

ABSTRACT
Palmitoylation is a reversible, posttranslational modification that helps target proteins to cellular membranes. The alphavirus small membrane proteins 6K and TF have been reported to be palmitoylated and to positively regulate budding. 6K and TF are isoforms that are identical in their N termini but unique in their C termini due to a −1 ribosomal frameshift during translation. In this study, we used cysteine (Cys) mutants to test differential palmitoylation of the Sindbis virus 6K and TF proteins. We modularly mutated the five Cys residues in the identical N termini of 6K and TF, the four additional Cys residues in TF's unique C terminus, or all nine Cys residues in TF. Using these mutants, we determined that TF palmitoylation occurs primarily in the N terminus. In contrast, 6K is not palmitoylated, even on these shared residues. In the C-terminal Cys mutant, TF protein levels increase both in the cell and in the released virion compared to the wild type. In viruses with the N-terminal Cys residues mutated, TF is much less efficiently localized to the plasma membrane, and it is not incorporated into the virion. The three Cys mutants have minor defects in cell culture growth but a high incidence of abnormal particle morphologies compared to the wild-type virus as determined by transmission electron microscopy. We propose a model where the C terminus of TF modulates the palmitoylation of TF at the N terminus, and palmitoylated TF is preferentially trafficked to the plasma membrane for virus budding.
IMPORTANCE Alphaviruses are a reemerging viral cause of arthritogenic disease. Recently, the small 6K and TF proteins of alphaviruses were shown to contribute to virulence in vivo. Nevertheless, a clear understanding of the molecular mechanisms by which either protein acts to promote virus infection is missing. The TF protein is a component of budded virions, and optimal levels of TF correlate positively with wild-type-like particle morphology. In this study, we show that the palmitoylation of TF regulates its localization to the plasma membrane, which is the site of alphavirus budding. Mutants in which TF is not palmitoylated display drastically reduced plasma membrane localization, which effectively prevents TF from participating in budding or being incorporated into virus particles. Investigation of the regulation of TF will aid current efforts in the alphavirus field searching for approaches to mitigate alphaviral disease in humans.
KEYWORDS: protein localization, virus budding, palmitoylation, 6K, TF, alphavirus
INTRODUCTION
Palmitoylation is the covalent attachment of a 16-carbon fatty acid via a thioester bond primarily to a cysteine residue (Cys) side chain of a protein. The fatty acid moiety can be dynamically added and removed by palmitoyl transferases and protein thioesterases, respectively (1). The increased hydrophobicity resulting from palmitoylation promotes the association of a protein with lipid membranes, allowing distinct and new subcellular localizations (1, 2). Cellular proteins use palmitoylation as a mechanism to regulate their localization and function (3–6). Similarly, viruses use palmitoylation as a means to efficiently localize viral proteins to specific cellular membranes during replication, assembly, and budding (7).
Enveloped viruses acquire their lipid membrane from the host during budding. Viruses bud at different cellular membranes, including the endoplasmic reticulum (ER) (flaviviruses and hepaciviruses) (8–11), the ER-Golgi intermediate complex (ERGIC) (coronaviruses) (12–14), and the plasma membrane (alphaviruses, retroviruses, and paramyxoviruses) (15–19). Regardless of where in the cell budding occurs, this process requires spatial and temporal regulation of viral and host factors to ensure that their concentration and localization within the cell will promote optimal assembly and budding. All the alphavirus structural proteins involved in budding, except capsid (CP), are palmitoylated as they traffic through the host cell secretory system to the plasma membrane, where budding occurs (20–23).
Alphaviruses such as Sindbis virus (SINV) are spherical, enveloped viruses with a positive-sense RNA genome. The viral genome contains two open reading frames (ORFs), and the second ORF encodes the structural polyprotein CP-E3-E2-6K-E1. In 2008, Firth et al. discovered that there was a heptanucleotide slip site, UUUUUUA, in the gene encoding the 6K protein, which caused the ribosome to shift to the −1 reading frame during translation (24). As a result, the polyprotein CP-E3-E2-TF is translated, and the E1 protein is not translated. The 6K and TF (for transframe) proteins have identical N-terminal domains but unique C-terminal domains. The frequency of translational frameshifting within the 6K gene varies among alphaviruses and has been reported to be ∼10:1 for Sindbis virus (24, 25). Cell lysates contain larger amounts of the 6K protein than of the TF protein (24), yet TF is predominantly incorporated into released virus particles (24, 26). The balance between 6K and TF protein translation presents an interesting conundrum with respect to alphavirus assembly. For optimal virus assembly, E1 and E2 heterodimerize, and heterodimers trimerize before being transported to the plasma membrane via the secretory pathway (27, 28). During frameshifting and TF translation, E1 translation is abrogated, altering the E2-to-E1 ratio, which can have consequences for subsequent spike assembly. It is not known if TF relies on other viral proteins to be transported to the plasma membrane or what regulates its incorporation into virus particles.
Alphavirus virions have T=4 icosahedral symmetry and contain three distinct layers (29). There is an inner nucleocapsid core consisting of 240 copies of CP and 1 copy of the genome, a lipid bilayer, and 240 copies of the spike proteins E2 and E1 on the particle surface. Because of the small size of the 6K and TF proteins (6 to 10 kDa) and their low copy number (10 to 30 copies) within the viral particle (20, 24, 30, 31), neither the 6K nor the TF protein has been identified in the virion structure, even in cryo-electron microscopy (cryo-EM) reconstructions at a subnanometer resolution (32–36).
In tissue culture, recombinant viruses lacking both 6K and TF (Δ6K) or only TF (6K only or ΔTF) or unpalmitoylated mutants negatively impact the infectious and total virus output from infected host cells compared to the wild type (WT) (20, 23, 26, 37–42). In addition, some purified particles in the mutants were multicored (20, 37, 43). The phenotypes of 6K and TF mutants are more severe when investigating pathogenesis in vivo. Snyder and colleagues demonstrated that mice survived when injected with a mutated TF strain, whereas injection of a WT strain was lethal (26). Taylor et al. demonstrated that the in-frame deletion of the entire gene encoding 6K and TF generated a nonvirulent strain of Ross River virus (RRV) suitable for vaccination against multiple alphaviruses in a mouse model (41). A Δ6K Chikungunya virus also serves as an effective vaccination in mouse models (83). These studies highlight the fact that the 6K and TF proteins play an important role in virus infection at the organismal level.
In this study, we used a panel of Cys mutants to demonstrate that WT TF protein palmitoylation occurs on the N-terminal Cys residues. In contrast, the 6K protein does not appear to be palmitoylated. The Cys mutants display altered levels of both the 6K and TF proteins inside infected cells compared to the WT. We found that palmitoylation directs the localization of the Sindbis virus TF protein to the plasma membrane and, indirectly, its subsequent incorporation into the particle during budding. In a tissue culture system, the Cys mutants were able to produce infectious virus. However, examination of the virions by transmission electron microscopy (TEM) showed that the Cys mutants had morphological abnormalities. Finally, we propose a model for the role of TF palmitoylation in alphavirus budding.
RESULTS
TF Cys mutations alter its steady-state levels.
We aligned the 6K and TF protein sequences to identify cysteines available for posttranslational modification. An amino acid alignment of 6K proteins reveals a pattern of conserved cysteines upstream of the frameshift slip site (Fig. 1A). The amino acid alignment of TF proteins additionally shows that the unique TF region contains nonconserved Cys residues (Fig. 1B). The atomic structures of 6K and TF are not known, but the topology of these two proteins has been deduced from hydrophobicity plots and the known membrane orientation of the structural polyprotein. The 6K protein is thought to be composed of two transmembrane domains connected by a short cytoplasmic loop. The first transmembrane domain and loop residues of 6K and TF are identical; however, TF has a unique extended cytoplasmic loop (Fig. 1C). We hypothesized that TF might be palmitoylated on the Cys residues in its cytoplasmic C terminus, anchoring it to the membrane. To test this, we constructed three Cys mutants, called 9C, 4C, and 5C, by replacing the Cys residues with Ala or Ser (Fig. 1D). In 9C, all nine Cys residues in the Sindbis virus TF protein were mutated; in 4C, the four Cys residues in the TF-unique C terminus were mutated; and in 5C, the five shared Cys residues in the portion of 6K and TF prior to the slip site were mutated.
FIG 1.

The 6K and TF proteins and their Cys residues. (A and B) Protein sequences were aligned by using ClustalOmega and manually adjusted around the slip site (boldface type). All residues N terminal to the slip site are identical in 6K and TF, while residues C terminal to the slip site are unique to each protein. All Cys residues theoretically available for palmitoylation are highlighted in green. The protein length is given in amino acids. Underlined residues indicate N-terminal or C-terminal antibody epitopes. Above the sequence, colored bars correspond to regions magnified in panel D. The percent frameshifting rate (%FS), as measured by Chung et al. in a cell-based dual-luciferase assay (indicated by *), is shown at the far right of panel B (25). Abbreviations: SINV, Sindbis virus; WEEV, Western equine encephalitis virus; CHIKV, Chikungunya virus; RRV, Ross River virus; SFV, Semliki Forest virus; EEEV, Eastern equine encephalitis virus; VEEV, Venezuelan equine encephalitis virus; AURAV, Aura virus; MIDV, Middelburg virus; EILV, Eilat virus; SESV, Southern elephant seal virus; SDV, sleeping disease virus. (C) Schematic of the theoretical membrane topology of the 6K and TF proteins. The blue regions are sequences shared between both 6K and TF isoforms. The dark blue and red regions are unique to the C terminus of 6K and TF, respectively. (D) The mutations made for constructing the TF Cys mutants 9C, 4C, and 5C are demonstrated below the WT sequence. Amino acid (aa) numbering across the top is for WT Sindbis virus TF. The blue regions on the left are sequences shared between both 6K and TF isoforms. The red regions to the right are unique to the C terminus of TF.
