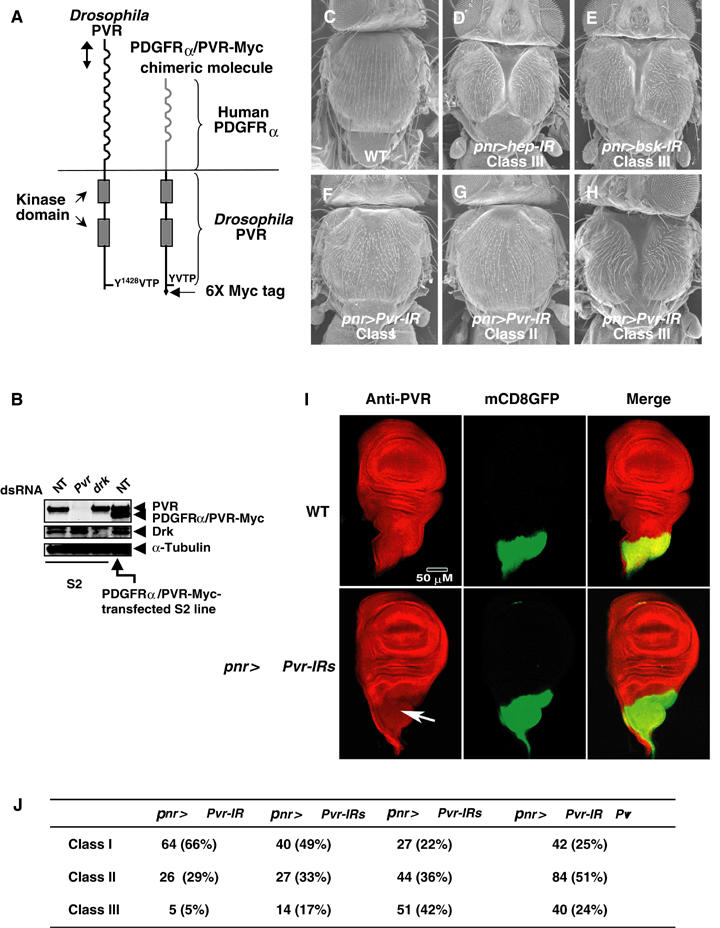
Interference of TC by Pvr RNAi. (A) Schematic structure of PVR (left) and PDGFRα/PVR-Myc chimeric molecule (right). Like mammalian PDGF and VEGF receptors, PVR is composed of Ig-like repeats, a transmembrane domain, and a split kinase domain. Corresponding region (approximately 700 bp in length) used as a template for in vitro synthesis of dsRNA and for the Pvr-IR construct is indicated by the vertical line with double arrowheads. The PDGFRα/PVR-Myc chimeric molecule is a chimera of an extracellular domain of human PDGFRα fused to a transmembrane domain and cytoplasmic domain of PVR with a 6X Myc-tag sequence at its C-terminus. (B) Treatment of S2 cells with dsRNA for Pvr (lane 2) inhibited the PVR expression, but had no effects on the Drk protein level (lane 3). On the other hand, dsRNA for drk had no effects on PVR expression (lanes 2 and 3), although Drk expression was almost completely abolished (a commercial mouse anti-Grb2 antibody was used for the detection of Drk). For biochemical analyses, we established the S2 cell line expressing PDGFRα/PVR-Myc chimeric molecules under the control of the metallothionein promoter. The chimeric molecule was expressed in an amount almost equal to that of endogenous PVR after overnight induction with 100 mM CuSO4 (lane 4). Blotting with anti-α-tubulin antibody was used for the control. NT: not treated with dsRNA. Top panel: anti-PVR immunoblot; middle panel: anti-Grb2 immunoblot; bottom panel: anti-α-tubulin immunoblot. (C–H) Morphology of WT (C), pnr-GAL4/UAS-hep-IR, abbreviated as pnr>hep-IR (Class I, 45%; Class II, 20%; Class III, 35%; total n value, 220) (D), pnr>bsk-IR (Class I, 21%; Class II, 33%; Class III, 46%; total n value, 243) (E), and pnr>Pvr-IR (F–H) adult nota. Severity of TC defects by hep, bsk, and Pvr RNAi was variable. Panels D, E, and H show a very strong phenotype (cleft nota, Class III). Other populations had nota, almost normal (Class I (F)) or with the midline devoid of bristles (Class II (G)). (I) A moderately reduced level of PVR by UAS-Pvr-IR driven with pnr-GAL4. Wing imaginal discs of a WT fly (genotype pnr-GAL4/UAS-mCD8∷GFP) and the PVR RNAi line (genotype pnr-GAL4/ 2X UAS-Pvr-IRs, UAS-mCD8∷GFP) were stained with anti-PVR antibody (Rosin et al, 2004). Pnr-GAL4 is expressed in the thoracic region of wing imaginal discs (marked by GFP in middle panels). As compared with a WT wing disc, expression of PVR was moderately reduced by Pvr RNAi (marked by an arrow). (J) Severity of TC defects by pnr>Pvr–IRs. Classes I–III correspond to phenotypes shown in panels F–H, respectively. Both n values and % values are included.
PVR controls JNK activity in vivo
Although three putative PVR ligands (PVF1–3) have been identified following Drosophila genome sequencing, we could not employ purified PVFs for our assay. Instead of using PVFs to stimulate PVR, we monitored signaling downstream of PVR by using S2 cell lines expressing a Myc-tagged chimeric molecule between human PDGFRα and PVR (PDGFRα/PVR-Myc; see Figure 1A and B, lane 4). When the chimeric receptors bound PDGF-BB (a dimerized human PDGF-B), they became autophosphorylated as expected (Figure 2A, fifth and sixth panels), creating docking sites for downstream signaling molecules. Stimulation of PDGFRα/PVR-Myc with PDGF-BB activated the MAPK pathway (Figure 2A, third and fourth panels), thus suggesting that our chimeric receptor transduces the same signal(s) as does the wild-type (WT) PVR (Duchek et al, 2001; Cho et al, 2002). However, we also found that PDGFRα/PVR-Myc triggered the activation of the JNK pathway (Figure 2A, top and second panels).
Figure 2.
