

Abstract
Sphingomyelin synthase (SMS) is the key enzyme for cross-talk between bioactive sphingolipids and glycerolipids. In mammals, SMS consists of two isoforms: SMS1 is localized in the Golgi apparatus, whereas SMS2 is localized in both the Golgi and plasma membranes. SMS2 seems to exert cellular functions through protein-protein interactions; however, the existence and functions of quaternary structures of SMS1 and SMS2 remain unclear. Here we demonstrate that both SMS1 and SMS2 form homodimers. The SMSs have six membrane-spanning domains, and the N and C termini of both proteins face the cytosolic side of the Golgi apparatus. Chemical cross-linking and bimolecular fluorescence complementation revealed that the N- and/or C-terminal tails of the SMSs were in close proximity to those of the other SMS in the homodimer. Homodimer formation was significantly decreased by C-terminal truncations, SMS1-ΔC22 and SMS2-ΔC30, indicating that the C-terminal tails of the SMSs are primarily responsible for homodimer formation. Moreover, immunoprecipitation using deletion mutants revealed that the C-terminal tail of SMS2 mainly interacted with the C-terminal tail of its homodimer partner, whereas the C-terminal tail of SMS1 mainly interacted with a site other than the C-terminal tail of its homodimer partner. Interestingly, homodimer formation occurred in the endoplasmic reticulum (ER) membrane before trafficking to the Golgi apparatus. Reduced homodimerization caused by C-terminal truncations of SMSs significantly reduced ER-to-Golgi transport. Our findings suggest that the C-terminal tails of SMSs are involved in homodimer formation, which is required for efficient transport from the ER.
Keywords: dimerization, glycerophospholipid, protein cross-linking, protein-protein interaction, sphingolipid, sphingomyelin synthase (SMS)
Introduction
Sphingomyelin (SM)2 is one of the major components of cell membranes and is especially enriched in lipid microdomains. SM is de novo synthesized from serine and palmitoyl coenzyme A by the sequential reactions of various enzymes. The final step of its synthesis is catalyzed by SM synthase (SMS). SMS transfers the phosphorylcholine moiety from phosphatidylcholine (PC) to the primary hydroxyl of ceramide (Cer), resulting in the production of SM and diacylglycerol (DAG) (1–2). Cer is involved in regulating proapoptotic cell responses that include growth arrest and apoptosis (3), whereas DAG is involved in regulating prosurvival cell responses that include cell survival and proliferation (4). PC and SM, another substrate and product, respectively, of SMS, are the most abundant glycero- and sphingophospholipids and are critical structural components of the cell membrane. The ratio of PC to SM is known to be responsible for both membrane lipid fluidity and osmotic fragility (5). It has been suggested that the ratios of PC/SM and DAG/Cer are intrinsically related (6). Thus, SMS is postulated to reciprocally regulate the amount of both sphingolipids and glycerolipids and to be the key enzyme mediating the cross-talk between these bioactive lipids.
In mammals, the SMS enzyme consists of two isoforms, SMS1 and SMS2 (SMSs) (1). Both isoforms are membrane proteins with multiple membrane-spanning domains. Presumably, SMSs are co-translationally integrated into the endoplasmic reticulum (ER) membrane and exported from the ER to the Golgi apparatus. SMS1 mainly localizes to the Golgi apparatus, whereas SMS2 is localized in both the Golgi apparatus and the plasma membrane (1).
Overexpression of SMS1 in Jurkat cells results in the suppression of photodamage-induced apoptosis by decreasing Cer production (7). SMS1/SMS2 double knockout cells revealed that SM regulates cell migration induced by chemokine CXCL12 through the repression of CXCR4 dimerization (8). Furthermore, SMSs have been implicated in DAG formation at the Golgi apparatus and, consequently, in the regulation of protein trafficking and secretion through protein kinase D recruitment (9).
Despite accumulating evidence of the functions of SMS1 and SMS2, the roles of each isoform are not fully understood. Mitsutake et al. (10) indicated that SMS2 is localized in lipid microdomains, where it interacts with the fatty acid transporter CD36/FAT and caveolin-1 to regulate caveola-dependent endocytosis. Our previous study also revealed a unique function of SMS2 in membrane fusion (11). We found that SMS2 serves as a modulator of the HIV, type 1 (HIV-1) receptor/co-receptor complex in the plasma membrane, promoting HIV-1 receptor/co-receptor-mediated Pyk2 phosphorylation in response to the HIV-1 envelope protein (Env). Pyk2 signaling induced F-actin polymerization at cell-cell contact sites, leading to augmented membrane fusion. SMS1 did not promote such fusion events; thus, this function is clearly specific to SMS2.
Based on the augmented actin polymerization in Sms2-expressing cells, we hypothesized that SMS2 can associate with polymerized actin or actin-interacting proteins, e.g. filamin, ezrin/radixin/moesin, and cofilin (12). To examine this hypothesis, SMS2-protein interactions were explored by chemical cross-linking. Although we did not detect any associations of β-actin and actin-interacting proteins with SMS2, we observed an additional band, as would be expected for an SMS2 homodimer. This was the first observation of oligomer formation of SMSs.
In this study, we further examined the mechanism and functions of the oligomerization of SMS1 and SMS2. Here we reveal that most SMSs exist as homodimers that are formed in the ER membrane before reaching their final destinations. Our analyses indicated that the C-terminal tails stabilized the SMS homodimers and that disruption of homodimer integrity by C-terminal truncations led to decreased ER-to-Golgi transport. Thus, homodimerization of SMSs is required for protein maturation and efficient transport from the ER.
Results
Homo-oligomers of SMSs Are More Stable Than Hetero-oligomers
As our previous study provided a clue to the existence of SMS2 homodimers in cells (11), we aimed to examine the physiological relevance of SMS oligomerization. To examine the oligomerization of SMSs in detail, we used co-immunoprecipitation and immunoblotting for the expression of differentially epitope-tagged SMS1 and SMS2. HEK293 cells stably expressing V5-tagged SMS1 were co-expressed with FLAG-tagged SMS1 or SMS2. After lysis of the cells with 1% Triton X-100, the extracts were immunoprecipitated using anti-FLAG beads. A significant amount of SMS1-V5 was detected in immunoprecipitates with SMS1-FLAG, whereas little SMS1-V5 was found in precipitates with the SMS2-FLAG complex (Fig. 1A, bottom row, right). Approximately 15% of the total input of SMS1-V5 was immunoprecipitated with SMS1-FLAG. Similar results were obtained in immunoprecipitation experiments with HEK293 cells expressing V5-tagged SMS2 (Fig. 1B). A significant amount of SMS2-V5 was detected in immunoprecipitates with SMS2-FLAG, which was largely reduced in precipitates with SMS1-FLAG (Fig. 1B, bottom row, right). Approximately 36% of total input of SMS2-V5 was immunoprecipitated with SMS2-FLAG.
FIGURE 1.