We probed the Cys mutants for steady-state 6K and TF protein levels in infected BHK cells (Fig. 2A). By using an antibody that recognizes a unique C-terminal TF epitope, no TF was detected in mock-infected cells or cells infected with Δ6K or 6K*, which translates 6K but not TF. However, at 8 and 16 h postinfection (hpi), TF was detected in the WT and the Cys mutants. At both times, the quantities and migration pattern of TF in the mutants were different from those of the WT. In the WT, TF migrates as a doublet with bands of similar intensities at all times. The 9C and 4C mutants, in particular, accumulate higher levels of the TF protein than does the WT, although the CP protein levels in the same samples are very similar. The 9C TF band migrates at a lower apparent molecular mass, as expected for a protein that is no longer palmitoylated. The 4C TF band is much more abundant than that of the WT but migrates at a position similar to that of the WT, suggesting a similar number of modifications. The 5C TF protein band migrates at the same position as 9C, suggesting that it is also not palmitoylated. A single palmitoyl moiety is ∼250 Da. While the shifts observed in these blots are greater than expected for palmitoylation based on molecular mass calculations alone, other small posttranslational modifications, such as phosphorylation, are commonly detected as shifted bands by gel electrophoresis (44, 45).
FIG 2.
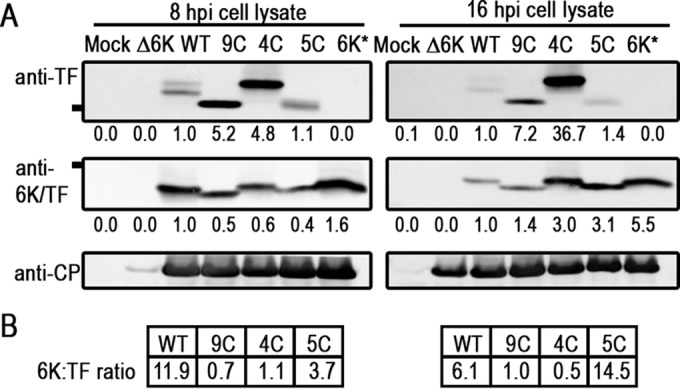
TF protein expression inside infected cells. (A) Lysates from infected BHK cells were analyzed for TF and 6K protein expression by Western blotting at 8 and 16 hpi, using the virus CP protein as a loading control. Mock and Δ6K are controls with no signal using anti-TF or anti-6K/TF antibodies. 6K* is a 6K-only mutant that does not translate TF. The line at the left indicates the position of the 10-kDa reference band. A blot representative of at least three repetitions is shown. (B) Ratio analysis of 6K and TF expression levels using values normalized to values for CP loading in each lane.
As shown in Fig. 2A, the 6K protein was also detected by Western blotting with antibodies directed against an N-terminal epitope called anti-6K/TF. The anti-6K/TF antibody poorly detects the TF protein but gives a strong signal for 6K. The 6K protein was detected in WT and all mutant infections but not in mock or Δ6K infections (Fig. 2A). At 8 hpi, the Cys mutants displayed lower levels of 6K than those of the WT, but at 16 hpi, 6K levels increased compared to those of the WT. Additionally, the 6K proteins in 9C and 5C infections migrated at a lower apparent molecular mass than that of WT 6K, suggesting that they might not possess a posttranslational modification that the WT, 4C, and 6K-only 6K proteins do.
The ratio of total 6K to TF protein in the cell (Fig. 2B) calculated here for the WT strain has a ratio close to 10:1, consistent with what has been reported for both in vivo calculations and in vitro assays (24, 25). The 9C and 4C mutants have a ratio of 6K to TF much closer to 1, and 5C has an elevated level of 6K compared to that of TF. Our data support the hypothesis that 6K and TF are potentially posttranslationally modified, that this modification occurs on the N-terminal Cys residues present and shared between the WT and 4C, and that the posttranslational modification state of TF and 6K may influence the amount of these proteins present in the cell.
Only the N-terminal Cys residues of TF are palmitoylated.
Having observed migration patterns in the Cys mutants that were consistent with reduced modifications, we next directly assayed the palmitoylation status of the virus proteins using click chemistry (46, 47). Infected cells were incubated with palmitic azide, which serves as a substrate for the cellular palmitoyl transferases, from 12 to 16 hpi. Upon harvesting, the lysates were split. One part of the lysate remained untreated, while another aliquot was further processed to react with a biotin-alkyne, covalently “clicking” it to the palmitic azide functional group. This method biotinylates all viral and cellular proteins that were palmitoylated during the 4-h labeling period. Next, the untreated and biotinylated lysate proteins were separated by SDS-PAGE and probed separately for the biotin-palmitic azide modification and viral proteins via Western blotting. As shown in Fig. 3A, a significant enrichment of palmitoylated protein at the position of virus glycoproteins E1 and E2, which are known to be palmitoylated, was observed in virus-infected samples (21, 22). For the CP protein which is known not to be palmitoylated, there was no enrichment of biotinylated protein. These two results demonstrate that this method specifically labels palmitoylated proteins. A comparison of the left and right panels of Fig. 3A indirectly addresses TF palmitoylation and suggests that palmitoylation of TF occurs in the shared N-terminal residues common between the WT and 4C rather than on the C-terminal Cys residue. In the biotinylated samples, there are several palmitoylated protein bands where TF migrates in the gel. The uppermost band is present in all samples, including the mock-infected sample, and therefore represents a palmitoylated host protein. The band indicated by an arrow at the bottom is present in all infected cell samples, including the 6K* sample, which does not produce TF (Fig. 2A), strongly suggesting that this is a host protein that is palmitoylated upon virus infection. The bands labeled with dots indicate where the WT (two bands) and 4C TF proteins are located (Fig. 3A), indicating that both of these proteins are palmitoylated. In contrast, the biotin signal in the region of the gel where 6K migrates does not correspond to the migration patterns of 6K (Fig. 3A). In 9C and 5C, the 6K protein has no Cys residues available for palmitoylation. In the WT and the 4C and 6K* mutants, there are five Cys residues in the protein available for palmitoylation. If the signal in the biotin blot (Fig. 3A, left) corresponds to 6K, there should be a higher-intensity band for WT, 4C, and 6K* infections but not for mock, 9C, or 5C infection. We observe that, in fact, the 5C lane has a signal in all the same positions as those for the WT and the other samples. Furthermore, the biotin signal intensity for the WT and the 4C and 5C mutants is the same, suggesting that if 6K is palmitoylated, it is palmitoylated at very low levels (Fig. 3A, left), although 6K is present and detectable in these lysates (right). Identical experiments were performed at 8 hpi, with similar results, except that the overall signal intensity was lower (data not shown).
FIG 3.
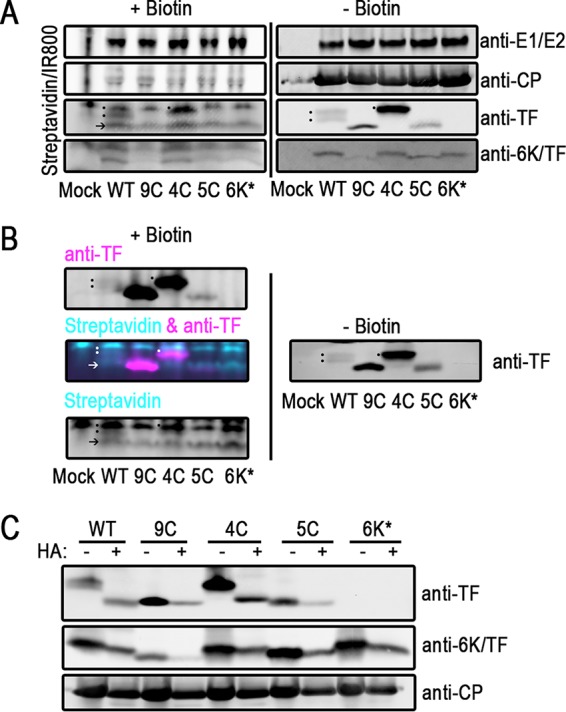
TF is palmitoylated on Cys residues in its shared N terminus. (A and B) Infected BHK cells were labeled with palmitic azide from 12 to 16 hpi. After harvesting of the cell lysate, proteins modified with palmitic azide were labeled via click chemistry with biotin. Proteins were separated by SDS-PAGE and transferred onto blotting paper. Biotinylated proteins were detected with fluorophore-conjugated streptavidin. Virus proteins were detected with specific antibodies. (A) On the same gel, untreated and biotinylated lysates were separated and detected. At the right are untreated virus proteins. On the left are biotin-conjugated palmitoylated proteins from the same lysates. The dots denote the palmitoylation signal from TF. The arrow indicates a host protein that is palmitoylated in virus infections, also indicated by an arrow in panel B (bottom). These experiments were repeated at least three times. (B) Click-modified samples were probed by using dual-color detection. Cyan indicates biotinylated protein, and magenta indicates virus protein. At the top left is the TF channel alone. At the bottom left is the streptavidin channel alone. At the middle left are overlaid TF and streptavidin channels. At the right, the same samples that were left untreated were electrophoresed on the same gel and detected only by anti-TF antibody. These experiments were repeated at least three times. (C) Infected BHK lysates were treated with hydroxylamine (HA) to remove palmitoyl moieties. Virus proteins were detected by Western blotting. One representative blot from at least three repetitions is shown.