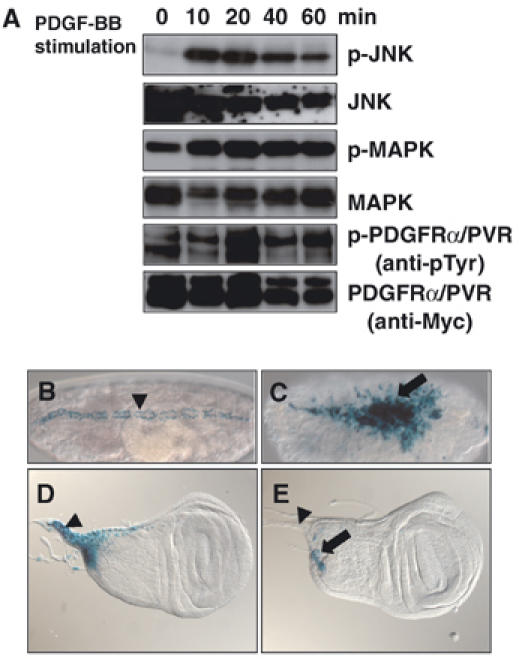
PVR controls JNK activity. (A) Human PDGF-BB stimulation of PDGFRα/PVR-Myc in S2 cell cultures. Cells were starved overnight in Schneider's medium+0.1% FCS supplemented with 100 mg/ml CuSO4 for the induction of the PDGFRα/PVR-Myc chimeric receptor. Then, the cells were incubated with 100 ng/ml PDGF-BB for the indicated amount of time before harvest. Cellular lysates were prepared and subjected to Western analysis. Expression of the chimeric molecule was detected by using anti-Myc antibody (bottom panel). By PDGF-BB stimulation, the chimeric molecule was tyrosine phosphorylated (anti-phosphotyrosine blotting, fifth panel). The lower band in the fifth panel might be a tyrosine-phosphorylated chimeric receptor whose phosphorylation is independent of the PDGF-BB stimulation. After ligand stimulation, the chimeric receptor might be additionally phosphorylated, so its mobility on an SDS–PAGE may be sifted. Phosphorylated Bsk and total Bsk protein were detected by antibodies against dual-phosphorylated JNK (p-JNK) and JNK1, respectively (top and second panels). Phosphorylated Rolled (Rl/Drosophila MAPK homolog) and total Rl were immunoblotted with anti-dual-phosphorylated ERK/MAPK (p-MAPK) and anti-MAPK, respectively (third and fourth panels). Maximal activation of both JNK and MAPK was observed 10–20 min after PDGF-BB stimulation. (B, C) Elevation of puc expression by the CA form of PVR (λPVR). (B) Control, normal puc expression, which was detected by X-gal staining of the puc-lacZ reporter (pucE69). During the DC process, puc is specifically expressed in the LE cells of WT embryos (marked with an arrowhead). (C) During germ band elongation, pnr-GAL4 drives expression of λPVR in 2–3 rows of epidermal cells including LE cells. pnr-GAL4 is also expressed in the amnioserosa after germ band retraction (see Supplementary Figure S1). Ectopic puc expression was induced in amnioserosa and ectodermal cells by pnr>λPvr (indicated with an arrow). Both embryos are dorsal views, with anterior to the left. (D, E) puc expression in wing discs was dependent on the PVR activity. (D) Control, normal puc expression. puc is expressed in the proximal part of a wing imaginal disc including stalk region (marked with an arrowhead). (E) Expression of puc in a wing imaginal disc of a third instar larva with pnr>3X Pvr-IRs. puc expression was greatly reduced by Pvr RNAi. No expression was observed in the stalk region (marked with an arrowhead). Remaining puc expression in a presumptive notum is marked with an arrow.
We also used a lacZ enhancer trap line inserted in the puckered (puc) gene, pucE69, to monitor JNK activity in vivo after ectopic expression of a CA form of PVR (λPVR) during embryogenesis (Duchek et al, 2001). puc transcription is under the control of the JNK signaling pathway (Martin-Blanco et al, 1998; Zeitlinger and Bohmann, 1999). When λPVR was driven by pnr-GAL4, which is expressed in the dorsal-most cells of the lateral ectoderm (including LE cells) and also in the amnioserosa during embryogenesis (Supplementary Figure S1), it induced a corresponding ectopic expression of puc-lacZ in a pnr pattern (Figure 2B and C). These data indicate that PVR could indeed activate the JNK pathway in vivo. During the third instar larval stage, puc-lacZ is specifically expressed in the proximal part of the WT wing imaginal disc, including the stalk region, where wing discs connect to the larval epidermis (Figure 2D; Agnes et al, 1999; Usui and Simpson, 2000). Pnr is normally expressed in wing discs in a broad domain corresponding to the central presumptive notum during metamorphosis (Figure 1I; Calleja et al, 1996). Driving three copies of UAS-Pvr-IR with pnr-GAL4 reduced puc-lacZ expression in the wing disc (Figure 2E), suggesting that PVR function is required in vivo for JNK activation during TC.
To determine the position of PVR function in the JNK pathway, we performed biochemical assays on RNAi-treated cells in culture (Figure 3A). When S2 cells expressing the PDGFRα/PVR-Myc transgene were treated with dsRNA for bsk, Bsk (JNK) expression was greatly reduced (data not shown). Cells treated with dsRNAs against hep and slipper (slpr, encoding JNKK-Kinase (JNKKK); Stronach and Perrimon, 2002) showed reduced Bsk (JNK) activation, although the amount of Bsk protein remained the same as in untreated cells (Figure 3A, lanes 1 and 2). These biochemical tests suggested that PVR functioned upstream of Slpr.
Figure 3.
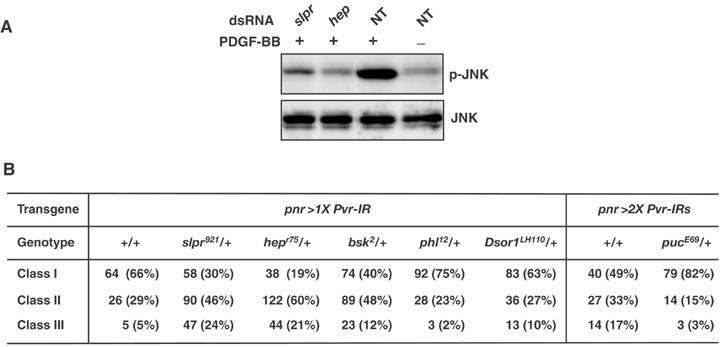
The position of PVR function in the JNK pathway. (A) The PDGFRα/PVR-Myc cell line was incubated in the absence (NT, lanes 3 and 4) or presence (lanes 1 and 2) of dsRNAs representing slpr or hep. After 2 days, the cells were serum-starved, and expression of the chimeric molecule was then induced with 100 mg/ml CuSO4 (after a medium change, the same dsRNA was supplied again to each sample). The next day, the cells were either left untreated (lane 4) or treated (lanes 1–3) with 100 ng/ml PDGF-BB for 20 min. Then, cellular extracts were prepared and subjected to Western analysis using anti-pJNK and anti-JNK1 antibodies. (B) Suppression or enhancement of the thorax phenotypes of pnr>Pvr-IRs by mutations implicated in the JNK pathway. Classes I–III correspond to phenotypes shown in Figure 1F–H, respectively.
Next, we examined genetic interactions between Pvr and components of the JNK signaling pathway during TC (Figure 3B). Reducing the gene dosage of slpr and hep to one copy dramatically enhanced the TC phenotype obtained by one copy of UAS-Pvr-IR driven by pnr-GAL4 (almost 24 and 21% of flies with a single copy of slpr and hep, respectively, had the Class III phenotype), whereas reducing that of bsk to one copy had a moderate enhancing effect (12% of flies were Class III; Figure 3B). In contrast, the Class III TC defect with two copies of UAS-Pvr-IR driven by pnr-GAL4 (Figure 3B) fell from 17 to 3% in flies with a single copy of puc, which encodes a Drosophila homolog of CL100, a VH-1 family MAPK phosphatase that negatively regulates the activities of the JNK pathway during both DC and TC (Martin-Blanco et al, 1998; Zeitlinger and Bohmann, 1999), suggesting that puc has genetic interaction with Pvr. Remarkable genetic interactions between Pvr and genes in the MAPK pathway, pole hole (phl, coding Raf) and Dsor 1 (coding MEK), were not observed for TC (Figure 3B). Taken together, the results of these biochemical and genetic studies strongly suggest that PVR serves as a receptor activating the JNK pathway during TC.