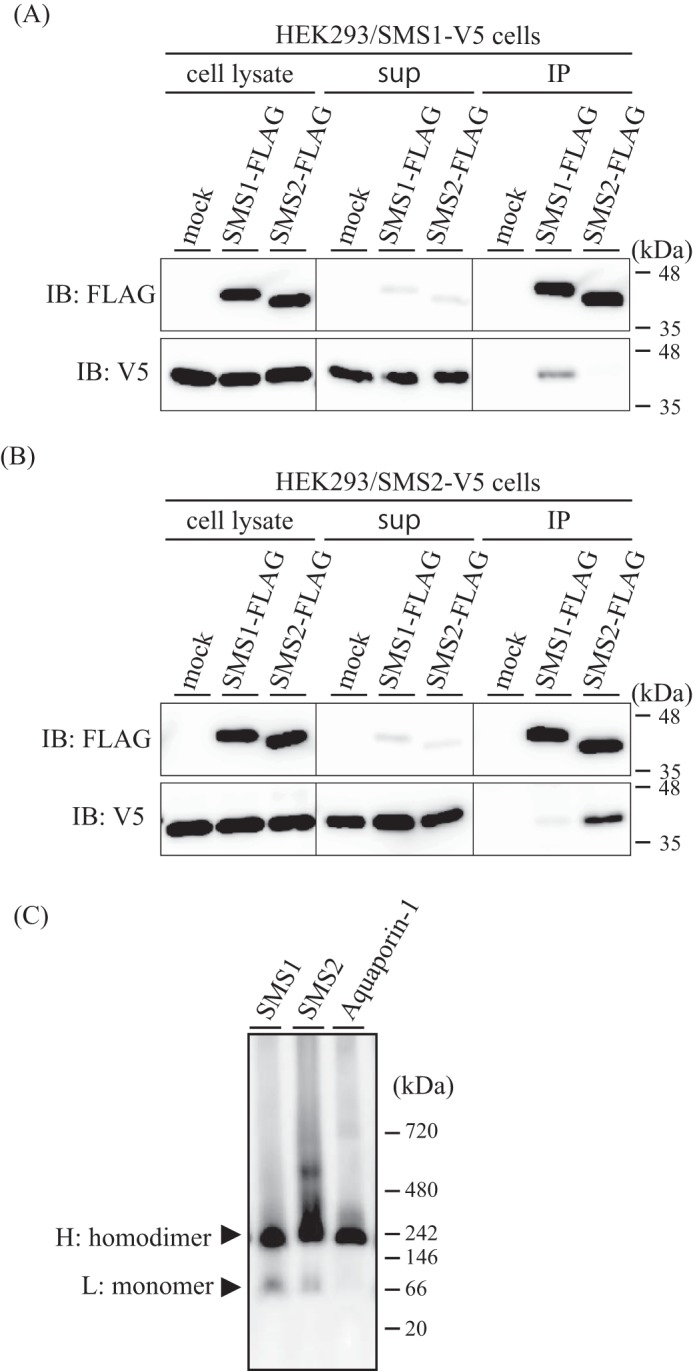
Homomeric complexes of SMSs are more stable than heteromeric ones. A and B, HEK293/SMS1-V5 cells (A) or HEK293/SMS2-V5 cells (B) were transfected with a plasmid encoding FLAG-tagged SMS1 or FLAG-tagged SMS2 or mock-transfected (empty vector). The cells were lysed in a buffer containing 1% Triton X-100, and immunoprecipitation (IP) was performed using anti-FLAG M2 beads. The cell lysates, supernatant (sup), and precipitated proteins were analyzed by immunoblotting (IB) with anti-FLAG or anti-V5 antibody. Left, cell lysate; center, supernatant; right, immunoprecipitates (anti-FLAG beads). C, membrane fractions from COS7 cells expressing V5-tagged SMS1, SMS2, or aquaporin-1 were extracted with 1% Triton X-100, separated by BN-PAGE, and immunoblotted with anti-V5 antibody. H, major band of high molecular mass; L, minor band of low molecular mass. NativeMark unstained protein standard was used for molecular mass estimation. Results are from one experiment representative of three independent experiments.
In contrast, significant hetero-oligomer formation was detected when SMS1 and SMS2 were immunoprecipitated in CHAPS solution (data not shown). These results suggest that SMSs can form both homo-oligomers and hetero-oligomers; however, the affinity of the homomeric complexes is much higher than that of the heteromer, as Triton X-100 is known as a more stringent detergent than CHAPS (13, 14). Therefore, we focused on the functions of SMS homo-oligomers.
To analyze the oligomer formation of SMSs further, membrane fractions from COS7 cells expressing V5-tagged SMS1 or SMS2 were extracted with 1% Triton X-100 and analyzed by blue native PAGE (BN-PAGE). Immunoblotting analysis with anti-V5 antibody revealed a major band of high molecular mass and a minor band of low molecular mass in both SMS1- and SMS2-expressing cells (Fig. 1C). The major and minor bands were ∼224 kDa and ∼103 kDa and ∼248 kDa and ∼115 kDa in SMS1- and SMS2-expressing cells, respectively. As the migration of membrane proteins is affected by several factors, including lipids, detergents, and Coomassie Blue G250 in BN-PAGE, calibration is crucial in mass estimation (15, 16). We used aquaporin-1, which is a well characterized membrane protein that forms tetramers (17), with a known molecular mass to calculate a correction factor. Based on the theoretical molecular mass of V5-tagged tetrameric aquaporin-1 of 120 kDa and the mass of ∼236 kDa estimated from BN-PAGE, the correction factor was calculated to be 1.97 (= 236/120). When applied to the SMSs, the corrected molecular masses of the minor and major bands were ∼52 kDa and ∼114 kDa, respectively, for SMS1-expressing cells and ∼58 kDa and ∼126 kDa, respectively, for SMS2-expressing cells. As the predicted molecular masses of V5-tagged SMS1 and SMS2 were 50 and 43.7 kDa, respectively, the minor bands were consistent with the sizes of both monomeric SMSs. The molecular masses of the major bands were approximately two times those of the minor bands, indicating that they represent homodimers. These results indicate that the majority of SMSs form homodimers.
Cysteine Residues in the N Terminus of SMS1 and the C Terminus of SMS2 Are Involved in Cross-linking
Chemical cross-linking is a well accepted method to determine protein-protein interactions. To confirm the oligomeric structures of SMSs, we used a membrane-permeable cross-linking reagent, bismaleimidohexane (BMH), to cross-link cysteine residues. When HEK293 cells expressing V5-tagged SMS1 or SMS2 were treated with BMH, we observed a band of approximately twice the molecular mass of each SMS monomer (Fig. 2, A and B, WT), confirming the existence of homodimeric SMS1 as well as SMS2.
FIGURE 2.

Cys50 of SMS1 and Cys343/Cys348 of SMS2 are involved in cross-linking by cysteine cross-linking reagents. A–F, COS7 cells were transfected with a plasmid encoding FLAG-tagged SMS1-WT or SMS1 with endogenous cysteine substitutions (C25S, C50S, C145S, C187S, C217S, C227S, C244S, C277S, C311S, or C321S) (A, C, and E) or SMS2-WT or SMS2 with endogenous cysteine substitutions (C160S, C171S, C188S, C221S, C255S, C265S, C331S and C332S, or C343S and C348S) (B, D, and F). 24 h post-transfection, the cells were treated with 20 μm BMH (spacer arm, 13.0 Å) (A and B) or 20 μm of BMB (spacer arm, 10.9 Å) or BMOE (spacer arm, 8.0 Å) (C and D) for 15 min. A–D, the cells were lysed and analyzed by immunoblotting with anti-FLAG antibody. Results are from one experiment representative of three independent experiments. E and F, SMS activity in the lysate of COS7 cells expressing WT or cysteine-substituted SMSs were determined using C6-NBD-Cer as a substrate. Reaction mixtures containing cell lysates (20 μg of protein/assay) were incubated at 37 °C for 30 min. Individual data points are shown as a scatterplot. Values represent the mean ± S.D. from three independent experiments. *, p < 0.01; **, p < 0.05.
Next, we examined the proximal regions within the SMS homodimer using serine substitution mutational analysis with cross-linking. SMS1 contains 10 endogenous cysteine residues (Cys25, Cys50, Cys145, Cys187, Cys217, Cys227, Cys244, Cys277, Cys311, and Cys321) and so does SMS2 (Cys160, Cys171, Cys188, Cys221, Cys255, Cys265, Cys331, Cys332, Cys343, and Cys348). We created several mutants in which either one or two cysteine residues were substituted with serine. After COS7 cells were transfected with each indicated mutant, they were treated with 20 μm BMH and analyzed by immunoblotting using anti-FLAG antibody. The cross-linking efficiencies were decreased only in SMS1-C50S and SMS2-C343S/C348S compared with the WT SMSs (Fig. 2, A and B). These results clearly indicate that Cys50 of SMS1 and Cys343/Cys348 of SMS2 are involved in the cross-linking by BMH; Cys50 in SMS1 and Cys343/Cys348 in SMS2 appear to be located in the interface of the homodimers.