To confirm that the palmitoylation-biotin signal observed was truly comigrating with TF, we used dual-color detection in our Western blot analysis to probe for palmitoylation (with biotin-streptavidin) and TF simultaneously (Fig. 3B). These results agree with the conclusions from the single-labeling experiment shown in Fig. 3A. In the WT and the 4C mutant, there is a biotin signal that exactly corresponds with the position of TF migration in the dual-color channel overlay. As a control, we show that the TF channel signal is the same when detected in a dual-color blot (Fig. 3B, top left) and when detected in the same untreated sample (Fig. 3B, right), although precipitated protein bands are not as sharp in Fig. 3B, left.
As a complementary method, we used hydroxylamine treatment to determine if the migration differences were due to palmitoylation (48, 49). Hydroxylamine catalyzes the hydrolysis of the thioester linkages between the Cys residue and the palmitate group. If the WT and the 4C mutant are palmitoylated, then treatment with hydroxylamine will result in a downward shift in the migration of 6K and TF on a gel. When WT- and 4C-infected lysates were treated with hydroxylamine, the TF protein bands shifted their migration to a lower molecular weight, confirming that they were palmitoylated (Fig. 3C). In contrast, the 9C and 5C TF bands, which have no Cys residue in their N terminus, did not shift upon hydroxylamine treatment, indicating that there is no palmitoylation in the C terminus of TF. Furthermore, the hydroxylamine-treated WT and 4C TF bands shifted to the same mobility as that of the 9C and 5C TF bands, confirming that palmitoylation alone accounts for the observed migration patterns of the WT and 4C TF proteins. Interestingly, we did not detect any lower-molecular-weight bands that might correspond to the unmodified TF protein in the WT or the 4C mutant without hydroxylamine treatment, suggesting that all the detectable TF protein in a native infection is palmitoylated. Taken together, our data show that TF is palmitoylated only on the Cys residue in the N terminus.
In contrast to TF, the 6K protein showed no sensitivity to hydroxylamine, suggesting that 6K may not be palmitoylated in Sindbis virus (Fig. 3C), consistent with our results using the palmitate-biotin labeling method (Fig. 3A). Although palmitoylation does not account for the migration differences observed in the 6K Cys mutants, the amino acid composition of low-molecular-weight proteins strongly influences their migration and may explain what we observe here.
Previous work showed that in Sindbis virus, residues C35, C36, and C39 in 6K are palmitoylated in chicken embryo fibroblasts (20, 23). The authors of that study also identified another smaller 4.2K band that was not palmitoylated. After the identification of TF, it was initially suggested that the 6K protein observed in palmitoylation studies was most likely TF and that the 4.2K protein was 6K. Our work is consistent with those previous studies suggesting that TF is palmitoylated at residues at the N terminus (which includes C35, C36, and C39) and that 6K has little to no palmitoylation in BHK cells.
Protein turnover does not account for higher levels of 6K or TF.
The observation that the Cys mutants have different steady-state levels of 6K and TF over time (Fig. 2A) raised the question of whether this was caused by a longer lifetime of the proteins. We reasoned that in the 4C mutant, which had higher levels of TF and 6K, each protein would have a longer half-life than that of the WT. Attempts to use [35S]Met-Cys labeling resulted in a low signal-to-noise ratio, preventing the TF bands from being accurately quantified above the host protein background level. Instead, we monitored protein turnover after treatment of infected cells with cycloheximide, an inhibitor of translation at the level of the ribosome.
6K and TF protein levels during cycloheximide treatment were monitored by Western blotting starting at 10 hpi (Fig. 4A). While absolute values of quantification differed between experiments, the general trends remained the same. When infected cells were treated with only the vehicle control, WT and 4C TF protein signals increased over 6 h (Fig. 4B, left). Both WT and 4C TF protein levels increased, but the amount of 4C TF was larger at all times measured, consistent with the data shown in Fig. 2A. In contrast, but consistent with the data shown in Fig. 2A, the 6K protein signal in the WT did not change dramatically over the period of treatment with only the dimethyl sulfoxide (DMSO) vehicle but increased slightly in 4C (Fig. 4B, right). In the presence of cycloheximide, TF and 6K protein levels in 4C and the WT decreased at similar rates, suggesting that the lifetimes of TF in the WT and the 4C mutant were similar (Fig. 4C). The decreasing levels of TF are most likely explained by degradation via the proteasome or proteases, but a decrease in levels due to budding cannot be ruled out. However, the level of the CP protein, which is budded from the cell at a higher ratio per particle, did not substantially decrease within the same time frame (Fig. 5A). We conclude that neither 6K nor TF has a significantly different turnover rate in the Cys mutant strain 4C compared to that in the WT, and therefore, protein lifetime does not explain the observed differences in steady-state TF levels.
FIG 4.

Protein turnover does not account for higher levels of 6K or TF. Infected BHK cells were treated with the DMSO vehicle (−) or 50 μg/ml cycloheximide (CHX) (+) at 10 hpi for up to 6 h. (A) Paired treated and untreated samples were lysed and probed by Western blotting for CP, TF, and 6K. Data from one representative experiment of three repetitions are shown. (B and C) TF and 6K protein levels were quantified over time (compared to WT levels of TF and 6K at time zero) after treatment with the DMSO vehicle (B) and CHX (C).
FIG 5.
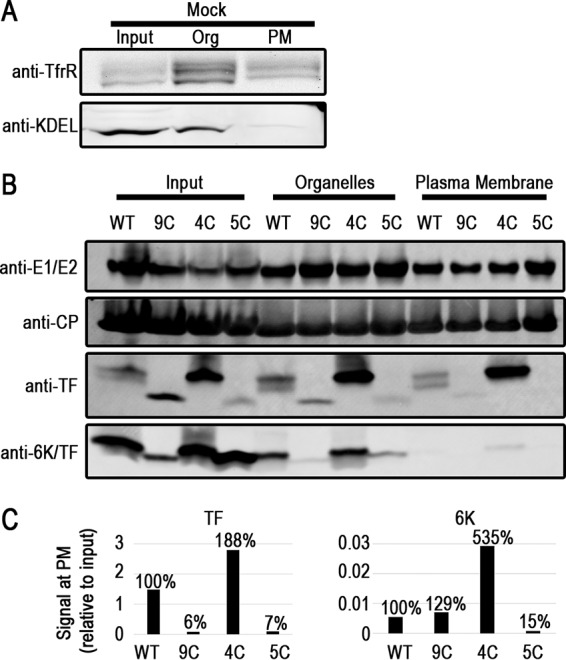
Differential TF subcellular localization in the Cys mutants. BHK cells were fractionated to separate subcellular compartments with a commercial kit. (A) Western blots on mock-infected cell lysates probing for a plasma membrane (PM) protein, the transferrin receptor (TfrR), and proteins containing KDEL, a marker for the endoplasmic reticulum. Org, organelles. (B) Infected-cell fractions at 16 hpi were probed by Western blotting for virus proteins. Similar results were obtained at least three times; one representative fractionation blot is shown. (C) Quantification of 6K and TF in the blot shown in panel B. The bars represent the signal in the plasma membrane fractions compared to the amount of input protein in that sample. Percentages represent localization efficiencies relative to the WT.
Localization of TF to the plasma membrane is dependent on its N-terminal palmitoylation.
Palmitoylation is a posttranslational modification usually invoked as a mechanism for precise control over protein localization to distinct subcellular membranes. We hypothesized that the palmitoylation state of the TF protein would provide a physical basis for the subcellular localizations that were previously observed by using immunofluorescence (24, 26, 41). The antibodies used in this study do not show specificity toward TF and/or 6K in an immunofluorescence assay (data not shown); therefore, we pursued subcellular localization by an alternative method. We separated membrane proteins derived from internal organelles and the plasma membrane proteins from each other and the rest of the intracellular soluble proteins. We verified the efficacy of the separation technique using two subcellular markers in uninfected BHK cells (Fig. 5A). The transferrin receptor purifies in organelle membranes, where it is synthesized and trafficked, and at the plasma membrane, where it functions as an iron uptake receptor. The KDEL signal, a retrieval motif found in ER-resident proteins, is enriched in the organellar fraction but not in the plasma membrane fraction.
Using the same separation method, we fractionated infected BHK cell lysates at 8 and 16 hpi, and at both times, WT TF reached the plasma membrane (Fig. 5B). Notably, the WT TF doublet was maintained both in the organelle membrane protein fraction and at the plasma membrane. Densitometry analysis of 4C TF demonstrates that the protein is present in the plasma membrane at higher levels than in the WT, consistent with its overall elevated levels in the cell (Fig. 2A). 9C and 5C TFs, on the other hand, were detected at very low levels at the plasma membrane despite being present in the organelle membrane fraction. When we monitored the localization of the other structural proteins, both CP and the glycoproteins E1 and E2 were observed at the plasma membrane, where budding occurs. In contrast to TF, though, we did not detect appreciable amounts of 6K at the plasma membrane in the WT or any of the Cys mutants, with the highest level of detection being found for 4C 6K. These data support our hypothesis that there is a correlation between the palmitoylation status of TF and 6K and subsequent plasma membrane localization.
Composition and morphology defects in Cys mutant virus particles do not influence infectivity.