PVR activates Rac1 and Cdc42 GTPases
Pvr interacts genetically with Rac1 and Mbc (Duchek et al, 2001). To see whether PDGFRα/PVR-Myc could activate Rac1, we used a PAK-p21 binding domain (PAK-PBD) pull-down assay. In PDGFRα/PVR-Myc-expressing S2 cells, treatment of the cells with PDGF-BB for 20 min promoted GTP loading of endogenous Rac1 (Figure 4A, left panels). Unexpectedly, it also elevated the amount of GTP-loaded Cdc42 (Figure 4B, left panels). These data suggest that PVR activates not only Rac1 but also Cdc42, at least in S2 cells. (Although Drosophila has two other Rac genes, Rac2 and Mtl, we could not evaluate their activation by PVR because of the unavailability of antibodies against them. Applicability of a pull-down assay using mammalian PAK-PBD for measuring the activity of Drosophila Rac1 and Cdc42 is reconfirmed in right panels of Figure 4A and B, respectively.)
Figure 4.
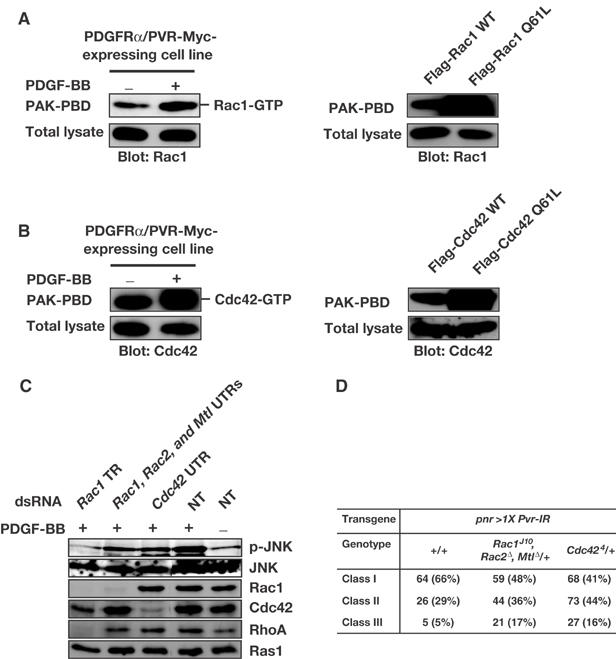
Involvement of Rac and Cdc42 in PVR function. (A, B) PDGF-BB stimulation of PDGFRα/PVR-Myc activates both Rac1 and Cdc42. The PDGFR α/PVR-Myc cell line was starved overnight in Schneider's medium containing 0.1% FCS and 100 mg/ml CuSO4 for induction of PDGFRα/PVR-Myc chimeric receptors. Then, the cells were incubated in the absence or presence of 100 ng/ml PDGF-BB for 20 min. Lysates were prepared and incubated with GST-PAK-PBD for 1 h. GST-PAK-PBD was precipitated and the amounts of co-precipitated endogenous Rac1-GTP and Cdc42-GTP were visualized by immunoblotting for Rac1 (A) and Cdc42 (B), respectively (left panels). The amounts of transiently expressed CA mutants of Rac1 and Cdc42 (Flag-Rac1 Q61L and Flag-Cdc42 Q61L, respectively) co-precipitated with GST-PAK-PBD were greater than those of WT Rac1 and Cdc42 (Flag-Rac1 WT and Flag Cdc42 WT, respectively), reconfirming that a pull-down assay using mammalian PAK-PBD can be applied for measuring the activity of Drosophila Rac1 and Cdc42 in S2 cells (right panels). (C) The PDGFRα/PVR-Myc cell line was incubated in the absence (NT, lanes 4 and 5) or presence (lanes 1–3) of dsRNA products directed against TR of Rac1 (lane 1), mixed UTRs of Rac1, Rac2, and Mtl (lane 2), or UTR of Cdc42 (lane 3). Cells were grown as described in the legend of Figure 2B. Subsequently, the cells were either treated (lanes 1–4) or left untreated (lane 5) with 100 ng/ml PDGF-BB for 20 min. Cellular extracts were prepared and subjected to Western analysis using anti-p-JNK, anti-JNK1, anti-Rac1, anti-Cdc42, anti-RhoA, and anti-Ras1 (pan-Ras) antibodies. (D) Increase in severity of the TC defect by pnr>Pvr-IR with heterozygosity for loss-of-function alleles of Rac and Cdc42. Classes I–III correspond to phenotypes shown in Figure 1F–H, respectively.
Rac and Cdc42 contribute to JNK activation and thorax closure downstream of PVR
We also analyzed the involvement of Rac and Cdc42 in JNK activation by using our PDGFRα/PVR-Myc chimeric receptor. dsRNA corresponding to the Rac1 translated region (TR) efficiently knocked down expression not only of Rac1 but also of other Rho family GTPases because of their extremely high nucleotide sequence similarities. By pretreating cells with Rac1 TR dsRNA, JNK activation was almost completely abolished (Figure 4C, lane 1). In contrast, mixed dsRNAs of Rac1-, Rac2-, and Mtl-untranslated regions (UTRs), which should specifically decrease the expression of each Rac protein, only weakly suppressed JNK activation by our chimeric receptor (Figure 4C, lane 2). This result suggested two possibilities: either RNAi using UTRs of Rac genes is less effective than that using Rac1 TR, or else other Rho family GTPase(s) also knocked down by Rac1 TR RNAi collaborate with Rac proteins for the full activation of the JNK pathway. In support of the second explanation, we found that stimulated PDGFRα/PVR-Myc activated both Rac1 and Cdc42 (left panels in Figure 4A and B) and that expression of CA forms of both Rac1 and Cdc42 strongly activated JNK in S2 cells (Supplementary Figure S2), as has been seen in mammalian cells (Bishop and Hall, 2000). dsRNA matching the 3′ UTR of Cdc42 (Cdc42 UTR) weakly interrupted JNK activation by PDGFRα/PVR-Myc (Figure 4C, lane 3). These data support the idea that both Rac molecule(s) and Cdc42 are required for the full activation of JNK by PVR, at least in S2 cells.
The results of genetic interaction experiments largely support the above conclusion. Simultaneous heterozygosity for loss-of-function alleles of Rac1, Rac2, and Mtl (Hakeda-Suzuki et al, 2002) increased the severity of TC defects by UAS-Pvr-IR driven with pnr-GAL4 (Class III, 17%; Figure 4D). Reduction of the Cdc42 gene dosage to one copy (Genova et al, 2000) also enhanced the TC defect (Class III, 16%; Figure 4D), which is also consistent with our biochemical data (Figure 4B). These experimental results, taken together, argue that Rac proteins and Cdc42 are involved in JNK activation by PVR during TC.