To further investigate the region proximal to the cross-linking residues of the SMS homodimers, cells transfected with each mutant were treated with cysteine cross-linkers having a shorter spacer length than BMH (spacer arm, 13.0 Å): bismaleimidobutane (BMB; spacer arm, 10.9 Å) or bismaleimidoethane (BMOE; spacer arm, 8.0 Å). Notably, essentially the same results were obtained as for the previous experiments; the cross-linking efficiencies were reduced only in SMS1-C50S and SMS2-C343S/C348S (Fig. 2, C and D). These results suggested that the regions around Cys50 in SMS1 and Cys343/Cys348 in SMS2 are flexible. To test whether SMS1-C50S and SMS2-C343S/C348S would maintain their normal conformation for SMS activity, we examined the SMS activity of each mutant. SMS1-C50S and SMS2-C343S/C348S had no effect on SMS activity, although SMS activity was severely decreased in SMS1-C227S, SMS1-C244S, SMS1-C277S, and SMS2-C188S and modestly decreased in SMS1-C311S, SMS1-C321S, and SMS2-C221S compared with the WT SMSs. These results suggest that the decreased cross-linking efficiencies of SMS1-C50S and SMS2-C343S/C348S are due to the loss of proximal cysteine residues for sufficient cross-linking and not due to disruption of the SMS conformation.
Topology of SMSs in Membranes
Next, we examined the orientation of the N and C termini in the SMSs and the transmembrane segments in SMS1 to identify the locations of Cys50 in SMS1 and Cys343/Cys348 in SMS2. The C termini of SMSs have been shown previously to have a cytoplasmic location by protease protection analysis (1); however, there are no experimental studies of the location of the N termini. The orientation of the V5-epitope at the N and C termini of the SMSs was determined and confirmed by antibody accessibility in differentially permeabilized cells. A low concentration of digitonin selectively permeabilizes the plasma membrane, whereas Triton X-100 can permeabilize all cellular membranes (18). As protein disulfide isomerase (PDI) resides in the ER lumen, selective membrane permeabilization was confirmed using an anti-PDI antibody. PDI signals were observed in Triton X-100-treated cells (Fig. 3, A–D; d, h, l, and p) but not digitonin-treated (Fig. 3; A–D, b, f, j, and n), showing that digitonin treatment does not allow the antibody to access the luminal side. Importantly, signals for the V5 epitope at the N and C termini of the SMSs were observed in Triton X-100-treated cells (Fig. 3, A–D; c, g, k, and o) as well as in digitonin-treated cells (Fig. 3, A–D; a, e, i, and m). These results indicate that both the N and C termini of the SMSs are oriented to the cytosolic side of the Golgi membrane.
FIGURE 3.

SMSs contain six transmembrane segments, and the N and C termini face the cytosolic side in the Golgi apparatus. A–D, COS7 cells were transfected with a plasmid encoding N-terminal V5-tagged SMS1 (V5-SMS1, A), C-terminal V5-tagged SMS1 (SMS1-V5, B), N-terminal V5-tagged SMS2 (V5-SMS2, C), or C-terminal V5-tagged SMS2 (SMS2-V5, D). After fixation and permeabilization with 0.002% digitonin or 0.1% Triton X-100 24 h post-transfection, the cells were stained with anti-V5 (SMS1 or SMS2) and anti-PDI (luminal marker) antibodies, followed by individual Alexa Fluor-conjugated secondary antibodies, and analyzed by confocal microscopy. SMS1 or SMS2, green; PDI, red. Scale bars = 40 μm. E, schematic showing the location of each of the eight cysteine residues that were individually introduced into the cysteine-null version of SMS1. The model has six membrane-spanning segments predicted by SOSUI, shown as a solid line, or four membrane-spanning segments, shown as a dashed line. The dashed line shows the only region different from a previous model (1). F, COS7 cells were transfected with a plasmid encoding the C-terminal V5-tagged SMS1-WT, Cys-null SMS1 (all Cys-to-Ser), or Cys-null with single Cys introductions (Cys50, Cys145, Cys187, Cys227, Cys259, Cys290, Cys311, or Cys333). After preparation of the membrane fraction, each fraction was treated with biotin maleimide and extracted. Precipitation was performed using NeutrAvidin agarose resin. The cell lysates and precipitated proteins were analyzed by immunoblotting (IB) with anti-V5 antibody. Top, cell lysate; bottom, precipitation. Results are from one experiment representative of three independent experiments.
Hydropathy analysis of SMSs has suggested two different transmembrane topology models. A model with six membrane-spanning segments was reported by Huitema et al. (1), and a model with four membrane-spanning segments is predicted by the SOSUI program (Fig. 3E). We employed the scanning cysteine accessibility method to identify the membrane segments of SMS1 from experimental data (19). We created several mutants in which all endogenous cysteines in SMS1 were substituted with serines (cysteine-null), and then a single cysteine was introduced into the cysteine-null SMS1 (S50C, S145C, S187C, S227C, I259C, T290C, S311C, or V333C) at the locations shown in Fig. 3E. Cell membrane fractions from COS7 cells transfected with each mutant were treated with 100 μm biotin maleimide, and the cell extracts were precipitated using NeutrAvidin agarose resins beads. Biotin maleimide, a membrane-permeable cysteine-reactive reagent, allows modification of cysteines at both the cytosolic/intracellular and luminal/extracellular sides but allows very low accessibility to residues in intramembrane segments (20). Precipitated bands were detected for the SMS1-C50 and SMS1-C259 mutants, suggesting that Cys50 and Cys259 were accessible for biotin maleimide modification, whereas there was no biotinylation in the cysteine-null mutant (Fig. 3F). This result is consistent with both a previous report (1) and location of Cys50 and Cys259 on the cytosolic and luminal side, respectively, as predicted by SOSUI. In contrast, precipitated bands were significantly reduced for the SMS1-C145, SMS1-C187, SMS1-C227, SMS1-C290, SMS1-C311, and SMS1-C333 mutants compared with the SMS1-C50 or SMS1-C259 mutants (Fig. 3F), suggesting that the corresponding residues are barely accessible to biotin maleimide. Although SOSUI predicted Cys290 and Cys311 to be located on the luminal side, our results clearly indicate that Cys145, Cys187, Cys227, Cys290, Cys311, and Cys333 are located within transmembrane segments. Taken together, we propose that SMS1 contains six transmembrane segments and that both the N and C termini face the cytosolic side, as shown in Fig. 3E, whereas Cys50 in SMS1 is expected to be located at the N-terminal tail in the cytosol.
As for SMS2, Cys333 in SMS1 corresponds to Val277 in SMS2, as indicated by sequence alignment analysis, suggesting that Val277 in SMS2 is also located in the sixth transmembrane segment. As the C terminus of SMS2 is oriented toward the cytosol, Cys343/Cys348 in SMS2 likely is located at the C-terminal tail in the cytosol.
N-terminal and/or C-terminal Tails of the SMS Homodimer Pairs Are Located in Proximity to Each Other
To clarify whether the N termini of SMS1 and the C termini of SMS2 homodimers are in close proximity, we employed a BiFC assay using several chimera proteins in which the N- or C-terminal half fragments of fluorescent protein Venus (VN or VC) were fused to the N or C terminus of either SMS (Fig. 4A). The BiFC assay is based on the complementation of VN and VC in chimeric SMS proteins; when VN and VC in the chimera are placed in close proximity (less than 15 nm) and allowed to interact, the complex emits fluorescence (21, 22). The chimera in which VN or VC is fused to the C terminus of SMS2, yielding SMS2-VN or SMS2-VC, respectively, was co-expressed in COS7 cells, and BiFC signals were observed using confocal microscopy. As shown in Fig. 4B, a BiFC signal was observed in the perinuclear region and plasma membrane, where the proteins co-localized (Fig. 4B, a and b). This result indicates that the C-terminal tail of SMS2 is in close proximity to the C-terminal tail of another SMS2 within the homodimer in the Golgi apparatus, supporting the results obtained by using the cysteine-specific cross-linkers (Fig. 2, B and D). Another chimeric protein, in which VC was fused to the N terminus of Cer glucosyltransferase (GlcT), VC-GlcT, was employed as a negative control to exclude the possibility of nonspecific interactions between VN and VC. The BiFC signal was not observed when SMS2-VN and VC-GlcT were co-expressed, despite SMS2 and GlcT co-localizing in the perinuclear region (Fig. 4B, b). This result suggests that the C-terminal tail of SMS2 and the N-terminal tail of GlcT are not close enough for the complementation of Venus.
FIGURE 4.