Having seen different levels of the TF protein at the plasma membrane in the WT and Cys mutants, as shown in Fig. 5, we next characterized the impact of the Cys mutations on released particles. Purified virions viewed by TEM were spherical and had similar diameters (Fig. 6A). Nevertheless, upon closer inspection, we saw that the Cys mutants demonstrated a markedly higher percentage of abnormal particles than did the WT (Fig. 6A, arrows, and B). The main defect observed was striking internal staining indicative of particles that had compromised structural integrity.
FIG 6.

Cys mutant virus particle morphology by TEM. (A) Released virions isolated from infected BHK cells by a low-speed spin and observed via TEM. Arrowheads indicate particles with abnormal morphology. Representative images from three experiments are shown. Bars, 100 nm. (B) Quantification by blind counting of virus morphology visualized by TEM, as described in Materials and Methods. (C) Virus proteins from purified virions were detected by Western blotting. (D) Growth curves of the TF Cys mutants in BHK cells. Determination of the growth curves for each virus strain was performed at least twice. A representative graph with two biological replicates is shown. Particle-to-PFU ratios measured for WT and Cys mutant viruses are also displayed.
To determine if the particle defects observed by TEM could be a result of differences in protein compositions between the WT and Cys mutants, we purified the virus and probed for the glycoproteins, CP, TF, and 6K. The incorporation of the structural proteins E1/E2 and CP was relatively unaffected in the Cys mutants compared to the WT, as determined by Western blotting (Fig. 6C). The TF protein, but not the 6K protein, was detected in virions by Western blotting as well (Fig. 6C); mass spectrometry analyses gave similar results (data not shown). The pattern of TF incorporation into particles reflects what was seen for TF levels at the plasma membrane (Fig. 5). WT TF was present at the plasma membrane and in the virion. 4C TF was present at even higher levels at the plasma membrane and in the virion. The 5C and 9C TF proteins were not present at high levels at the cell surface, nor were they detectable in virions. Our data suggest that the palmitoylation state of TF modulates its transport to the plasma membrane (Fig. 5), thereby indirectly affecting the amount of TF that is incorporated into the released virion particle (Fig. 6C).
While the particle morphologies and compositions of the WT and the Cys mutants are different, the viruses do not display drastic differences in infectivity. We measured the output of infectious virus over time in BHK cells (Fig. 6D). The 9C and 4C mutants produced ∼1 log unit less infectious virus than the WT at all times. The 5C mutant produced ∼2 log units less infectious particles than the WT until 12 hpi, and by 24 h, when all the host cells were dead, 5C produced the same number of infectious particles as the WT. The Cys mutant phenotypes are not as severe as the growth phenotype of Δ6K. Furthermore, the Cys mutants have a particle-to-PFU ratio that is essentially the same as that of the WT (Fig. 6D). The particle morphology and composition of the Cys mutants are different from those of the WT virus, but neither their growth nor their infectivity, as observed in a tissue culture system, is severely reduced.
DISCUSSION
Palmitoylation is a targeting mechanism used by virus proteins involved in budding.
To transit successfully from cell to cell, viruses very precisely target their assembly proteins within the cell to build a new virion. For enveloped viruses, both cytoplasmic and transmembrane structural proteins use palmitoylation as a mechanism to achieve or enhance membrane association during budding. Palmitoylation of influenza virus hemagglutinin, a transmembrane protein, increases its association with membrane microdomains, where it interacts with the M2 protein during budding (50–52). In herpes simplex virus, the tegument protein UL51 relies on palmitoylation for association with the Golgi membrane, where secondary envelopment occurs (53, 54). In hepatitis C virus, palmitoylation in the core protein that encapsulates the genome has been reported to increase its association with ER membranes, where budding occurs (55).
Several families of enveloped viruses contain proteins that are not required for budding but whose presence greatly enhances it. Characteristically, these proteins are <20 kDa and are transmembrane or membrane associated. Examples include Vpu in HIV (56–58), M2 in influenza virus (59–61), E in coronaviruses (14, 62, 63), and 6K/TF in alphaviruses (20, 23). Of these, M2 (64, 65), E (66), and TF (20, 23) are also palmitoylated, suggesting that posttranslational modification might be important for either cellular localization, function to enhance budding, or both. However, in currently documented cases, palmitoylation of the M2 (67) and E (68) proteins is not required for proper localization. Palmitoylation of M2 enhances its function in overall influenza virus maturation and exit (59, 61). Palmitoylation of E enhances the budding of coronaviruses from the Golgi apparatus (66, 69). Our work here demonstrates that the palmitoylation of TF, in contrast to that of M2 and E, is important for targeting the TF protein to the plasma membrane (Fig. 5) while also influencing optimal particle assembly (Fig. 6).
Model where palmitoylation regulates TF localization, controlling its participation in budding.
A driving force behind this study was to characterize the regulatory mechanisms governing the functions of proteins that participate in budding. All sequenced alphavirus species encode the small membrane protein isoforms 6K and TF, which are palmitoylated and positively affect budding (70, 71). Initially, we hypothesized that alternative palmitoylation states between 6K and TF would provide a physical explanation for how the TF protein, and not 6K, is usually incorporated into alphavirus particles at low stoichiometric levels (20, 24, 26, 30, 31). However, our data suggest that palmitoylation regulates intracellular localization rather than virus particle incorporation during budding (Fig. 5 and 6).
One mechanism supported by our data is that palmitoylation in the N terminus renders TF competent to participate in virion assembly by localizing it to the site of budding (Fig. 7). The high level of TF palmitoylation results in its translocation to the plasma membrane, while 6K, with its lower palmitoylation levels, remains at the interior of the host cell. It is interesting that in the WT and the 4C mutant, the Cys residues that are palmitoylated on the TF protein are also Cys residues that are present in the 6K protein, yet any palmitoylation on the 6K residues occurs at very low levels if at all. This suggests that the C terminus of TF has a role in promoting the regulated addition of palmitoylation, as shown by the two stable forms of palmitoylated WT TF proteins. When the C-terminal regulatory domain is disrupted in its Cys residues, as occurs in 4C, the addition of palmitoylation is not regulated, resulting in one stable population of palmitoylated TF. In the WT and the 4C mutant, TF is shuttled to the plasma membrane, because N-terminal Cys residues are palmitoylated, and TF can then participate in virus budding. In 5C, the C terminus can still theoretically regulate palmitoylation levels, but there are no Cys residues to be modified, so 5C is not transported to the plasma membrane. In 9C, even though the C terminus may not regulate palmitoylation levels, because there are no Cys residues, no palmitoylation and minimal plasma membrane localization occur. When TF is not located at the plasma membrane, it does not participate in budding. From this study, we propose that in Sindbis virus, the Cys residues play a role in the regulatory function of the C terminus; however, in other alphaviruses lacking these C-terminal Cys residues, there are likely other features of the cytoplasmic tail or alternate regulatory mechanisms that govern protein modification status and localization.
FIG 7.

Model for regulation of the participation of palmitoylated TF in alphavirus assembly. TF is targeted to the site of budding via a 2-step regulatory mechanism. First, the C-terminal domain regulates the palmitoylation state of TF. The conserved Cys residues in the N-terminal shared cytoplasmic region are palmitoylated. Second, palmitoylation on TF targets the protein to the plasma membrane, where it participates in the assembly and budding of WT particles. When the C-terminal domain lacks the Cys residues, TF is still targeted to the plasma membrane by N-terminal palmitoylation, as in 4C. When palmitoylation is missing as in 5C and 9C, TF is not targeted to the plasma membrane efficiently.
Another possible role for the TF C-terminal domain is self-feedback that regulates its own frameshifting rate. By using a plasmid-based reporter system, Chung et al. demonstrated that the secondary structure of RNA downstream of the slip site influences frameshifting rates (25). The mechanism controlling TF synthesis can be investigated by using C-terminal mutants with altered levels of TF.
Palmitoylation levels differentiate the roles of 6K and TF in budding.
The original studies that investigated the 6K protein and its alternative palmitoylated form (called 4.2K and 6K) were reported in the early 1990s (20, 23). Since Firth et al. demonstrated that TF is in fact a different protein than 6K, it has been inferred (and we agree) that the 4.2K protein band corresponds to the 6K protein and that the 6K protein band corresponds to the TF protein (24). Gaedigk-Nitschko et al. found that their 4.2K protein was not palmitoylated and did not shift to a lower-molecular-weight band upon hydroxylamine treatment, consistent with our findings for the 6K protein. Furthermore, Gaedigk-Nitschko et al. found that their 6K band was palmitoylated and sensitive to hydroxylamine, consistent with our findings for the TF protein. The same group, using [3H]palmitic acid, showed that their 6K band (the TF protein) in virions is palmitoylated (20). In studies of the specific residues that were palmitoylated, one of the viruses was mutated at Cys35 and Cys36, and another virus was mutated at Cys39, both of which are a subset of the 5C mutant described here (23). Those researchers saw that in the double mutant, C35S C36S, there were aberrant virus particle morphologies. It is important to note that the presence of TF (which has 4 additional Cys residues) was not known at the time of those studies. Our mutants account for the presence of the additional Cys residues found in TF.
One reason why studies on 6K and TF have been difficult to pursue for other alphaviruses is that there is a lack of tools to detect such a low-abundance protein. Even by using the specific antibodies generated for this study, small proteins like 6K and TF are challenging to resolve by SDS-PAGE. The gel composition strongly influences the number and resolution of the bands. In the past, 6K was best resolved on high-percentage-acrylamide gels with urea (23). In this study, we detected the WT TF protein as a sharp palmitoylated doublet using Tricine gradient gels (see Materials and Methods). The TF protein is present as two populations of differentially palmitoylated proteins. The TF doublet is maintained even in the presence of reducing agent, ruling out alternate disulfide bonding between Cys residues (data not shown). The existence of two pools of functionally distinct small membrane proteins has been noted for the coronavirus E protein (72). There are clearly detectable shifts in the size of TF upon mutation of the Cys residues, indicating that the presence of palmitoylation, and, likely, the specific amino acid composition, greatly affects its migration in gels. The 4C TF band is not a clear doublet but appears to correspond to the upper (more modified) WT band. The 5C and 9C TF bands represent completely unmodified proteins. While it is possible that the TF protein has another posttranslational modification, we note that previous studies probed for but did not detect any glycosylation or phosphorylation on 6K or TF (23, 73).