Crk, Mbc, and Drosophila CED-12/ELMO homolog are required for thorax closure
Next, we sought to identify the molecule(s) that transfers signals from PVR to downstream Rac and Cdc42. Myc-tagged PVR (PVR-Myc) was transiently expressed in S2 cells with various Flag-tagged SH2- or PTB-domain-containing proteins whose mammalian homologs directly bind to PDGFRs. PVR-Myc itself was highly expressed and consequently autophosphorylated (Figure 5B, fifth panel). We found that several SH2 molecules, that is, Crk, Drk/Grb2, Small wing (Sl)/PLC-γ, and p60/a regulatory subunit of PI3K, and one PTB-domain-containing adaptor, Shc, bound to the autophosphorylated PVR-Myc (Supplementary Figure S3). We focused on Crk, because a functional relationship between Crk/CED-2 and Dock180/CED-5 has already been defined (Albert et al, 2000; Reddien and Horvitz, 2000). Crk is an adaptor molecule composed of only SH2 and SH3 domains (Figure 5A). Drosophila Crk (unlike mammalian Crk) has two isoforms (caused by alternative splicing), differing in the size of their SH2 domains (called CG1587-PA and -PB by Berkley Drosophila Genome Project (BDGP), which we renamed as Crk (SH2L) and Crk (SH2S), respectively; see Supplementary Figure S4A). The SH2 domain of Crk (SH2L) is 18 amino acids longer than that of Crk (SH2S) (Supplementary Figure S4B). We found that Crk (SH2S), but not Crk (SH2L), bound to the C-terminal phosphotyrosine residue on PVR (Y1428VTP in Figure 1A; see Figure 5B and Supplementary Figure S3). Mutation of the tyrosine 1428 to phenylalanine (Y1428F) greatly reduced the level of phosphorylated tyrosine in PVR and abolished Crk (SH2S) binding to PVR (Figure 5B).
Figure 5.
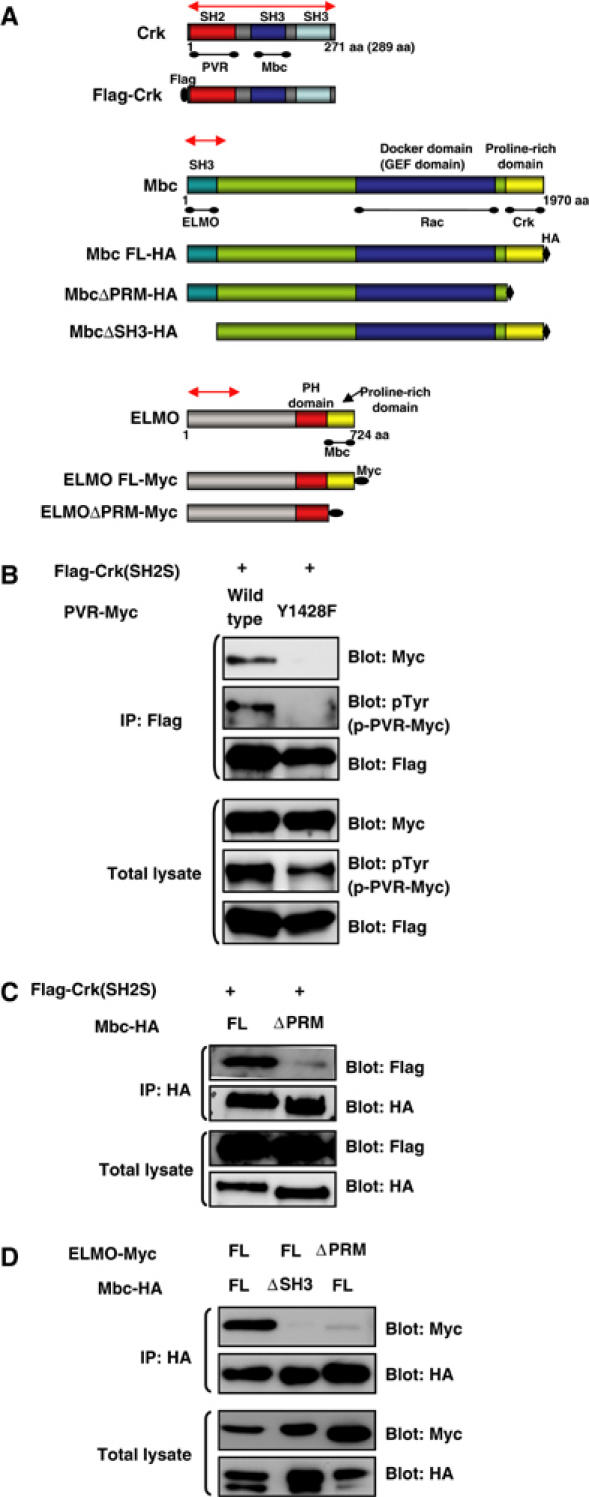
Physiological interactions among PVR, Crk, Mbc, and ELMO. (A) Schematic diagram of WT and modified forms of Crk, Mbc, and ELMO constructs used in this study. For the biochemical interaction experiments, Crk, Mbc, and ELMO were Flag, HA, and 6X Myc tagged, respectively. A corresponding region (approximately 700 bp in length) of each gene was used as a template for in vitro synthesis of dsRNA and for the IR construct, indicated by a red line with double arrowheads. PRM: proline-rich motif, FL: full length. (B) The C-terminal tyrosine phosphorylation site of PVR (Y1428VTP in Figure 1A) is required for interaction with Crk (SH2S). S2 cells were transfected with Myc-tagged WT or Y1428F mutant constructs of PVR cloned in the pUAST vector together with pUAST Flag-Crk (SH2S) and pWAGAL4, the latter of which supplied the GAL4 protein. After 48 h, the cells were lysed, immunoprecipitated with anti-Flag (rabbit polyclonal) antibody, and immunoblotted with anti-Myc, anti-pTyr, or anti-Flag (mouse monoclonal) antibodies. The total lysates from the same experiment were immunoblotted with the same antibodies to see the expression of PVR and Crk and the tyrosine phosphorylation level of PVR molecules. (C) C-terminal region of Mbc is required for interaction with Crk. S2 cells were transfected with the indicated plasmids coding for Flag-Crk (SH2S) along with Mbc-HA (lane 1) or Mbc ΔPRM-HA (lane 2). The lysates were immunoprecipitated with anti-HA antibody and immunoblotted with anti-Flag antibody. The same blot was then stripped and reprobed with anti-HA. Immunoblotting of total lysates indicated the expression of HA-tagged Mbc molecules and Flag-Crk. (D) ELMO interacts with the SH3 domain of Mbc through its C-terminal PRM. S2 cells were transfected with the indicated plasmids coding for HA-tagged Mbc FL or Mbc ΔSH3 along with 6X Myc-tagged ELMO FL or ELMO ΔPRM. After anti-HA immunoprecipitation, the co-precipitation of Myc-tagged ELMO molecules was analyzed by anti-Myc immunoblotting. The same blot was then stripped and reprobed with anti-HA. Immunoblotting of total lysates indicated the expression of HA-tagged Mbc and Myc-tagged ELMO molecules.