The two N-terminal tails, the N- and C-terminal tails, and the two C-terminal tails in the SMS homodimers are in close proximity. A, schematic of the chimeric proteins used for BiFC assays. The chimeras included VN (N-terminal residues 1–173 of the Venus protein) and V5 epitope or VC (C-terminal residues 155–238 of the Venus protein) and FLAG epitope. B–D, COS7 cells were transfected with a plasmid encoding V5-tagged SMS2-VN and FLAG-tagged SMS2-VC or V5-tagged SMS2-VN and FLAG-tagged VC-GlcT. 24 h post-transfection, the cells were fixed and permeabilized with 0.1% Triton X-100. For confocal microscopy (B), the cells were stained with anti-V5 and anti-FLAG antibodies, followed by appropriate Alexa Fluor-conjugated secondary antibodies. For flow cytometry (C and D), the cells were stained with anti-V5 antibody, followed by PerCP/Cy5.5-conjugated secondary antibody and APC-conjugated anti-FLAG antibody. B, SMS2-VN, blue; SMS2-VC or VC-GlcT, red; Venus, green. B, a close-up view of the plasma membrane in the squared region in a. Scale bar = 10 μm. C, representative dot plot from cells expressing SMS2-VN/SMS2-VC (left panel) and SMS2-VN/VC-GlcT (right panel). The gating of the double-positive cell population expressing both VN- and VC-fused proteins is shown as a bold square. D, FACS histogram representing the intensities of Venus fluorescence detected by the FITC channel in double-positive cells (PerCP-Cy5.5/APC). Histograms represent the extent of FITC quantified in cells expressing SMS2-VN/SMS2-VC (filled) or SMS2-VN/VC-GlcT (open). E and F, the intensity of Venus fluorescence in COS7 cells co-expressing the indicated combinations of VN- or VC-fused SMS1 (E) or SMS2 (F) was quantified by flow cytometry. Individual data points are shown as a scatterplot. Values represent the mean ± S.D. from at least three independent experiments. *, p < 0.01; **, p < 0.05. G and H, schematics of SMS homodimers, viewed as a section through the Golgi or plasma membrane (G) and viewed from the cytosolic space (H). Individual monomers of transmembrane segments in SMSs are shown in black and gray. N- and C-terminal tails are represented by blue and red lines, respectively. 1–6 and I-VI indicated the transmembrane helices. H, SMS1 and SMS2 have the following intermolecular interactions: between the N-terminal tails, between the N-terminal tail and C-terminal tail, and between the C-terminal tails, shown as dashed arrow lines.
Next, we quantified fluorescence complementation by measuring the Venus fluorescence intensity of cells by flow cytometry (23). To compare the fluorescence intensity among different combinations of chimeric proteins, we selected populations expressing equivalent levels of V5-tagged VN proteins (PerCP-Cy5.5) and FLAG-tagged VC proteins (APC) (Fig. 4C). The gated APC/PerCP-Cy5.5 double-positive cells showed that Venus fluorescence intensity generated by SMS2-VN and SMS2-VC was ∼130-fold higher than that from SMS2-VN and VC-GlcT (Fig. 4D). Next, we examined the proximity of the N-terminal tails between the N-terminal and C-terminal tails and between the C-terminal tails in SMS homodimers. Interestingly, a Venus signal was detected when VN-SMS1/VC-SMS1, SMS1-VN/SMS1-VC, or VN-SMS1/SMS1-VC were co-expressed (Fig. 4E, column 1, 2, or 3), indicating that the N/C-terminal tail in SMS1 is in close proximity to the N/C-terminal tail of another SMS1 in the homodimer (Fig. 4, G and H). Given that the measured BiFC signal for VN-SMS1/SMS1-VC was somewhat higher than that for the other combinations, the distance between the N-terminal and C-terminal tails was less than that between the two N-terminal tails or between the two C-terminal tails in the SMS1 homodimer. Similar results were observed for SMS2; a signal was detected when VN-SMS2/VC-SMS2, SMS2-VN/SMS2-VC, or VN-SMS2/SMS2-VC were co-expressed (Fig. 4F, column 1, 2, or 3). Collectively, the results demonstrate that the two N-terminal tails, the N-terminal tail and C-terminal tail, and the two C-terminal tails in a SMS2 homodimer are all in close proximity to each other (Fig. 4, G and H).
C-terminal Truncation of SMSs Impairs Homodimer Formation
To examine whether the N- and/or C-terminal tails of SMSs are involved in the interactions to form homodimers, we created N- and/or C-terminal truncation mutants of the SMSs (Fig. 5A) and then analyzed homodimerization by BN-PAGE. Interestingly, the ratios of monomer to homodimer were significantly increased in SMS1-ΔC22 and SMS1-ΔN68C22 compared with SMS1-WT (Fig. 5, B and C). Given that there were no obvious differences in monomer amounts between SMS1-WT and SMS1-ΔN68 as well as between SMS1-ΔC22 and -ΔN68C22, the C-terminal tail, comprised of amino acids Pro392 to Thr413, is involved in homodimer formation whereas the N-terminal tail, comprised of amino acids Met1 to Lys68, is not, despite the close proximity of the N-terminal tails and N-terminal and C-terminal tails in the SMS1 homodimer. Similar results were observed for SMS2. The ratios of monomer to homodimer were significantly increased in SMS2-ΔC30 compared with SMS2-WT (Fig. 5, D and E). This result indicates that the C-terminal tail, comprised of amino acids Pro336 to Thr365, in SMS2 is responsible for homodimerization.
FIGURE 5.

C-terminal truncations of SMSs decrease homodimer formation. A, schematic of the N- and/or C-terminal truncations of SMS1 and SMS2. Cys50 in SMS1 and Cys343/Cys348 in SMS2 are represented as C in the circle and were identified as sites cross-linked by cysteine cross-linking reagents (Fig. 2). The boxes represent transmembrane (TM) segments TM1 to TM6 as indicated. All constructs contain a FLAG or V5 tag at the C terminus. Both the C-terminal deletion mutants SMS1-ΔC22 and SMS2-ΔC30 are expected to have 44-amino acid extensions from the end of TM6. B–E, COS7 cells were transfected with a plasmid encoding V5-tagged SMS1-WT, SMS1-ΔN68, SMS1-ΔC22, or SMS1-ΔN68C22 (B and C) or V5-tagged SMS2-WT or SMS2-ΔC30 (D and E). 24 h post-transfection, the membrane fractions were separated by BN-PAGE and immunoblotted with anti-V5 antibody (B and D). The monomer-to-homodimer ratio was determined from the intensities of monomer and homodimer in the immunoblot assay (C and E). Individual data points are shown as a scatterplot. Values represent the mean ± S.D. from at least three independent experiments. *, p < 0.01.
To further examine the oligomeric states of the N- and/or C-terminal truncation mutants of SMSs, immunoprecipitation assays were performed. Immunoprecipitation using the combinations SMS1-ΔC22/SMS1-ΔC22 and SMS1-ΔN68C22/SMS1-ΔN68C22 generated very little signal compared with SMS1-WT/SMS1-WT and SMS1-ΔN68/SMS1-ΔN68 (Fig. 6, A, bottom panel, right, and B). These results indicate that the C-terminal tail, but not the N-terminal tail, is responsible for homodimer formation, supporting the results of BN-PAGE (Fig. 5, B and C). Interestingly, the interaction affinity of SMS1-WT/SMS1-ΔC22 and SMS1-ΔN68/SMS1-ΔC22 was somewhat lower than that of SMS1-WT/SMS1-WT but not comparable with that of SMS1-ΔC22/SMS1-ΔC22 (Fig. 6, C, bottom panel, right, and D). These results indicate that the C-terminal tails of SMS1-WT and SMS1-ΔN68 can interact weakly with the C-terminal tail within a homodimer but that the interaction is stronger with other segments in SMS1 (Fig. 6E). This suggested that the interactions between the C-terminal tails are not primarily responsible for homodimerization of SMS1.
FIGURE 6.

The C-terminal tails of SMS1 are important for homodimeric interactions. A–D, COS7 cells were transfected with a plasmid encoding FLAG- or V5-tagged SMS1-WT, SMS1-ΔN68, SMS1-ΔC22, or SMS1-ΔN68C22 as indicated. A and C, left, cell lysates; right, immunoprecipitates (anti-FLAG beads). IB, immunoblot; IP, immunoprecipitation. B and D, the quantities of V5-tagged SMS1 precipitated with FLAG-tagged SMS1 are represented as the ratios of the band intensities. Results are from one experiment representative of at least three independent experiments. Individual data points are shown as a scatterplot. Values represent the mean ± S.D. from at least three independent experiments. *, p < 0.01; **, p < 0.05. E, the interaction between the C-terminal tails in SMS1, shown as a thin arrow line, is not primarily responsible for the formation of the SMS1 homodimer. The bold arrow line shows the interaction between the C-terminal tail and another, unidentified region in SMS1 (U in the box), which seems to be primarily responsible for the formation of the SMS1 homodimer.