6K migrates as a single band in the WT and not a doublet like TF. The size of the 6K protein also becomes smaller in the 5C and 9C mutants. There is no clear signal for modified 6K with palmitic azide labeling (Fig. 3A), nor does the protein size shift to a lower mobility with hydroxylamine treatment (Fig. 3C). It is also possible that levels of palmitoylated 6K are transient or cell type dependent and not in high abundance in our experimental system. The size shift could be due to an unidentified posttranslational modification or the amino acid composition causing differential migration. From this work, it is clear that the 6K and TF proteins localize differently, with TF localizing to the plasma membrane and in a position to participate in budding and 6K being internal, where it functions in an unknown fashion to influence the cellular state to promote budding.
Future directions for delineating the roles of 6K and TF in alphavirus budding.
All studies using tissue culture reveal only small growth defects in 6K and TF mutants, but in vivo experiments using mouse models showed that 6K and TF mutants cause disease that is less severe than that caused by the WT (26, 41), emphasizing the important role that these proteins play in virus infection at the organismal level. By corollary, we might expect that the morphological defects in Cys mutant virions will impact infection in an animal. Studying the role of TF during infection at the molecular level will help draw mechanistic connections to the phenotypes observed at the organismal level. While TF is a minor component of the budded virus, here we show that both its palmitoylated form and WT incorporation levels are important for proper virus assembly. Investigations of the molecular role of TF in released virions, cell localization studies to determine how alphaviruses temporally and spatially regulate the production and modification status of TF, and identification of the differences in TF regulation in vertebrate and invertebrate host cells will help us understand how the failure of TF to efficiently traffic to the plasma membrane helps drive budding. Further studies of the 6K protein will also be needed to understand the role that it plays in promoting budding from the interior of the infected host cell.
MATERIALS AND METHODS
Cells and virus.
All tissue culture was performed in the baby hamster kidney line BHK-21, here referred to as BHK cells. Cells were passaged in 1× minimum essential medium (MEM) supplemented with nonessential amino acids, penicillin-streptomycin, and l-glutamine in 10% fetal bovine serum (Corning Cellgro, Manassas, VA) and grown in a 37°C incubator in a controlled-humidity environment with 5% CO2.
The viruses in this study were all derivatives of the Sindbis virus TE12 infectious cDNA clone (74). 6K* is a virus with three silent nucleotide mutations at the frameshift slip site (U UUU UUA to G UUC CUA) that abolish frameshifting and, hence, TF production. The Δ6K virus is a clean deletion of the 6K gene between E2 and E1. Cys mutant strains were generated according to recommendations for the QuikChange Lightning site-directed mutagenesis kit (Agilent, Santa Clara, CA). The three Cys mutant strains are 9C, with mutations of all 9 Cys residues to Ser residues; 4C, with mutations of the 4 unique TF Cys residues (C46, C59, C62, and C65) to Ser residues; and 5C, with mutations in the 6K/TF shared coding sequence region at C23 to Ala and at C35, C36, C38, and C39 to Ser. Due to overlapping reading frames, TF C46S causes an A46L mutation in the 6K reading frame, TF C59S causes an A4L mutation in the E1 reading frame, and TF C65S causes a V10Q mutation in the E1 reading frame. All oligonucleotides in this study were obtained from Integrated DNA Technologies (Coralville, IA). The primers used to mutate the TF Cys residues were forward primer 5′-CTTCTGGGTCCAGTTGAGCATACCTTTGGCCGCTTTC-3′ and reverse primer 5′-GAAAGCGGCCAAAGGTATGCTCAACTGGACCCAGAAG-3′ for C23S, forward primer 5′-GTTCTTCTGGGTCCAGTTGGCCATACCTTTGGCCGCTTTC-3′ and reverse primer 5′-GAAAGCGGCCAAAGGTATGGCCAACTGGACCCAGAAGAAC-3′ for C23A, forward primer 5′-GCCTGCCGTTCCTAGTGGTTGCCGGCGCCTA-3′ and reverse primer 5′-TAGGCGCCGGCAACCACTAGGAACGGCAGGC-3′ for C35 and C36, forward primer 5′-CTAATGCGCTCATCATCCTCATCACTGCCTTTTTTAGTGGTTGCCGGC-3′ and reverse primer 5′-GCCGGCAACCACTAAAAAAGGCAGTGATGAGGATGATGAGCGCATTAG-3′ for C38 and C39, forward primer 5′-GGTAGGCGCCGAGAACCACTAAAAAAGGCAGGCA-3′ and reverse primer 5′-TGCCTGCCTTTTTTAGTGGTTCTCGGCGCCTACC-3′ for C46, and forward primer 5′-GGTATCTGTGGCTGATTTGGAACTGTGGTCAGATGTTCGTAGGCGTCTACC-3′ and reverse primer 5′-GGTAGACGCCTACGAACATCTGACCACAGTTCCAAATCAGCCACAGATACC-3′ for C59, C62, and C65. All clones were validated to contain the designed mutations via Sanger sequencing.
Peptide synthesis.
Two peptides were synthesized, with the sequences ETFTETMSYLWSC, from the shared N terminus of 6K and TF, and RLPGEGRRLRTC, from the unique TF C terminus (Fig. 1). The full peptides were created according to typical solid-phase synthesis procedures and techniques (75). Reagents were purchased from Midwest Biotech (Indianapolis, IN) except where otherwise noted. In this scheme, the C-terminal residue is anchored to phenylacetamidomethyl (PAM) resin beads, and the growing peptide chain is constructed by the addition of N-tert-butoxycarbonyl (Boc)-protected amino acids and 3-(diethoxy-phosphoryloxy)-3H-benzo[d][1,2,3]triazin-4-one (DEPBT) (purchased from National Biochemicals, Twinsburg, OH) as a coupling reagent. Coupling reactions were performed for each residue with the Boc-protected amino acid and DEPBT present at a 10-fold molar excess. By adding each amino acid with a 10-fold molar excess of the reagent, quantitative incorporation of each amino acid at a given position is ensured. Deprotection of the Boc-protected amino acid was performed by adding trifluoroacetic acid (TFA) to the vessels containing the resin with the growing peptide chain. After the deprotection step was completed, coupling solution (Boc-protected amino acid, DEPBT, N,N-ethyldiisopropylamine [DIPEA], and dimethyl fluoride [DMF]) was added to the growing peptide chain. After all amino acids were added, the peptides on the resin were washed with DMF and dichloromethane (DCM). Once ready for cleavage, the resin-peptide chain was washed with DCM, allowing 30 min of continuous drying time on a vacuum-sealed manifold apparatus. Cleavage of the dried peptide from the PAM resin was conducted by using a hydrogen fluoride (HF) cleavage protocol. The HF cleavage protocol used includes a scavenger para-cresol, where the approximate ratio is 95% HF to 5% para-cresol. The retrieved peptides were purified via lyophilization.
Antibody generation.
The N-terminal 6K/TF and C-terminal TF peptides were conjugated to mariculture KLH (mcKLH) protein as a carrier by using the Imject Maleimide Activated mcKLH spin kit (Thermo Scientific, Waltham, MA). Conjugated peptides were injected into rabbits for antibody generation by Covance, Inc. (Princeton, NJ). Crude rabbit sera were concentrated and purified over a peptide column, where the original synthesized 6K/TF or TF peptides were conjugated to an Affi-gel support (Bio-Rad, Hercules, CA). Antibodies against the virus glycoproteins E1/E2 and CP were generated by our laboratory and described previously (76–78).
Sequence alignment.
Representatives from all the major clades and alphaviruses that are well studied were chosen for alignment (70). Sequences were retrieved from the following GenBank accession numbers and used to translate the TF protein sequence: NC_001547 for SINV, NC_003908.1 for Western equine encephalitis virus (WEEV), NC_004162.2 for Chikungunya virus (CHIKV), HM234643.1 for RRV, NC_003215.1 for Semliki Forest virus (SFV), NC_003899 for Eastern equine encephalitis virus (EEEV), NC_001449 for Venezuelan equine encephalitis virus (VEEV), NC_003900.1 for Aura virus (AURAV), AF339486 for Middelburg virus (MIDV), NC_018615.1 for Eilat virus (EILV), NC_016960.1 for Southern elephant seal virus (SESV), and NC_003433.1 for sleeping disease virus (SDV). Sequences were aligned by using default parameters in ClustalOmega (1.2.1) (79) and then manually adjusted around the conserved frameshift slip site.
Virus growth and purification.
Infectious viruses were generated from a cDNA clone, similarly to previous descriptions (76). Briefly, the linearized TE12 cDNA plasmid or its 6K/TF mutant derivatives were transcribed into infectious RNA in vitro, with a synthetic cap analog (New England BioLabs, Ipswich, MA). RNA was electroporated (1500 V, 25 μF, 200 Ω) into BHK cells resuspended in phosphate-buffered saline (PBS) in a 2-mm cuvette. Upon display of significant cytopathic effect, the media were harvested, and cellular debris were pelleted at 5,000 × g for 15 min. Infectious virus was measured as PFU on BHK monolayers by using standard procedures, where serial dilutions were adhered to cells for 1 h at room temperature. Cells were overlaid with 1% low-melt agarose–1× complete MEM–5% fetal bovine serum. Plaques were detected at 48 hpi by formaldehyde fixation and crystal violet staining.