The Drosophila genome also encodes one CED-12/ELMO homolog (CG5336, which we renamed ELMO; Figure 5A). CED-12/ELMO molecules have a pleckstrin homology (PH) domain and a proline-rich-motif (PRM) domain near their C-terminus (Figure 5A). We confirmed interactions between Drosophila Crk and Mbc and between Mbc and ELMO by co-immunoprecipitation following expression of full-length or truncated proteins in S2 cells (Figure 5A, C, and D). Consequently, we presume that Crk bound to the C-terminal PRM of Mbc and Mbc bound to the C-terminal PRM of ELMO through its N-terminal SH3 domain.
We then examined whether Crk, mbc, and ELMO were required for TC by conducting RNAi knock-down experiments. When three copies of UAS-Crk-IR, three copies of UAS-mbc-IR, and two copies of UAS-ELMO-IR were driven by pnr-GAL4, we observed TC defects (Figure 6A–C, respectively). Whereas one copy of UAS-mbc-IR and one copy of UAS-ELMO-IR driven by pnr-GAL4 also caused mild TC defects with low frequencies, we never observed any TC defects with one or two copies of UAS-Crk-IR (data not shown). The similarity between the adult phenotypes obtained with Crk, mbc, and ELMO RNAi and those with Pvr, hep, and bsk RNAi imply that Crk, Mbc, and ELMO are involved in the signaling downstream of PVR to activate JNK during TC.
Figure 6.
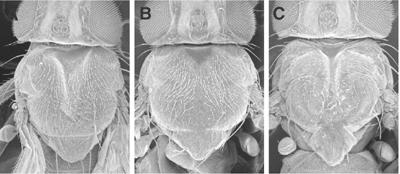
Crk, Mbc, and ELMO are implicated in TC. (A–C) Crk, Mbc, and ELMO RNAi caused TC defects. Morphology of pnr>3X Crk-IRs (A), pnr>3X mbc-IRs (B), and pnr>2X ELMO-IRs (C) adult nota is seen. Severity of TC defects caused by Crk, mbc, and ELMO RNAi was variable, and panels A–C show a strong phenotype (cleft nota, Class III). The corresponding frequencies are as follows: pnr>3X Crk-IRs (Class I, 75%; Class II, 24%; Class III, 1%; total n value, 272), pnr>3X mbc-IRs (Class I, 31%; Class II, 55%; Class III, 14%; total n value, 241), pnr>2X ELMO-IRs (Class I, 69%; Class II, 19%; Class III, 12%; total n value, 185).
As Pvr genetically interacts with mbc for border cell migration (Duchek et al, 2001), we decided to examine the genetic interaction of Crk and mbc with Pvr during TC. We identified a P-element insertion, KG00336 (isolated by BDGP), as a lethal mutation in the Drosophila Crk gene, which we renamed CrkKG00336 (see Supplementary data and Supplementary Figure S4A and B). Performing the Pvr RNAi (one copy of UAS-Pvr-IR driven with pnr-GAL4) on flies with the CrkKG00336/+ heterozygous background moderately enhanced TC defects (Class III, 11%; Figure 7A). Similarly, Crk RNAi from two copies of UAS-Crk-IR driven with pnr-GAL4 enhanced them (Class III, 26%; Figure 7A). Heterozygosity for a loss-of-function allele of mbc (mbcD11.2) moderately increased the severity of TC defects by UAS-Pvr-IR driven with pnr-GAL4 (Class III, 11%; Figure 7A). From these data, we concluded that Crk and mbc are involved in TC downstream of Pvr.
Figure 7.
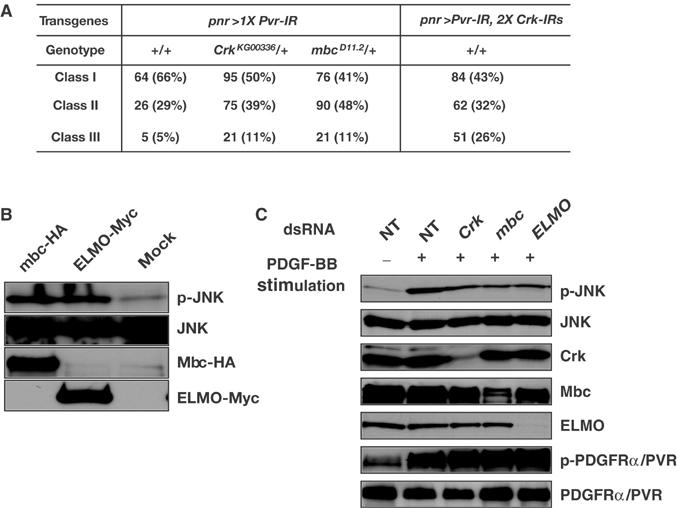
Involvement of Crk, Mbc, and ELMO in JNK activation downstream of PVR. (A) Genetic interaction of Crk and mbc with Pvr. The severity of the TC defect caused by pnr>Pvr-IR was increased by heterozygosity for loss-of-function alleles of Crk and mbc and Crk RNAi. Classes I–III correspond to phenotypes shown in Figure 1F–H, respectively. (B) S2 cell lines expressing Mbc-HA or ELMO-Myc (see Figure 5A) under the control of the metallothionein promoter were established. Cells were serum-starved overnight, and then transgenic proteins were induced for 5 h by the addition of CuSO4 to the medium. Compared with the vector-transfected (mock) cells (lane 3), S2 cells expressing Mbc-HA (lane 1) or ELMO-Myc (lane 2) have elevated JNK activity. (C) The PDGFRα/PVR-Myc cell line was incubated in the absence (NT, lanes 1 and 2) or presence (lanes 3–5) of dsRNA products directed against Crk (lane 3), mbc (lane 4), or ELMO (lane 5). Cells were grown as described in the legend of Figure 2B. Subsequently, the cells were either left untreated (lane 1) or treated (lanes 2–5) with 100 ng/ml PDGF-BB for 20 min. Cellular extracts were prepared and subjected to Western analyses using anti-p-JNK, anti-JNK1, anti-Crk, anti-Mbc, anti-ELMO, anti-pTyr, and anti-Myc antibodies.
Ectopic expression of Mbc and ELMO triggers JNK activation
Dock180, the mammalian homolog of Mbc, can trigger the JNK signaling cascade, but this JNK activation is blocked by DN Rac1 (Rac1N17), suggesting that Dock180 can regulate JNK signaling through its activation of Rac (Kiyokawa et al, 1998). On the other hand, mbc mutant fly embryos showed DC defects and occasional modest reduction in JNK activity, suggesting that Mbc is not absolutely required for JNK activation and that it may play a distinct role in DC (Nolan et al, 1998). Using S2 cells expressing HA-tagged Mbc (Mbc-HA), we found that JNK activity in Mbc-HA-expressing cells was higher than that in mock-transfected cells (Figure 7B, compare lanes 1 and 3). We also confirmed that an ectopic expression of Mbc activated Rac but had no effects on Cdc42 activity in S2 cells (Supplementary Figure S5). Interestingly, ectopic expression of Myc-tagged ELMO also raised JNK activity in S2 cells (Figure 7B, compare lanes 2 and 3). These data strongly indicate that both Mbc and ELMO have competence to activate JNK in Drosophila cells.