In the case of SMS2, immunoprecipitation with SMS2-ΔC30/SMS2-ΔC30 yielded little signal compared with SMS2-WT/SMS2-WT (Fig. 7, A, bottom panel, right, and B). Furthermore, the interaction affinity of SMS2-WT/SMS2-ΔC30 was higher than that of SMS2-ΔC30/SMS2-ΔC30 but significantly lower than that of SMS2-WT/SMS2-WT. These results indicate that the interaction between the C-terminal tails in the SMS2 homodimer is primarily responsible for homodimer formation, although the C-terminal tail of SMS2 can interact weakly with other segments within the homodimer (Fig. 7C).
FIGURE 7.

The C-terminal tails of SMS2 are important for homodimer interactions. A and B, COS7 cells were transfected with a plasmid encoding FLAG- or V5-tagged SMS2-WT or SMS2-ΔC30 as indicated. A, left, cell lysates; right, immunoprecipitates (anti-FLAG beads). IB, immunoblot; IP, immunoprecipitation. B, the quantities of V5-tagged SMS2 precipitated with the FLAG-tagged SMS2 are represented as ratios between the band intensities. Individual data points are shown as a scatterplot. Values represent the mean ± S.D. from three independent experiments. *, p < 0.01. C, the interaction between the C-terminal tails in SMS2, shown as a bold arrow line, is primarily responsible for SMS2 homodimerization. The thin arrow line shows the interaction between the C-terminal tail and another, unidentified portion in SMS2 (U in the box), which seems not to be primarily responsible for SMS2 homodimerization. D, COS7 cells were transfected with plasmids encoding V5-tagged SMS2-WT together with FLAG-tagged SMS2-WT or SMS2-C343S/C348S as indicated. Results are from one experiment representative of three independent experiments.
Cysteine residues mediate dimerization via intermolecular disulfide bonds (24). We examined whether Cys343 and Cys348 in SMS2 are important for homodimer formation, although it is unclear whether these cysteines might form disulfide bonds. When COS7 cells co-expressed V5-tagged SMS2 and either FLAG-tagged SMS2-WT or SMS2-C343S/C348S, the amount of V5-tagged SMS2 co-immunoprecipitated with FLAG-tagged SMS2-WT was comparable with that precipitated with FLAG-tagged SMS2-C343S/C348S (Fig. 7D). This result suggests that Cys343/Cys348 are not involved in disulfide bond and homodimerization formations of SMS2.
Furthermore, SMS2 is known to be palmitoylated; Cys343/Cys348 are major palmitoylation sites (25). Our results also suggest that palmitoylation of Cys343/Cys348 in SMS2 is not responsible for homodimerization.
SMSs Form Homodimers in the ER Membrane
To establish where SMSs assemble as homodimers during synthesis and trafficking, the effect of brefeldin A (BFA), an inhibitor of ER-to-Golgi traffic, on homodimer formation was examined. After COS7 cells were treated with BFA, they were transfected with V5-tagged SMS1 or SMS2. Immunofluorescence staining of non-BFA-treated cells demonstrated that SMS1 was mainly localized in the perinuclear region (Fig. 8A, a), whereas SMS2 was localized on the cell surface and concentrated structures in the perinuclear region (Fig. 8A, c). These locations were consistent with the findings of previous studies (1, 11). The subcellular localizations of SMSs were altered by BFA treatment; most co-localized with PDI, a marker of the ER (Fig. 8A, b and d). However, no significant differences in SMS homodimer formation were observed in BN-PAGE after treatment with BFA (Fig. 8B). These results indicate that homodimerization of SMSs occurs in the ER membrane.
FIGURE 8.

SMSs form homodimers in the ER membrane. A and B, COS7 cells were incubated for 14 h in the presence or absence of 50 nm BFA. Then the cells were transfected with a plasmid encoding V5-tagged SMS1 or SMS2 and cultured for an additional 24 h. A, the cells were stained with anti-V5 (SMS1 or SMS2) and anti-PDI (an ER marker) antibodies, followed by appropriate Alexa Fluor-conjugated secondary antibodies, and analyzed by confocal microscopy. SMS1 or SMS2, green; PDI, red. Scale bar = 40 μm. B, the membrane extracts were resolved by BN-PAGE and detected by immunoblotting with anti-V5 antibody. The results are from one experiment representative of three independent experiments.
Reduced Golgi Localization of C-terminal Truncation Mutants
There are several reports that protein oligomerization is required for proper subcellular localization (26–31); therefore, we examined the subcellular localization of N- and/or C-terminal truncation mutants to obtain insights into the functions of SMS homodimers. Confocal microscopy showed that SMS1-WT co-localized with the Golgi marker GM130 (Fig. 9A, a) but not with PDI (Fig. 9B, b). SMS1-ΔN68 showed a localization pattern similar to that of SMS1-WT (Fig. 9, A, c; B, d; and E), suggesting that N-terminal truncation of SMS1 does not influence its subcellular location. Interestingly, SMS1-ΔC22 and SMS1-ΔN68C22 were observed to have different distributions, showing an elliptical morphology that co-localized with PDI (Fig. 9B, f and h) but not with GM130 (Fig. 9A, e and g). These structures were morphologically similar to organized smooth ER (OSER), which forms by expression of particular ER-resident transmembrane proteins with weak homodimerization (32, 33). OSER formation in cells expressing SMS1-ΔC22 and SMS1-ΔN68C22 may be due to a decreased affinity for homodimer formation by the C-terminal truncation.
FIGURE 9.

C-terminal truncation mutants decrease the Golgi localization of SMSs. A–F, COS7 cells were transfected with a plasmid encoding C-terminal V5-tagged SMS1-WT, SMS1-ΔN68, SMS1-ΔC22, or SMS1-ΔN68C22 (A, B, and E), or SMS2-WT or SMS2-ΔC30 (C, D, and F). 24 h post-transfection, the cells were stained with anti-V5 antibody (SMS1 or SMS2), followed by appropriate Alexa Fluor-conjugated secondary antibodies, and analyzed by confocal microscopy. Localization was confirmed by co-staining with antibodies against the Golgi marker GM130 (A and C) or the ER marker PDI (B and D). SMS1 or SMS2, green; GM130 or PDI, red. Scale bars = 40 μm. Arrows and arrowheads indicate the localization of SMS1 at the Golgi apparatus and OSER, respectively. E and F, SMS localization was assessed in at least 150 cells using confocal microscopy, and the localization ratio was calculated by dividing the number of cells showing subcellular protein localization by the total number of observed cells. Individual data points are shown as a scatterplot. Values represent the mean ± S.D. from four independent experiments. *, p < 0.01. G and H, SMS activity in the lysates of COS7 cells expressing WT or truncated mutants were determined using C6-NBD-Cer as a substrate. Reaction mixtures containing cell lysates (10 μg of protein/assay) were incubated at 37 °C for 30 min. Individual data points are shown as a scatterplot. Values represent the mean ± S.D. from four independent experiments. *, p < 0.01; NS, not significant.
Confocal microscopy corroborated that SMS2-WT mainly co-localized with GM130 (Fig. 9C, i) but not with PDI (Fig. 9D, j). Interestingly, SMS2-ΔC30 showed a different distribution, displaying clear ER localization (Fig. 9, C, k; D, l; and F). Importantly, the results were essentially the same as those obtained for SMS1; the C-terminal truncations of SMS1 and SMS2 induced ER confinement of SMSs. Similar results were obtained in N-terminal V5-tagged SMS1-ΔC22 or SMS2-ΔC30, which were mainly localized in the OSER and ER, respectively (data not shown).
To exclude the possibility that the altered localization of the C-terminal truncation mutants in SMSs was caused by protein misfolding, we examined the SMS activity of each mutant. All N- and/or C-terminal truncation mutants had comparable activity in an in vitro SMS assay using C6-NBD-Cer (Fig. 9, G and H). These results suggest that the altered subcellular localization of C-terminally truncated SMSs is not due to protein misfolding or aggregation.