For all experiments, a multiplicity of infection (MOI) of 5 PFU per cell was used to infect a confluent monolayer of BHK cells for 1 h at room temperature. Upon the removal of the virus inoculum and the addition of fresh medium, the cells were incubated at 37°C in 5% CO2 until harvest. Virus samples for growth curves were collected at each time point by removing and replacing the entire medium volume. Infectious-particle concentrations were measured at each time point via a plaque assay as described above. For infections where cells were collected, media were removed, and monolayers were washed with PBS prior to being lysed in SDS lysis buffer (10 mM Tris [pH 7.4] and 1% SDS supplemented with 1 mM fresh phenylmethylsulfonyl fluoride [PMSF] and 1 μg/ml leupeptin).
To purify high-concentration virus, confluent BHK cells were infected with WT, 9C, 4C, and 5C viruses at an MOI of 5 PFU per cell. After rocking at room temperature for an hour, the cell monolayers were washed with 1× PBS three times and then incubated in Virus Production Serum-Free medium (Gibco, Waltham, MA) supplemented with 2 mM l-glutamine and 1× MEM-nonessential amino acids at 37°C in 5% CO2. After 16 to 24 h, the medium was harvested and clarified by a 5-min centrifugation at 5,000 × g. Finally, the virus was pelleted via low-speed centrifugation at 5,300 × g for 16 h. The pelleted virus was resuspended in HNE (20 mM HEPES [pH 7.4], 150 mM NaCl, 0.1 mM EDTA).
Western blotting.
To ensure equal loading, cell lysate protein concentrations were measured by using a bicinchoninic acid (BCA) assay (Pierce, Waltham, MA). Samples were mixed to a 1× final concentration with 2× Tricine sample buffer (450 mM Tris HCl [pH 8.45], 12% [vol/vol] glycerol, 4% [wt/vol] SDS, 0.0025% [wt/vol] Coomassie blue G250, 0.0025% [wt/vol] phenol red) and heated for 2 min at 85°C or 95°C before gel electrophoresis on a precast 10 to 20% Tricine gel (Thermo Fisher Scientific-Novex, Waltham, MA) in Tricine running buffer (100 mM Tris base [pH 8.3], 100 mM Tricine, 0.1% SDS). The PageRuler prestained protein ladder (Thermo Fisher Scientific-Invitrogen, Waltham, MA) was used in all studies. The gel was transferred to 0.2-μm polyvinylidene difluoride (PVDF) blotting paper by wet transfer at 25 V for 30 min, and then at 100 V for 1 h, in Tris-glycine transfer buffer (12 mM Tris base [pH 8.3], 96 mM glycine, 20% methanol). Blots probed for 6K, TF, or biotin-streptavidin were blocked in 1% bovine serum albumin (BSA) in Tris-buffered saline (TBS); all other blots were blocked in 5% nonfat dry milk in TBS. Primary antibodies directed against virus protein targets, purified anti-TF at a 1:1,000 dilution, purified anti-6K/TF at a 1:200 dilution, anti-CP at a 1:5,000 dilution, and anti-E1/E2 at a 1:2,500 dilution, were incubated with blots in 2% milk–TBS. The blots were then incubated with the Alexa Fluor 750 goat anti-rabbit IgG(H+L) fluorescent secondary antibody (Thermo Fisher Scientific-Life Technologies, Waltham, MA) at a 1:20,000 dilution in 2% milk–TBS. After washing in TBS and drying, the blot was scanned in the 700- and 800-nm channels on a Li-Cor Odyssey Classic infrared imaging system. Where applicable, the protein bands were quantified with ImageStudioLite v.4.0.21 software (Li-Cor, Lincoln, NE).
Click chemistry labeling of palmitoylated proteins.
BHK cells were infected at an MOI of 5 PFU per cell and then labeled with 50 μM palmitic azide (Invitrogen, Carlsbad, CA) at 4 or 12 hpi for 4 h. Cells were lysed in SDS lysis buffer (10 mM Tris [pH 7.4] and 1% SDS supplemented with 1 mM fresh PMSF and 1 μg/ml leupeptin). Equal amounts of protein measured by the BCA assay were input into a click kit (Invitrogen, Carlsbad, CA) according to the manufacturer's recommendations. In the reaction, the azide group was covalently conjugated to an alkyne-biotin compound. After methanol-chloroform precipitation, the entire clicked lysate sample was analyzed by Western blotting as described above. The biotin-conjugated proteins were detected by probing with an IR800CW-conjugated streptavidin reagent (Li-Cor, Lincoln, NE).
Hydroxylamine treatment.
BHK cells were infected at an MOI of 5 PFU per cell. At 16 hpi, cells were washed with PBS and then harvested in SDS lysis buffer. Equal amounts of protein from each lysate were treated with 1 M hydroxylamine at pH 7.5 for 1 h at room temperature in 50 mM HEPES (pH 7.5). Reactions were terminated by trichloroacetic acid precipitation similarly to methods described previously (80), except for the following modifications. Trichloroacetic acid was added to 20% (as one-fifth of the reaction volume of 100% trichloroacetic acid), and the protein pellet was washed once with methanol and once with acetone. Samples were resuspended in Tricine sample buffer diluted to 1× with SDS lysis buffer, heated at 95°C for 2 min, and then analyzed by Western blotting as described above.
Subcellular fractionation.
BHK cells were infected at an MOI of 5 for 16 h. A total of 20 million to 35 million cells were harvested according to instructions provided with the 101Bio plasma membrane protein extraction kit (101Bio, Palo Alto, CA). This non-detergent-based separation generates an internal membrane protein fraction and a plasma membrane protein fraction. The fractions were analyzed by Western blotting as detailed above. Fractions from uninfected cells were also probed with anti-transferrin receptor (Abcam, Cambridge, MA) at a 1:1,000 dilution as a plasma membrane protein and anti-KDEL (Enzo Life Sciences, Farmingdale, NY) at a 1:200 dilution as an ER marker.
Cycloheximide translation block.
BHK cells were infected at an MOI of 5. At 10 hpi, the cells were treated with the DMSO vehicle control or 50 μg/ml cycloheximide (Sigma-Aldrich, St. Louis, MO). Samples with and without cycloheximide were collected at 0, 2, 4, and 6 h posttreatment by lysis in NP-40 lysis buffer (20 mM Tris [pH 7.4], 0.5% NP-40, and 200 mM NaCl supplemented with fresh 1 mM PMSF and 1 μg/ml leupeptin). The NP-40 lysis buffer used here extracts the TF protein as well as the SDS lysis buffer described above. Samples were then analyzed by Western blotting as indicated above.
Transmission electron microscopy.
The high-concentration virus samples were prepared for TEM by incubation on a 300-mesh Formvar- and carbon-coated copper grid (Ted Pella, Inc., Redding, CA) for 1 min. The samples were stained for 1 min with 1% uranyl acetate. The grids were observed with a JEM 1010 transmission electron microscope (JEOL, Tokyo, Japan). Images were acquired by using a Gatan charged-coupled-device camera. For quantification of virion morphology, the particles were counted with ImageJ 1.44o (81). The authors were blind to the names of the image files so that they would not be biased in their counting. In each image, three categories were counted: total particles fully in the view, WT-like particles, and abnormal particles. WT-like morphology was defined as whole round virions with no blemishes, measuring between 65 and 75 nm in diameter. Abnormal particles were defined as particles that were larger or smaller than WT particles, stained internally, broken, or not spherical.
Reverse transcription and quantitative real-time PCR.
BHK cells were infected with the WT or the Cys mutants at an MOI of 5 PFU per cell for 16 h. Media were collected and clarified at 5,000 × g for 15 min for three biological replicates. Reverse transcription and quantitation were performed similarly to methods described previously (82). Each replicate was analyzed in triplicate by heating 5 μl of clarified medium for 45 s at 94°C with excess (500 ng) E1 reverse primer 5′-ATTGACCTTCGCGGTCGGATACAT-3′ and then for 5 min at 70°C and then chilling the mixture on ice. The required components for reverse transcription with ProtoScript II reverse transcriptase (New England BioLabs, Ipswich, MA) were added for a 20-μl reaction mixture, and samples were incubated at 25°C for 5 min, at 42°C for 45 min, and then at 70°C for 15 min to generate single-stranded DNA complementary to the positive-sense RNA genome of our Sindbis virus samples.
Quantitative real-time PCRs were set up in triplicate according to the manufacturer's recommendations provided with the Sensi-Fast SYBR Hi-ROX kit (Bioline, London, UK) with E1 forward primer 5′-TCAGATGCACCACTGGTCTCAACA-3′ and the E1 reverse primer described above. Twenty-five-microliter reaction mixtures with 2 μl each of cDNA in 96-well plates were cycled on an ABI StepOne instrument. The samples were compared to a paired standard curve prepared as a serial dilution series of TE12 plasmid DNA extending below and above the concentration of the medium samples. Negative controls included mock-infected samples and samples without a template to monitor for reagent contamination. The final quantification was done by relating the threshold cycle (CT) values for medium samples to the exponential fit of the standard curve.
ACKNOWLEDGMENTS
We thank the Mukhopadhyay, Danthi, and Hardy laboratory members for critical and productive discussions.