Involvement of Crk, Mbc, and ELMO in JNK activation downstream of PVR
Finally, we examined the requirement of Crk, Mbc, and ELMO for JNK activation specifically downstream of PVR. S2 cells express Crk, Mbc, and ELMO endogenously (Figure 7C), but treatment with dsRNAs directed against Crk, Mbc, and ELMO moderately reduced Bsk (JNK) activation by PDGFRα/PVR-Myc after PDGF-BB stimulation (Figure 7C). These observations implicate Crk, Mbc, and ELMO in JNK activation by PDGFRα/PVR-Myc. However, their contribution to the activation might not be absolute (see Discussion).
Discussion
Our study suggests that PVR is required not only for border cell migration and hemocyte migration, but also for imaginal cell migration and/or imaginal cell shape changes during TC (Figure 1F–H). PVR is the first receptor tyrosine kinase (RTK) in Drosophila to our knowledge with a role in JNK activation during development. For border cell migration, genetic evidence has implicated Rac and Mbc as mediators of the guidance signal downstream of PVR (Duchek et al, 2001). Our genetic and biochemical data suggest that PVR functions to activate the JNK pathway not only through Rac but also through Cdc42 both during the TC process and in S2 cells (see summarized scheme in Supplementary Figure S6). Our data have implicated Crk, Mbc, and ELMO in Rac activation by PVR during TC. Since the GEF activity of Mbc is specific for Rac, other unidentified proteins may be involved in Cdc42 activation downstream of PVR. We discuss below a model by which the PVR pathway controls JNK activation and explore its implications.
Involvement of PVR signaling in thorax closure
TC and DC in Drosophila represent excellent model systems for the analysis of the mechanisms involved in coordinating epithelial sheet spreading and cell reorganization during development. Although several findings support the existence of an evolutionally related molecular program that regulates both TC and DC (Stronach and Perrimon, 1999; Harden, 2002), PVR seems to be involved in TC only. Although the Pvr mutation affected migration and survival of hemocytes (Cho et al, 2002; Bruckner et al, 2004), we could not see DC defects in PvrC2195 mutant embryos (data not shown). We suppose that other receptors such as integrins (e.g., Myospheroid/βPS and Scab/αPS3) or RTKs (e.g., DInR, an insulin receptor homolog; Fernandez et al, 1995) might be involved in JNK activation during DC (Harden, 2002; Stronach and Perrimon, 2002).
PDGF-BB stimulation of our PDGFRα/PVR-Myc activated MAPK (Figure 2A). However, a requirement for MAPK signaling downstream of PVR is not clear. The MAPK pathway does not affect border cell migration (Duchek and Rørth, 2001). Zygotic loss-of-function mutations in genes encoding adaptor proteins (drk, shc) or a Ras GEF (sos) had no effects on hemocyte migration, although expression of DN Ras1 (Ras1N17) in hemocytes caused migration defects, implicating Ras1 in the process (Cho et al, 2002). Whereas the results of our genetic interaction experiments also suggested no (i.e., phl) or a little (i.e., Dsor1) involvement of the MAPK signaling pathway in TC, we observed enhancement of the Pvr RNAi phenotype by a reduction of the Ras1 gene dosage to one copy (Figure 2C and data not shown). Thus, Ras1 function might be required for effectors that are involved in actin reorganization and/or JNK activation downstream of PVR.
JNK activation by PVR
JNK pathway is supposedly required in a specific subset of peripodial cells for morphogenesis during TC. JNK signals affect the adhesion between imaginal and larval epidermal cells (Agnes et al, 1999). Involvement of PVR in JNK activation during TC implies that PVR is also required in those peripodial cells, although a role of PVR in driving cell migration and cell shape changes during TC is not directly demonstrated.
Our data suggested that PVR activated JNK through both Rac and Cdc42. We demonstrated that the Crk–Mbc–ELMO complex contributed to Rac activation but not Cdc42 activation (Supplementary Figure S5). Other unknown molecules are supposedly required for Cdc42 activation, which would indicate that the Crk–Mbc–ELMO complex is not the sole player involved in JNK activation by PVR.
It is possible that Rac and Cdc42 activate the JNK cascade through direct contact with Slrp/JNKKK, since Slpr, belonging to the MLKs, contains sequences with homology to the PBD motif (Stronach and Perrimon, 2002) and we observed TC defects by Slpr RNAi (data not shown). However, the Drosophila gene misshapen (msn), which encodes a homolog of mammalian Nck-interacting kinase (NIK), a supposed JNKKKK (Su et al, 1998), might be also required for TC (see Supplementary Figure S6).
Although expression of CA form of RhoA had no effects on JNK activation in S2 cells (data not shown), involvement of RhoA in JNK activation is still obscure. The function of RhoA might be required for JNK activation by PVR, because sequential Rac1 and RhoA activation is known to be necessary for polarized cell migration and phagocytosis.
In mammalian established cell lines such as NIH3T3, JNK is activated by PDGF stimulation (Lallemand et al, 1998). PDGF not only acts as a growth factor promoting cell proliferation but also functions as a chemoattractant (Kundra et al, 1994; Wennstrom et al, 1994). Mice carrying a targeted null mutation of the PDGFRα gene exhibit cephalic closure defect (Soriano, 1997). In addition, mammalian Crk has been shown to bind directly and with high affinity to PDGFRα (Heldin and Westermark, 1999). Recently, fibroblasts derived from jnk−/− mouse embryos were found to exhibit significantly lower ability to close mechanically induced wounds made in a cell layer than their WT counterparts. Wound closure by mouse fibroblasts was also dramatically impaired by the specific JNK inhibitor SP600125 (Javelaud et al, 2003). Mice conditionally deleted for c-jun in their epidermis are born, but with open eyes (eyelid closure defects) and with defects in epidermal wound healing. Keratinocytes lacking c-jun are unable to migrate or elongate properly in culture at the border in scratch assays (Li et al, 2003). Taken together, the available data suggest that the signaling pathway involved in JNK activation by the PDGF receptor family has been functionally conserved throughout phylogeny. The JNK signaling pathway might be required for activation of the AP-1 complex.
Function of the Crk–Mbc–ELMO complex
The role of Crk/CED-2 in the regulation of the Dock180/CED-5- and ELMO/CED-12-mediated Rac/CED-10 activation is still obscure. In mammalian cells in culture, the αvβ5 integrin heterodimer recruits the p130Cas–Crk–Dock180 complex when the αvβ5 receptor binds apoptotic cells to cause their internalization (Albert et al, 2000). Dock180 and ELMO1 also cooperate to promote Rac-dependent cell migration (Grimsley et al, 2004). In C. elegans, it was genetically demonstrated that ced-2, ced-5, ced-12, and ced-10 (encoding Rac) mediate cytoskeletal rearrangements during phagocytosis of apoptotic cells and cell motility (Wu and Horvitz, 1998; Reddien and Horvitz, 2000; Gumienny et al, 2001; Zhou et al, 2001). Recently, Wang et al (2003) found that psr-1, encoding the C. elegans homolog of a mammalian putative phosphatidylserine receptor, acts upstream of ced-2, ced-5, ced-12, and ced-10 to control the engulfment of cell corpses. Curiously, the intracellular domain of PSR-1 directly interacted with CED-5 and CED-12, but not with CED-2. On the other hand, we unambiguously demonstrated that Crk (SH2S) directly bound to PVR when PVR was autophosphorylated (Figure 5B). We found that all members of the Crk–Mbc–ELMO complex were involved in TC (Figure 6A–C). The results of our genetic interaction experiments suggested that both Crk and Mbc transduced signals downstream of PVR during TC (Figure 7A). Furthermore, RNAi for Crk, mbc, and ELMO moderately reduced JNK activation by PDGFRα/PVR-Myc (Figure 7C). These data strongly suggest that Crk mediates Rac activation by PVR through the recruitment of the Mbc–ELMO complex to the receptor and that the activated Rac molecule(s) then transduces a signal to the JNK pathway. Further studies are needed to determine whether integrin receptors and/or a phosphatidylserine receptor activate Rac through the Crk–Mbc–ELMO complex in response to different upstream signals in Drosophila.