C-terminal Tails of SMSs Do Not Function as an ER Export Motif
Protein trafficking between the ER and Golgi apparatus is mediated by the coat protein complex (COP). COPI mediates retrograde transport (34), whereas COPII mediates anterograde transport (35). In general, selective transport depends on the interaction between a motif in the cytoplasmic region of the cargo and the cargo recognition site of COP. We examined the possibility that C-terminal truncations in SMSs expose a hidden motif for retrograde transport or remove the endogenous motif for anterograde transport, which might lead to an alteration in subcellular localization. Although there are no known ER retention/retrieval motifs for COPI in the residual C-terminal tails of SMS1-ΔC22 and SMS2-ΔC30, a diacidic motif (360DNE362) for COPII was found in SMS2-ΔC30 (Fig. 10A). A DXE motif, where X stands for any amino acid, has been shown to interact with the coat component Sec24 and mediate efficient ER export of vesicular stomatitis virus glycoprotein (36). To test whether the DNE sequence in SMS2 could function as an ER export motif, we generated a V5-tagged SMS2-ΔC6 that deleted Asp360 to Thr365 of SMS2. The ratio of SMS2-ΔC6 monomer to homodimer was not obviously different from that of SMS2-WT, whereas there was a significant decrease for SMS2-ΔC30 (Fig. 10, B and C). Importantly, SMS2-ΔC6 mostly co-localized with the Golgi marker GM130 (Fig. 10D, a) but not with the ER marker PDI (Fig. 10D, b), showing a subcellular distribution similar to that of SMS2-WT (Figs. 9F and 10E). These results suggest that 360DNE362 in SMS2 is not an ER export motif to interact with the coat component Sec24.
FIGURE 10.

C-terminal tails of SMSs do not function as ER export motifs. A, amino acid sequence alignment of the C-terminal truncated regions of SMS1-ΔC22 (from Pro392 to Thr413) and SMS2-ΔC30 (from Pro336 to Thr365). The asterisks indicate identical residues. The DXE sequence, where X is any amino acid, known to be an ER export motif, is highlighted in black. B–E, COS7 cells were transfected with a plasmid encoding C-terminal V5-tagged SMS2-WT, SMS2-ΔC6 (truncation of the DXE motif), or SMS2-ΔC30. B, membrane extracts were resolved by BN-PAGE and immunoblotted with anti-V5 antibody. C, the SMS2 monomer-to-homodimer ratio was determined from the intensities of SMS2 monomer and homodimer bands in BN-PAGE. *, p < 0.01; NS, not significant. D, after fixation and permeabilization, cells were stained with anti-V5 (SMS2-ΔC6) and anti-GM130 (a Golgi marker) or anti-PDI (an ER marker) antibodies, followed by Alexa Fluor-conjugated secondary antibodies, and analyzed by confocal microscopy. SMS2-ΔC6, green; GM130 or PDI, red. Scale bar = 40 μm. E, SMS2-ΔC6 localization was assessed in 270 cells using confocal microscopy, and the localization ratio was calculated by dividing the number of cells showing subcellular protein localization by the total number of observed cells. Individual data points are shown as a scatterplot. Values represent the mean ± S.D. from three independent experiments. F and G, the constructs were designed with the amino acid sequences from Pro392 to Thr413 of SMS1 or from Pro336 to Thr365 of SMS2 fused to the C terminus of MBOAT5. COS7 cells were transfected with a plasmid encoding C-terminal V5-tagged MBOAT5-SMS1392–413 or MBOAT5-SMS2336–365 chimeric protein. After fixation and permeabilization, cells were stained with anti-V5 (MBOAT5 or chimeric protein) and anti-PDI (an ER marker) (F) or anti-GM130 (a Golgi marker) (G) antibodies, followed by appropriate Alexa Fluor-conjugated secondary antibodies. MBOAT5 and chimeric protein, green; PDI or GM130, red. Scale bars = 20 μm.
To examine whether the C-terminal tails of the SMSs contain an ER export motif, we generated chimeric proteins in which the Pro392 to Thr413 of SMS1 or Pro336 to Thr365 of SMS2 were fused to the C terminus of the ER-resident protein MBOAT5 (LPCAT3, lysophosphatidylcholine acyltransferase) (37). MBOAT5-WT mainly co-localized with PDI (Fig. 10F, a) but not with GM130 (Fig. 10G, b), which is consistent with a previous report (38). Importantly, the subcellular localization of the chimera of MBOAT5 with the C-terminal tail of SMS1 or of SMS2 (MBOAT5-SMS1–395-413 or MBOAT5-SMS2–336-365) showed similar expression patterns as MBOAT5-WT (Fig. 10, F, c and e, and G, d and f). These results indicate that the C-terminal tails of SMSs do not function as an ER export motif and that the altered subcellular localizations of the proteins are not due to the removal of an endogenous motif for ER export.
Fluorescent Protein Tagging Compensates for Reduced Homodimer Formation and Subcellular Trafficking of SMSs by C-terminal Truncation
Previously, Yeang et al. (39) reported that N-terminal enhanced GFP-tagged SMS1-ΔC27 or SMS2-ΔC32 had no obvious impact on export from the ER. To investigate the inconsistency, we examined the effect of fluorescent protein tagging on SMS dimerization. To this end, we generated a chimera in which strongly enhanced GFP (SGFP2) was fused to the C terminus of SMSs-WT or C-terminal truncation mutants and then analyzed homodimer formation by BN-PAGE. There were no obvious differences in homodimer formation between SGFP2-tagged SMS1-WT and SMS1-ΔC22 as well as SMS2-WT and SMS2-ΔC30 (Fig. 11A). These results indicate that the reduced homodimerization of SMS1-ΔC22 or SMS2-ΔC30 by C-terminal truncation is restored by SGFP2 tagging. Confocal microscopy showed that SGFP2-tagged SMS1-WT and SMS2-WT mainly co-localized with the Golgi marker GM130 (Fig. 11B, a and c). Interestingly, SGFP2-tagged SMS1-ΔC22 and SMS2-ΔC30 showed a subcellular distribution similar to that of the respective SMS-WT, displaying a clear Golgi pattern (Fig. 11B, b and d). The confocal images are consistent with the results reported by Yeang et al. (39), regardless of the differences in position (N- or C terminus of SMSs) and type of fluorescent protein tag (enhanced GFP or SGFP2). Given that augmented interaction by SGFP2-tagging of C-terminal truncation mutants (Fig. 11A) restored export from the ER (Fig. 11B), SMS homodimer formation was thought to be an important determinant for ER export of each SMS isoform.
FIGURE 11.

The fluorescent protein tag compensates the reduced homodimerization and ER-to-Golgi trafficking of SMSs by C-terminal truncation. A and B, COS7 cells were transfected with a plasmid encoding C-terminal SGFP2-tagged SMS1-WT and SMS2-WT and their truncation mutants (SMS1-ΔC22 and SMS2-ΔC30, respectively). A, membrane extracts were resolved by BN-PAGE and immunoblotted with anti-GFP antibody. B, after fixation and permeabilization, cells were stained with anti-GM130 (a Golgi marker) antibody, followed by appropriate Alexa Fluor-conjugated secondary antibodies, and analyzed by confocal microscopy. SMS1 or SMS2, green; GM130, red. Scale bar = 40 μm.
Collectively, our results indicate that the C-terminal tails of SMSs contribute significantly to intermolecular interactions and stabilize SMS homodimers (Figs. 6E and 7C). Deletion of the C-terminal tail disturbs these interactions within homodimers, which might lead to a reduction in homodimer formation and inefficient ER-to-Golgi transport of both SMS1 and SMS2.
Discussion
In this study, BN-PAGE clearly demonstrated that most SMS1 and SMS2 exist as homodimers (Fig. 1C). Cysteine substitution analysis revealed that the two N-terminal tails of SMS1 and the two C-terminal tails of SMS2 are in close proximity within each homodimer (Fig. 2). Furthermore, a BiFC assay suggested that the two N-terminal tails, the N-terminal tail and C-terminal tail, and the two C-terminal tails in an SMS homodimer are all in close proximity to each other (Fig. 4, G and H).