This work was funded by National Science Foundation award MCB-1157716 to S.M. Also, J.R. was supported by National Institutes of Health grant T32 GM007757.
REFERENCES
- 1.Aicart-Ramos C, Valero RA, Rodriguez-Crespo I. 2011. Protein palmitoylation and subcellular trafficking. Biochim Biophys Acta 1808:2981–2994. doi: 10.1016/j.bbamem.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 2.Stepanek O, Draber P, Horejsi V. 2014. Palmitoylated transmembrane adaptor proteins in leukocyte signaling. Cell Signal 26:895–902. doi: 10.1016/j.cellsig.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Konitsiotis AD, Jovanovic B, Ciepla P, Spitaler M, Lanyon-Hogg T, Tate EW, Magee AI. 2015. Topological analysis of Hedgehog acyltransferase, a multipalmitoylated transmembrane protein. J Biol Chem 290:3293–3307. doi: 10.1074/jbc.M114.614578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin BR, Wang C, Adibekian A, Tully SE, Cravatt BF. 2011. Global profiling of dynamic protein palmitoylation. Nat Methods 9:84–89. doi: 10.1038/nmeth.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, Kuhlmann J, Waldmann H, Wittinghofer A, Bastiaens PI. 2005. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 307:1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 6.Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates JR III, Davis NG. 2006. Global analysis of protein palmitoylation in yeast. Cell 125:1003–1013. doi: 10.1016/j.cell.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veit M. 2012. Palmitoylation of virus proteins. Biol Cell 104:493–515. doi: 10.1111/boc.201200006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hourioux C, Ait-Goughoulte M, Patient R, Fouquenet D, Arcanger-Doudet F, Brand D, Martin A, Roingeard P. 2007. Core protein domains involved in hepatitis C virus-like particle assembly and budding at the endoplasmic reticulum membrane. Cell Microbiol 9:1014–1027. doi: 10.1111/j.1462-5822.2006.00848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones DM, McLauchlan J. 2010. Hepatitis C virus: assembly and release of virus particles. J Biol Chem 285:22733–22739. doi: 10.1074/jbc.R110.133017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Popescu CI, Rouille Y, Dubuisson J. 2011. Hepatitis C virus assembly imaging. Viruses 3:2238–2254. doi: 10.3390/v3112238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukhopadhyay S, Kuhn RJ, Rossmann MG. 2005. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol 3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 12.Klumperman J, Locker JK, Meijer A, Horzinek MC, Geuze HJ, Rottier PJ. 1994. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J Virol 68:6523–6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tooze J, Tooze S, Warren G. 1984. Replication of coronavirus MHV-A59 in sac− cells: determination of the first site of budding of progeny virions. Eur J Cell Biol 33:281–293. [PubMed] [Google Scholar]
- 14.Ujike M, Taguchi F. 2015. Incorporation of spike and membrane glycoproteins into coronavirus virions. Viruses 7:1700–1725. doi: 10.3390/v7041700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Acheson NH, Tamm I. 1967. Replication of Semliki Forest virus: an electron microscopic study. Virology 32:128–143. doi: 10.1016/0042-6822(67)90261-9. [DOI] [PubMed] [Google Scholar]
- 16.Baker RF, Gordon I, Rapp F. 1960. Electron-dense crystallites in nuclei of human amnion cells infected with measles virus. Nature 185:790–791. doi: 10.1038/185790a0. [DOI] [PubMed] [Google Scholar]
- 17.Cadd TL, Skoging U, Liljestrom P. 1997. Budding of enveloped viruses from the plasma membrane. Bioessays 19:993–1000. doi: 10.1002/bies.950191109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gottlinger HG. 2001. The HIV-1 assembly machine. AIDS 15(Suppl 5):S13–S20. doi: 10.1097/00002030-200100005-00003. [DOI] [PubMed] [Google Scholar]
- 19.Morgan C, Howe C, Rose HM. 1961. Structure and development of viruses as observed in the electron microscope. V. Western equine encephalomyelitis virus. J Exp Med 113:219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaedigk-Nitschko K, Ding MX, Levy MA, Schlesinger MJ. 1990. Site-directed mutations in the Sindbis virus 6K protein reveal sites for fatty acylation and the underacylated protein affects virus release and virion structure. Virology 175:282–291. doi: 10.1016/0042-6822(90)90210-I. [DOI] [PubMed] [Google Scholar]
- 21.Ivanova L, Schlesinger MJ. 1993. Site-directed mutations in the Sindbis virus E2 glycoprotein identify palmitoylation sites and affect virus budding. J Virol 67:2546–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt M, Schmidt MF, Rott R. 1988. Chemical identification of cysteine as palmitoylation site in a transmembrane protein (Semliki Forest virus E1). J Biol Chem 263:18635–18639. [PubMed] [Google Scholar]
- 23.Gaedigk-Nitschko K, Schlesinger MJ. 1990. The Sindbis virus 6K protein can be detected in virions and is acylated with fatty acids. Virology 175:274–281. doi: 10.1016/0042-6822(90)90209-A. [DOI] [PubMed] [Google Scholar]
- 24.Firth AE, Chung BY, Fleeton MN, Atkins JF. 2008. Discovery of frameshifting in alphavirus 6K resolves a 20-year enigma. Virol J 5:108. doi: 10.1186/1743-422X-5-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung BY, Firth AE, Atkins JF. 2010. Frameshifting in alphaviruses: a diversity of 3′ stimulatory structures. J Mol Biol 397:448–456. doi: 10.1016/j.jmb.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 26.Snyder JE, Kulcsar KA, Schultz KL, Riley CP, Neary JT, Marr S, Jose J, Griffin DE, Kuhn RJ. 2013. Functional characterization of the alphavirus TF protein. J Virol 87:8511–8523. doi: 10.1128/JVI.00449-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molinari M, Helenius A. 1999. Glycoproteins form mixed disulphides with oxidoreductases during folding in living cells. Nature 402:90–93. doi: 10.1038/47062. [DOI] [PubMed] [Google Scholar]
- 28.Mulvey M, Brown DT. 1995. Involvement of the molecular chaperone BiP in maturation of Sindbis virus envelope glycoproteins. J Virol 69:1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng RH, Kuhn RJ, Olson NH, Rossmann MG, Choi HK, Smith TJ, Baker TS. 1995. Nucleocapsid and glycoprotein organization in an enveloped virus. Cell 80:621–630. doi: 10.1016/0092-8674(95)90516-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liljestrom P, Garoff H. 1991. Internally located cleavable signal sequences direct the formation of Semliki Forest virus membrane proteins from a polyprotein precursor. J Virol 65:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lusa S, Garoff H, Liljestrom P. 1991. Fate of the 6K membrane protein of Semliki Forest virus during virus assembly. Virology 185:843–846. doi: 10.1016/0042-6822(91)90556-Q. [DOI] [PubMed] [Google Scholar]
- 32.Akahata W, Yang ZY, Andersen H, Sun S, Holdaway HA, Kong WP, Lewis MG, Higgs S, Rossmann MG, Rao S, Nabel GJ. 2010. A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat Med 16:334–338. doi: 10.1038/nm.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kostyuchenko VA, Jakana J, Liu X, Haddow AD, Aung M, Weaver SC, Chiu W, Lok SM. 2011. The structure of Barmah Forest virus as revealed by cryo-electron microscopy at a 6-angstrom resolution has detailed transmembrane protein architecture and interactions. J Virol 85:9327–9333. doi: 10.1128/JVI.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukhopadhyay S, Zhang W, Gabler S, Chipman PR, Strauss EG, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. 2006. Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. Structure 14:63–73. doi: 10.1016/j.str.2005.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang J, Jose J, Chipman P, Zhang W, Kuhn RJ, Baker TS. 2011. Molecular links between the E2 envelope glycoprotein and nucleocapsid core in Sindbis virus. J Mol Biol 414:442–459. doi: 10.1016/j.jmb.2011.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang R, Hryc CF, Cong Y, Liu X, Jakana J, Gorchakov R, Baker ML, Weaver SC, Chiu W. 2011. 4.4 A cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J 30:3854–3863. doi: 10.1038/emboj.2011.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaedigk-Nitschko K, Schlesinger MJ. 1991. Site-directed mutations in Sindbis virus E2 glycoprotein's cytoplasmic domain and the 6K protein lead to similar defects in virus assembly and budding. Virology 183:206–214. doi: 10.1016/0042-6822(91)90133-V. [DOI] [PubMed] [Google Scholar]
- 38.Ivanova L, Lustig S, Schlesinger MJ. 1995. A pseudo-revertant of a Sindbis virus 6K protein mutant, which corrects for aberrant particle formation, contains two new mutations that map to the ectodomain of the E2 glycoprotein. Virology 206:1027–1034. doi: 10.1006/viro.1995.1025. [DOI] [PubMed] [Google Scholar]
- 39.Liljestrom P, Lusa S, Huylebroeck D, Garoff H. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J Virol 65:4107–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loewy A, Smyth J, von Bonsdorff CH, Liljestrom P, Schlesinger MJ. 1995. The 6-kilodalton membrane protein of Semliki Forest virus is involved in the budding process. J Virol 69:469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor A, Melton JV, Herrero LJ, Thaa B, Karo-Astover L, Gage PW, Nelson MA, Sheng KC, Lidbury BA, Ewart GD, McInerney GM, Merits A, Mahalingam S. 2016. Effects of an in-frame deletion of the 6k gene locus from the genome of Ross River virus. J Virol 90:4150–4159. doi: 10.1128/JVI.03192-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao JS, Strauss EG, Strauss JH. 1996. Interactions between PE2, E1, and 6K required for assembly of alphaviruses studied with chimeric viruses. J Virol 70:7910–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ivanova L, Le L, Schlesinger MJ. 1995. Characterization of revertants of a Sindbis virus 6K gene mutant that affects proteolytic processing and virus assembly. Virus Res 39:165–179. doi: 10.1016/0168-1702(95)00083-6. [DOI] [PubMed] [Google Scholar]