What triggers PVR during TC?
Three PVFs related to PDGFs and VEGFs are encoded by the Drosophila genome (Pvf1/Vegf17E, PVF2/Vegf27Cb, and PVF3/Vegf27Ca; Duchek et al, 2001; Cho et al, 2002). Concerning TC, we have no suggestive data about the stimulus for PVR activation as yet. Does the source of the proposed signal come from tissue neighboring the leading edge, such as squamous tissues (peripodial membranes) or columnar epithelia in imaginal discs or larval tissues? Which PVF serves as a ligand for PVR during TC? Obviously, future studies are needed to explore the extracellular signal(s) triggering PVR activation during TC. Further in vivo studies providing the direct targets and pathways of PVR during TC are also necessary to delineate the role of PVR in TC.
Materials and methods
DNA manipulations
Genetics
Targeted expression of hairpin-loop RNAs (Kennerdell and Carthew, 2000) and λPVR was induced by using the pnr-GAL4 driver line (Calleja et al, 1996). See Supplementary Experimental procedures concerning amorphic or hypomorphic alleles used for genetic interaction experiments.
Cell culture and biochemical analyses
S2 cells were propagated in Schneider's medium (GIBCO) supplemented with 10% FCS (Hyclone), 100 U/ml penicillin, and 100 μg/ml streptomycin (GIBCO). Cells were transfected with 2 μg of construct(s) by using Cellfectin (Invitrogen). Constructs in the pUAST vector were used for cotransfection with pWAGAL4, which expressed GAL4 in S2 cells. For the details of the PDGFRα/PVR-Myc transfectant S2 cell lines, see Supplementary Experimental procedures. Also, see Supplementary Experimental procedures concerning Western analysis, in vitro GEF assay, and immunoprecipitation assay.
dsRNA production and RNAi in Drosophila S2 cells
PCR fragments (700–1000 bp) containing coding sequence of targeted genes were used to produce dsRNA as described (Clemens et al, 2000). For the details, see Supplementary Experimental procedures.
Antibodies
See Supplementary Experimental procedures concerning antibodies used for Western analyses and immunoprecipitation.
Fixation and staining of embryos and larvae
puc expression was monitored by using the puc-lacZ enhancer trap line pucE69 (Martin-Blanco et al, 1998). Embryos were fixed with 8% paraformaldehyde for 20 min at room temperature, devitellinized with heptane/ethanol, and washed with PBS+0.1% Tween 20. Dissected larval imaginal discs were fixed for 2 h in 4% paraformaldehyde on ice and washed with PBS+0.1% Triton X-100. Staining for β-galactosidase activity was performed according to standard protocols. For immunohistochemical staining, dissected larval imaginal discs were fixed for 2 h in PLP at room temperature and washed with PBS+0.1% Triton X-100. Anti-PVR antibody staining was according to a previous paper (Rosin et al, 2004).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Data
Acknowledgments
We thank A Fujita for technical assistance and Drs R Karess, C Desplan, and T Akagi for critical reading and comments on the manuscript. Also, we thank Dr S Kondo for providing the facility for experiments using scanning electron microscopy and Dr R Yagi for his technical help and suggestions during affinity purification of anti-Crk antibody and for confocal laser microscopy, as well as members of the Ueda laboratory for making transgenic flies. We are indebted to Drs A Martinez-Arias, S Hakeda-Suzuki, RG Fehon, N Perrimon, P Rørth, SM Abmayr, and F Karim (Exelixis), and B-Z Shilo for gifts of Drosophila stocks and antibodies. This work was supported by a grant from the program Grants-in-Aid for Specially Promoted Research of the Ministry of Education, Culture, Sports, Science, and Technology of Japan and by a grant from Osaka City. This work was also supported in part by a grant from the Senri Life Science Foundation to SI.
References
- Agnes F, Suzanne M, Noselli S (1999) The Drosophila JNK pathway controls the morphogenesis of imaginal discs during metamorphosis. Development 126: 5453–5462 [DOI] [PubMed] [Google Scholar]
- Albert ML, Kim JI, Birge RB (2000) αvβ5 integrin recruits the CrkII–Dock180–Rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol 2: 899–905 [DOI] [PubMed] [Google Scholar]
- Bishop AL, Hall A (2000) Rho GTPases and their effector proteins. Biochem J 348: 241–255 [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415 [DOI] [PubMed] [Google Scholar]
- Bruckner K, Kockel L, Duchek P, Luque CM, Rørth P, Perrimon N (2004) The PDGF/VEGF receptor controls blood cell survival in Drosophila. Dev Cell 7: 73–84 [DOI] [PubMed] [Google Scholar]
- Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC, Macara IG, Madhani H, Fink GR, Ravichandran KS (2002) Unconventional Rac-GEF activity is mediated through the Dock180–ELMO complex. Nat Cell Biol 4: 574–582 [DOI] [PubMed] [Google Scholar]
- Calleja M, Moreno E, Pelaz S, Morata G (1996) Visualization of gene expression in living adult Drosophila. Science 274: 252–255 [DOI] [PubMed] [Google Scholar]
- Cho NK, Keyes L, Johnson E, Heller J, Ryner L, Karim F, Krasnow MA (2002) Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell 108: 865–876 [DOI] [PubMed] [Google Scholar]
- Clemens JC, Worby CA, Simonson-Leff N, Muda M, Maehama T, Hemmings BA, Dixon JE (2000) Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc Natl Acad Sci USA 97: 6499–6503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchek P, Rørth P (2001) Guidance of cell migration by EGF receptor signaling during Drosophila oogenesis. Science 291: 131–133 [DOI] [PubMed] [Google Scholar]
- Duchek P, Somogyi K, Jekely G, Beccari S, Rørth P (2001) Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell 107: 17–26 [DOI] [PubMed] [Google Scholar]
- Erickson MR, Galletta BJ, Abmayr SM (1997) Drosophila myoblast city encodes a conserved protein that is essential for myoblast fusion, dorsal closure, and cytoskeletal organization. J Cell Biol 138: 589–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez R, Tabarini D, Azpiazu N, Frasch M, Schlessinger J (1995) The Drosophila insulin receptor homolog: a gene essential for embryonic development encodes two receptor isoforms with different signaling potential. EMBO J 14: 3373–3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galletta BJ, Niu XP, Erickson MR, Abmayr SM (1999) Identification of a Drosophila homologue to vertebrate Crk by interaction with MBC. Gene 228: 243–252 [DOI] [PubMed] [Google Scholar]
- Genova JL, Jong S, Camp JT, Fehon RG (2000) Functional analysis of Cdc42 in actin filament assembly, epithelial morphogenesis, and cell signaling during Drosophila development. Dev Biol 221: 181–194 [DOI] [PubMed] [Google Scholar]