Based on the BN-PAGE (Fig. 5, B and D) and immunoprecipitation results (Figs. 6A and 7A), we expected the C-terminal tails of SMSs to contribute to these intermolecular interactions. However, immunoprecipitation using SMSs-WT and the C-terminal truncation mutants suggested that SMS1 and SMS2 considerably differ in terms of the affinity of the homodimer interaction via the C-terminal tails (Figs. 6C and 7A). The interaction between the C-terminal tail and a site outside of the C-terminal tail is involved in the formation of the SMS1 homodimer, whereas the interaction between C-terminal tails is primarily responsible for SMS2 homodimerization. The second site involved in the homodimerization of SMS1 was not elucidated. However, given that SMS1-ΔN119, in which almost the entire N-terminal tail was truncated (Fig. 12), showed no significant decrease in interaction affinity in immunoprecipitation experiments (data not shown), the cytosolic loop(s) might be involved in the interaction with the C-terminal tail in SMS1.
FIGURE 12.

Predicted membrane topology of human SMS1 and the positions of Cys substitutions and N- or C-terminal truncations of SMS1 mutants in this study. Snake diagram depicting the predictions of a previous report (1) and this study (Fig. 3). Cys substitutions in SMS1 that result in decreases of >80% and >20% in SMS activity compared with WT (Fig. 2E) are indicated by red filled circles and yellow filled squares, respectively. His285, a catalytic acid-base residue (48), is shown as a black filled circle. Residues in the intramembrane and non-transmembrane segments identified by the scanning cysteine accessibility method (Fig. 3F) are indicated by blue open squares and red open squares, respectively. The Cys residue responsible for homodimer formation by cysteine cross-linking (Fig. 2, A and C) is indicated by a green filled square. Arrows indicate the sites of truncation to form SMS1-ΔN68, SMS1-ΔN119, or SMS1-ΔC22.
Furthermore, C-terminal truncations of SMS1 and SMS2 reduced but did not eliminate homodimer formation (Fig. 5, B–E), suggesting that other interactions may be involved. Lateral interactions between transmembrane helices in membrane proteins are known to be important for oligomer formation (40). The intermolecular helix-helix interactions between transmembrane domains in SMSs require further examination to elucidate their functions in homodimer formation.
The obtained set of evidence for homodimer formation of SMS1 and SMS2 prompted us to explore the function and/or physiological significance of homodimerization. We evaluated whether decreased homodimerization might impact the subcellular localization of SMSs (Fig. 9, A–F). Although most of SMS1-WT was localized in the Golgi apparatus, the C-terminal truncation mutant SMS1-ΔC22 localized less to the Golgi but accumulated in the OSER. Similarly, SMS2-ΔC30 was mainly localized in the ER, whereas SMS2-WT localized to the Golgi apparatus and plasma membrane. OSER structures are known to form as a result of the overexpression of some ER-resident transmembrane proteins such as cytochrome b5 (32), calnexin (41), vesicle-associated membrane protein-associated protein B (VAPB) (42), and Lap2β (43). The attachment of GFP, which retains the ability to form weak homodimers, to the cytoplasmic region of an ER-resident protein is sufficient for generating OSER structures. Given that the C-terminal tail of SMS1 faced the cytosolic side (Fig. 3B) and that the truncation of the C-terminal tail significantly decreased homodimer formation (Fig. 5, B and C) and immunoprecipitation (Fig. 6, A and B), OSER formation through the expression of SMS1-ΔC22 appears to be due to the attenuation of homodimeric interactions in the cytosolic region. Why the expression of SMS2-ΔC30 did not cause OSER formation remains unclear, but this observation might be attributed to slight differences in interaction affinities in the residual C-terminal tails of SMS1-ΔC22 and SMS2-ΔC30. Importantly, essentially the same results were obtained for SMS1-ΔC22 and SMS2-ΔC30. The C-terminal truncations of both isoforms caused subcellular mislocalization. It is unlikely that the altered subcellular localization of C-terminally truncated SMSs was the result of protein misfolding because these mutations had no effect on SMS activity in vitro (Fig. 9, G and H). Hence, we examined whether this subcellular mislocalization was due to the exposure/removal of motifs for COPI/COPII. We did not detect ER retention/retrieval motifs for COPI, such as KXKXX and KKXX (34), RR, RXR, and RXXR (44), in the newly exposed C-terminal tails of SMS1-ΔC22 and SMS2-ΔC30. In addition, the C-terminal tails of SMSs contain no functional unit for ER export (Fig. 10).
We found that fluorescent protein tagging compensated for the reduced dimerization and ER exit by C-terminal truncation (Fig. 11, A and B). Furthermore, even half-size fluorescent protein (VC) could compensate for the defect of SMS dimerization by truncation of the C-terminal tail and lead to restored ER export (data not shown). Fluorescent proteins provide a very useful experimental tool; however, they sometimes alter protein functional behavior and subcellular localization because of their relatively large molecular mass and dimer formation (45, 46). We reckon that augmented interaction by dimerization of the fluorescent proteins increases the stability of homodimer of SMSs, leading to restored ER export. The results are consistent with the idea that SMS homodimerization is an important determinant for ER export of both SMS1 and SMS2.
As we could not detect SMSs in the immunoprecipitates of COPII component Sec24 isoforms (data not shown), SMSs might be exported from the ER in a COPII-independent manner. There are several reports that protein oligomerization is required for export from the ER (26–31); however, the precise mechanisms of intracellular trafficking of oligomerized membrane proteins are not fully understood. Interestingly, Springer et al. (47) have proposed a model in which oligomerization can generate a local membrane curvature that promotes vesicle formation, and, thus, the oligomeric form itself can act as an ER export signal without direct interaction with COPII components. Based on the present results, we propose that proper homodimerization of SMSs is necessary for efficient ER-to-Golgi transport.
Another insight was drawn from data on the catalytic activity of cysteine-substituted SMSs. SMS activity was significantly decreased in the mutants SMS1-C244S, SMS1-C277S, and SMS2-C188S compared with WT SMSs (Fig. 2, E and F). Interestingly, although sequence alignment of SMS1 with SMS2 revealed that Cys244 and Cys277 in SMS1 correspond to Cys188 and Cys221 in SMS2, respectively, SMS2-C221S had little effect on enzyme activity. The different contributions of corresponding Cys residues in SMSs may reflect the unique active-site configuration of each isoform. Indeed, Cys244/Cys277 in SMS1 and Cys188/Cys221 in SMS2 are evolutionarily conserved between human SMSs and Caenorhabditis elegans SMS (Swiss-Prot accession number Q9U3D4.2). His285 in SMS1 and His229 in SMS2 were identified as the catalytic acid-base residues corresponding to those in the lipid phosphate phosphatase family (48). Given that these cysteines are located in the same second luminal loop and close to the catalytic acid-base residue, they might determine the overall stability of the substrate-binding domain (Figs. 12 and 13). Further investigations, such as X-ray crystallography and NMR studies, are required to reveal how these cysteines contribute to the structures of SMSs.
FIGURE 13.

Predicted membrane topology of human SMS2 and the positions of Cys substitutions and C-terminal truncations of SMS2 mutants in this study. A snake diagram depicting the predictions of a previous report (1) and this study (Fig. 3). Cys substitutions in SMS2 that result in decreases of >80% and >20% in SMS activity compared with WT (Fig. 2F) are indicated by red filled circle and yellow filled squares, respectively. His229, a catalytic acid-base residue (48), is shown as a black filled circle. Cys residues that are responsible for homodimer formation by cysteine cross-linking (Fig. 2, B and D) are indicated by green filled squares. Arrows indicate the sites of truncation to form SMS2-ΔC6 or SMS2-ΔC30.
In summary, this study, for the first time, shows that both SMS1 and SMS2 form homodimers. The C-terminal tails of both isoforms are largely responsible for homodimerization. Reduced homodimer formation causes inefficient transport from the ER of both SMS1 and SMS2. As the subcellular localization of SMSs is considered an important determinant of enzyme function, this study might provide useful clues for the functional characterization of both SMS1 and SMS2.