- 44.Barbieri CM, Stock AM. 2008. Universally applicable methods for monitoring response regulator aspartate phosphorylation both in vitro and in vivo using Phos-tag-based reagents. Anal Biochem 376:73–82. doi: 10.1016/j.ab.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wayne KJ, Li S, Kazmierczak KM, Tsui HC, Winkler ME. 2012. Involvement of WalK (VicK) phosphatase activity in setting WalR (VicR) response regulator phosphorylation level and limiting cross-talk in Streptococcus pneumoniae D39 cells. Mol Microbiol 86:645–660. doi: 10.1111/mmi.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, Shamir E, Hang HC. 2009. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc 131:4967–4975. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]
- 47.Hannoush RN, Sun J. 2010. The chemical toolbox for monitoring protein fatty acylation and prenylation. Nat Chem Biol 6:498–506. doi: 10.1038/nchembio.388. [DOI] [PubMed] [Google Scholar]
- 48.Magee AI, Koyama AH, Malfer C, Wen D, Schlesinger MJ. 1984. Release of fatty acids from virus glycoproteins by hydroxylamine. Biochim Biophys Acta 798:156–166. doi: 10.1016/0304-4165(84)90298-8. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt MF, Bracha M, Schlesinger MJ. 1979. Evidence for covalent attachment of fatty acids to Sindbis virus glycoproteins. Proc Natl Acad Sci U S A 76:1687–1691. doi: 10.1073/pnas.76.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidt MF. 1982. Acylation of viral spike glycoproteins: a feature of enveloped RNA viruses. Virology 116:327–338. doi: 10.1016/0042-6822(82)90424-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scolari S, Imkeller K, Jolmes F, Veit M, Herrmann A, Schwarzer R. 2016. Modulation of cell surface transport and lipid raft localization by the cytoplasmic tail of the influenza virus hemagglutinin. Cell Microbiol 18:125–136. doi: 10.1111/cmi.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Veit M, Kretzschmar E, Kuroda K, Garten W, Schmidt MF, Klenk HD, Rott R. 1991. Site-specific mutagenesis identifies three cysteine residues in the cytoplasmic tail as acylation sites of influenza virus hemagglutinin. J Virol 65:2491–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nozawa N, Daikoku T, Koshizuka T, Yamauchi Y, Yoshikawa T, Nishiyama Y. 2003. Subcellular localization of herpes simplex virus type 1 UL51 protein and role of palmitoylation in Golgi apparatus targeting. J Virol 77:3204–3216. doi: 10.1128/JVI.77.5.3204-3216.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nozawa N, Kawaguchi Y, Tanaka M, Kato A, Kato A, Kimura H, Nishiyama Y. 2005. Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J Virol 79:6947–6956. doi: 10.1128/JVI.79.11.6947-6956.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Majeau N, Fromentin R, Savard C, Duval M, Tremblay MJ, Leclerc D. 2009. Palmitoylation of hepatitis C virus core protein is important for virion production. J Biol Chem 284:33915–33925. doi: 10.1074/jbc.M109.018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bour S, Strebel K. 2003. The HIV-1 Vpu protein: a multifunctional enhancer of viral particle release. Microbes Infect 5:1029–1039. doi: 10.1016/S1286-4579(03)00191-6. [DOI] [PubMed] [Google Scholar]
- 57.Guo F, Liang C. 2012. Transmembrane interactions of HIV-1 Vpu and tetherin. Curr HIV Res 10:292–297. doi: 10.2174/157016212800792450. [DOI] [PubMed] [Google Scholar]
- 58.Terwilliger EF, Cohen EA, Lu YC, Sodroski JG, Haseltine WA. 1989. Functional role of human immunodeficiency virus type 1 vpu. Proc Natl Acad Sci U S A 86:5163–5167. doi: 10.1073/pnas.86.13.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rossman JS, Jing X, Leser GP, Balannik V, Pinto LH, Lamb RA. 2010. Influenza virus M2 ion channel protein is necessary for filamentous virion formation. J Virol 84:5078–5088. doi: 10.1128/JVI.00119-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rossman JS, Lamb RA. 2011. Influenza virus assembly and budding. Virology 411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rossman JS, Jing X, Leser GP, Lamb RA. 2010. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ruch TR, Machamer CE. 2012. The coronavirus E protein: assembly and beyond. Viruses 4:363–382. doi: 10.3390/v4030363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye Y, Hogue BG. 2007. Role of the coronavirus E viroporin protein transmembrane domain in virus assembly. J Virol 81:3597–3607. doi: 10.1128/JVI.01472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sugrue RJ, Belshe RB, Hay AJ. 1990. Palmitoylation of the influenza A virus M2 protein. Virology 179:51–56. doi: 10.1016/0042-6822(90)90272-S. [DOI] [PubMed] [Google Scholar]
- 65.Veit M, Klenk HD, Kendal A, Rott R. 1991. The M2 protein of influenza A virus is acylated. J Gen Virol 72(Part 6):1461–1465. [DOI] [PubMed] [Google Scholar]
- 66.Corse E, Machamer CE. 2002. The cytoplasmic tail of infectious bronchitis virus E protein directs Golgi targeting. J Virol 76:1273–1284. doi: 10.1128/JVI.76.3.1273-1284.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thaa B, Siche S, Herrmann A, Veit M. 2014. Acylation and cholesterol binding are not required for targeting of influenza A virus M2 protein to the hemagglutinin-defined budozone. FEBS Lett 588:1031–1036. doi: 10.1016/j.febslet.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 68.Lopez LA, Riffle AJ, Pike SL, Gardner D, Hogue BG. 2008. Importance of conserved cysteine residues in the coronavirus envelope protein. J Virol 82:3000–3010. doi: 10.1128/JVI.01914-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fischer F, Stegen CF, Masters PS, Samsonoff WA. 1998. Analysis of constructed E gene mutants of mouse hepatitis virus confirms a pivotal role for E protein in coronavirus assembly. J Virol 72:7885–7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Forrester NL, Palacios G, Tesh RB, Savji N, Guzman H, Sherman M, Weaver SC, Lipkin WI. 2012. Genome-scale phylogeny of the alphavirus genus suggests a marine origin. J Virol 86:2729–2738. doi: 10.1128/JVI.05591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nasar F, Palacios G, Gorchakov RV, Guzman H, Da Rosa AP, Savji N, Popov VL, Sherman MB, Lipkin WI, Tesh RB, Weaver SC. 2012. Eilat virus, a unique alphavirus with host range restricted to insects by RNA replication. Proc Natl Acad Sci U S A 109:14622–14627. doi: 10.1073/pnas.1204787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Westerbeck JW, Machamer CE. 2015. A coronavirus E protein is present in two distinct pools with different effects on assembly and the secretory pathway. J Virol 89:9313–9323. doi: 10.1128/JVI.01237-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Welch WJ, Sefton BM. 1980. Characterization of a small, nonstructural viral polypeptide present late during infection of BHK cells by Semliki Forest virus. J Virol 33:230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lustig S, Jackson AC, Hahn CS, Griffin DE, Strauss EG, Strauss JH. 1988. Molecular basis of Sindbis virus neurovirulence in mice. J Virol 62:2329–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lebl M, Krchnak V. 1997. Synthetic peptide libraries. Methods Enzymol 289:336–392. doi: 10.1016/S0076-6879(97)89055-6. [DOI] [PubMed] [Google Scholar]
- 76.Cheng F, Mukhopadhyay S. 2011. Generating enveloped virus-like particles with in vitro assembled cores. Virology 413:153–160. doi: 10.1016/j.virol.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 77.Parrott MM, Sitarski SA, Arnold RJ, Picton LK, Hill RB, Mukhopadhyay S. 2009. Role of conserved cysteines in the alphavirus E3 protein. J Virol 83:2584–2591. doi: 10.1128/JVI.02158-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Snyder AJ, Sokoloski KJ, Mukhopadhyay S. 2012. Mutating conserved cysteines in the alphavirus E2 glycoprotein causes virus-specific assembly defects. J Virol 86:3100–3111. doi: 10.1128/JVI.06615-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. 2013. Analysis tool Web services from the EMBL-EBI. Nucleic Acids Res 41:W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wan J, Roth AF, Bailey AO, Davis NG. 2007. Palmitoylated proteins: purification and identification. Nat Protoc 2:1573–1584. doi: 10.1038/nprot.2007.225. [DOI] [PubMed] [Google Scholar]
- 81.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sokoloski KJ, Hayes CA, Dunn MP, Balke JL, Hardy RW, Mukhopadhyay S. 2012. Sindbis virus infectivity improves during the course of infection in both mammalian and mosquito cells. Virus Res 167:26–33. doi: 10.1016/j.virusres.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hallengärd D, Kakoulidou M, Lulla A, Kümmerer BM, Johansson DX, Mutso M, Lulla V, Fazakerley JK, Roques P, Le Grand R, Merits A, Liljeström P. 2014. J Virol 88:2858–2866. Novel attenuated Chikungunya vaccine candidates elicit protective immunity in C57BL/6 mice. doi: 10.1128/JVI.03453-13. [DOI] [PMC free article] [PubMed] [Google Scholar]