- Glise B, Bourbon H, Noselli S (1995) hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell 83: 451–461 [DOI] [PubMed] [Google Scholar]
- Glise B, Noselli S (1997) Coupling of Jun amino-terminal kinase and Decapentaplegic signaling pathways in Drosophila morphogenesis. Genes Dev 11: 1738–1747 [DOI] [PubMed] [Google Scholar]
- Grimsley CM, Kinchen JM, Tosello-Trampont A-C, Brugnera E, Haney LB, Lu M, Chen Q, Klingele D, Hengartner MO, Ravichandran KS (2004) Dock180 and ELMO1 proteins cooperate to promote evolutionarily conserved Rac-dependent cell migration. J Biol Chem 279: 6087–6097 [DOI] [PubMed] [Google Scholar]
- Gumienny TL, Brugnera E, Tosello-Trampont AC, Kinchen JM, Haney LB, Nishiwaki K, Walk SF, Nemergut ME, Macara IG, Francis R, Schedl T, Qin Y, Van Aelst L, Hengartner MO, Ravichandran KS (2001) CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 107: 27–41 [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S, Ng J, Tzu J, Dietzl G, Sun Y, Harms M, Nardine T, Luo L, Dickson BJ (2002) Rac function and regulation during Drosophila development. Nature 416: 438–442 [DOI] [PubMed] [Google Scholar]
- Harden N (2002) Signaling pathways directing the movement and fusion of epithelial sheets: lessons from dorsal closure in Drosophila. Differentiation 70: 181–203 [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, Shibuya M, Kurata T, Matsuda M (1996) DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol 16: 1770–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79: 1283–1316 [DOI] [PubMed] [Google Scholar]
- Hoch RV, Soriano P (2003) Roles of PDGF in animal development. Development 130: 4769–4784 [DOI] [PubMed] [Google Scholar]
- Javelaud D, Laboureau J, Gabison E, Verrecchia F, Mauviel A (2003) Disruption of basal JNK activity differentially affects key fibroblast functions important for wound healing. J Biol Chem 278: 24624–24628 [DOI] [PubMed] [Google Scholar]
- Kennerdell JR, Carthew RW (2000) Heritable gene silencing in Drosophila using double-stranded RNA. Nat Biotechnol 18: 896–898 [DOI] [PubMed] [Google Scholar]
- Kiyokawa E, Hashimoto Y, Kobayashi S, Sugimura H, Kurata T, Matsuda M (1998) Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev 12: 3331–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundra V, Escobedo JA, Kazlauskas A, Kim HK, Rhee SG, Williams LT, Zetter BR (1994) Regulation of chemotaxis by the platelet-derived growth factor receptor-beta. Nature 367: 474–476 [DOI] [PubMed] [Google Scholar]
- Lallemand D, Ham J, Garbay S, Bakiri L, Traincard F, Jeannequin O, Pfarr CM, Yaniv M (1998) Stress-activated protein kinases are negatively regulated by cell density. EMBO J 17: 5615–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Gustafson-Brown C, Hanks SK, Nason K, Arbeit JM, Pogliano K, Wisdom RM, Johnson RS (2003) c-Jun is essential for organization of the epidermal leading edge. Dev Cell 4: 865–877 [DOI] [PubMed] [Google Scholar]
- Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A (1998) puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev 12: 557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Blanco E, Pastor-Pareja JC, Garcia-Bellido A (2000) JNK and decapentaplegic signaling control adhesiveness and cytoskeleton dynamics during thorax closure in Drosophila. Proc Natl Acad Sci USA 97: 7888–7893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan KM, Barrett K, Lu Y, Hu KQ, Vincent S, Settleman J (1998) Myoblast city, the Drosophila homolog of DOCK180/CED-5, is required in a Rac signaling pathway utilized for multiple developmental processes. Genes Dev 12: 3337–3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noselli S (1998) JNK signaling and morphogenesis in Drosophila. Trends Genet 14: 33–38 [DOI] [PubMed] [Google Scholar]
- Noselli S, Agnes F (1999) Roles of the JNK signaling pathway in Drosophila morphogenesis. Curr Opin Genet Dev 9: 466–472 [DOI] [PubMed] [Google Scholar]
- Reddien PW, Horvitz HR (2000) CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat Cell Biol 2: 131–136 [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar JR, Hafen E (1997) Common and distinct roles of DFos and DJun during Drosophila development. Science 278: 669–672 [DOI] [PubMed] [Google Scholar]
- Rosin D, Schejter E, Volk T, Shilo B-Z (2004) Apical accumulation of the Drosophila PDGF/VEGF receptor ligands provides a mechanism for triggering localized actin polymerization. Development 131: 1939–1948 [DOI] [PubMed] [Google Scholar]
- Soriano P (1997) The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 124: 2691–2700 [DOI] [PubMed] [Google Scholar]
- Stronach B, Perrimon N (2002) Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev 16: 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stronach BE, Perrimon N (1999) Stress signaling in Drosophila. Oncogene 18: 6172–6182 [DOI] [PubMed] [Google Scholar]
- Su YC, Treisman JE, Skolnik EY (1998) The Drosophila Ste20-related kinase misshapen is required for embryonic dorsal closure and acts through a JNK MAPK module on an evolutionarily conserved signaling pathway. Genes Dev 12: 2371–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno M, Nishida Y, Adachi-Yamada T (2000) Regulation of JNK by Src during Drosophila development. Science 287: 324–327 [DOI] [PubMed] [Google Scholar]
- Usui K, Simpson P (2000) Cellular basis of the dynamic behavior of the imaginal thoracic discs during Drosophila metamorphosis. Dev Biol 225: 13–25 [DOI] [PubMed] [Google Scholar]
- Wang X, Wu YC, Fadok VA, Lee MC, Gengyo-Ando K, Cheng LC, Ledwich D, Hsu PK, Chen JY, Chou BK, Henson P, Mitani S, Xue D (2003) Cell corpse engulfment mediated by C. elegans phosphatidylserine receptor through CED-5 and CED-12. Science 302: 1563–1566 [DOI] [PubMed] [Google Scholar]
- Wennstrom S, Siegbahn A, Yokote K, Arvidsson AK, Heldin CH, Mori S, Claesson-Welsh L (1994) Membrane ruffling and chemotaxis transduced by the PDGF β-receptor require the binding site for phosphatidylinositol 3′ kinase. Oncogene 9: 651–660 [PubMed] [Google Scholar]
- Wu YC, Horvitz HR (1998) C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature 392: 501–504 [DOI] [PubMed] [Google Scholar]
- Zeitlinger J, Bohmann D (1999) Thorax closure in Drosophila: involvement of Fos and the JNK pathway. Development 126: 3947–3956 [DOI] [PubMed] [Google Scholar]
- Zhou Z, Caron E, Hartwieg E, Hall A, Horvitz HR (2001) The C. elegans PH domain protein CED-12 regulates cytoskeletal reorganization via a Rho/Rac GTPase signaling pathway. Dev Cell 1: 477–489 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Data