Experimental Procedures
Antibodies
Mouse IgG1 monoclonal anti-FLAG (catalog no. F1804, lot no. 124K6106), rabbit polyclonal anti-V5 (catalog no. V8137, lot no. 112M4850V), and rabbit anti-PDI (catalog no. P7372, lot no. 084K4826) antibodies and anti-FLAG affinity gel (catalog no. A2220, lot no. SLBG5784V) were obtained from Sigma. Mouse anti-GM130 antibody (catalog no. 610822, lot no. 70228) was from BD Biosciences. Mouse IgG2a monoclonal anti-V5 (catalog no. 46–0705, lot no. 1718556), goat HRP-conjugated anti-mouse IgG (catalog no. 62–6520, lot no. QG215721), anti-mouse IgG-Alexa Fluor 488 (catalog no. A11001, lot no. 774904), anti-rabbit IgG-Alexa Fluor 488 (catalog no. A11008, lot no. 751094), anti-mouse IgG-Alexa Fluor 546 (catalog no. A11030, lot no. 1345046), anti-rabbit IgG-Alexa Fluor 546 (catalog no. A11010, lot no. 1600212), and anti-rabbit IgG-Alexa Fluor 405 (catalog no. A31556, lot no. 1145173) antibodies were obtained from Invitrogen). Rat PerCP/Cy5.5-conjugated anti-mouse IgG2a antibody (catalog no. 407111, lot no. B207066) was from BioLegend. Mouse APC-conjugated anti-FLAG antibody (catalog no. ab72569, lot no. GR193571–8) and anti-GFP antibody (catalog no. ab290, lot no. GR278073-1) were obtained from Abcam.
Plasmids
We used the following nomenclature for epitope-tagged enzymes: TagA-(enzyme)-TagB indicates that TagA and TagB are located at the N- and C terminus, respectively. SMS1-V5 stands for SMS1 with V5-tag at the C terminus, VN-SMS1 stands for SMS1 with N-terminal residues 1–173 of Venus at the N terminus. Plasmids for retroviral expression of SMSs (pQCXIP/SMS1-V5 and pQCXIP/SMS2-V5) were constructed as described previously (11). SMS expression vectors (pcDNA4TO/SMS1-FLAG pcDNA4TO/SMS1-V5, pcDNA4TO/V5-SMS1, pcDNA4TO/SMS2-FLAG, pcDNA4TO/SMS2-V5, and pcDNA4TO/V5-SMS2) were constructed by PCR with pQCXIP/SMS1-V5 or pQCXIP/SMS2-V5 as a template, using specific primers containing sequences corresponding to the FLAG epitope in front of the stop codon or V5 epitopes in front of the stop codon or start methionine. The PCR products were subcloned into pcDNA4TO (Invitrogen). The single- or double-endogenous cysteines in the open reading frames of SMSs were mutated to serine (SMS1-C25S, -C50S, -C145S, -C187S, -C217S, -C227S, -C244S, -C277S, -C311S, or -C321S and SMS2-C160S, -C171S, -C188S, -C221S, -C255S, -C265S, -C331S/C332S, or -C343S/C348S) using the QuikChange site-directed mutagenesis kit (Stratagene) with pcDNA4TO/SMS1-FLAG or pcDNA4TO/SMS2-FLAG as a template. QuikChange site-directed mutagenesis was also used to change all endogenous cysteine residues in SMS1 to serine with pcDNA4TO/SMS1-V5 as a template. The resultant cysteine-null SMS1 was subjected to site-directed mutagenesis to introduce individual cysteines at single amino acid positions (Cys50, Cys145, Cys187, Cys227, Cys259, Cys290, Cys311, or Cys333). Deletion mutants of SMS1 (deletion of 68 amino acids from N terminus (ΔN68), deletion of 22 amino acids from C terminus (ΔC22), or deletion of both ends (ΔN68/ΔC22)) and SMS2 (deletion of 6 or 30 amino acids from C terminus (ΔC6 or ΔC30)) were constructed by PCR with primers containing sequences corresponding to the FLAG or V5 epitope in front of the stop codon designed to truncate each region. The PCR products were subcloned into pcDNA4TO. Chimeric genes, with the amino acid sequences from Pro392 to Thr413 of SMS1 or from Pro336 to Thr365 of SMS2 fused to the C terminus of membrane-bound O-acyl transferase 5 (MBOAT5), were constructed by PCR with overlapping primers as described previously (49). The PCR products were subcloned into pcDNA4TO. The following chimeric gene constructs for use in the BiFC assay were created: VN (N-terminal residues 1–173 of Venus)-SMS1-V5, VC (C-terminal residues 155–238 of Venus)-SMS1-FLAG, SMS1-V5-VN, SMS1-FLAG-VC, VN-SMS2-V5, VC-SMS2-FLAG, SMS2-V5-VN, SMS2-FLAG-VC, or VC-GlcT-FLAG. The expression vector for human aquaporin-1 was constructed by PCR with cDNA derived from HEK293 and Molt4 cells as a template using specific primers containing sequences corresponding to the V5 epitopes in front of the stop codon. The PCR products were subcloned into pcDNA4TO. To yield the expression vector for SMS1-SGFP2, SMS1-ΔC22-SGFP2, SMS2-SGFP2, or SMS2-ΔC30-SGFP2, the PCR products were cloned into pSGFP2-N1.
Cell Culture and cDNA Transfection
V5-tagged SMS1 and SMS2 were stably expressed in HEK293 cells with a retrovirus-based gene transfer system (Clontech) using the pQCXIP/SMS1-V5 or pQCXIP/SMS2-V5 vector according to the instructions of the manufacturer. The obtained cells, which stably expressed SMS1 or SMS2, were named HEK293/SMS1-V5 and HEK293/SMS2-V5. HEK293/SMS1-V5, HEK293/SMS2-V5, and COS7 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 100 μg/ml streptomycin, and 100 units/ml penicillin at 37 °C in a humidified incubator containing 5% CO2. For cDNA transfection, the cells were plated at 2 × 105 cells in 6-well plates for all experiments, except for the confocal microscopy analysis, in which cells were seeded at 4 × 104 cells in 35-mm-diameter glass-bottom dishes (MatTek). After 24 h, the cells were transfected with 1 μg of plasmid in total for 6-well plates or 100 ng of plasmid in total per 35-mm dish using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. After transfection, the cells were cultured for an additional 24 h and used in subsequent experiments.
Co-immunoprecipitation Analysis of SMSs
HEK293/SMS1-V5 cells, HEK293/SMS2-V5 cells, and COS7 cells transfected with plasmids were washed three times in PBS and lysed in buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, CompleteTM protease inhibitor (Roche), and 1% Triton X-100 or 1% CHAPS. Postnuclear supernatants were prepared by centrifugation at 900 × g for 10 min and stored at −30 °C. Co-immunoprecipitation was performed with anti-FLAG M2 beads at 4 °C for 4 h. Then the beads were washed four times with lysis buffer and eluted in SDS sample buffer by incubation at 65 °C for 4 min. Co-immunoprecipitated proteins were subjected to SDS-PAGE with Dr. Western (Oriental Yeast) or WIDE-VIEWTM Prestained Protein Size Marker III (Wako). After the proteins were transferred to a nitrocellulose membrane, the membrane was incubated with specific antibodies, such as anti-FLAG or anti-V5 antibodies, and HRP-conjugated secondary antibody. Signals were detected with Western BLoT Quant HRP substrate (Takara) and analyzed and quantified using a Amersham Biosciences Imager 600 with ImageQuant TL software (GE Healthcare).
Preparation of the Cell Membrane Fraction
COS7 cells transfected with plasmids were washed three times in PBS, suspended in STE buffer (250 mm sucrose, 50 mm Tris-HCl (pH 8.0), 1 mm EDTA, 1.5 mm MgCl2, and CompleteTM protease inhibitor), and then passed 15 times through a 22-gauge needle for homogenization. After the removal of cell debris by 900 × g centrifugation for 10 min, the supernatant was centrifuged at 105,000 × g for 60 min. The pellet was resuspended in the appropriate buffer for each experiment and was used as the cell membrane fraction.
BN-PAGE
The procedures were modified from those described by Schägger and von Jagow (50). Briefly, the pellet prepared as the cell membrane fraction was resuspended in NativePAGE sample buffer (Invitrogen) containing 1% Triton X-100 and CompleteTM protease inhibitor and incubated on ice for 5 min. After 20,000 × g centrifugation for 5 min, the supernatant was mixed with 2.5 μl of NativePAGE 5% G-250 sample additive (Invitrogen) and stored at −30 °C. BN-PAGE was performed in NativePAGE Novex 4–16% gels (Invitrogen) using NativeMark unstained protein standard (Invitrogen) as a marker for molecular mass. Resolved proteins were transferred to PVDF membranes and immunoblotted with specific antibodies.
Immunocytochemistry and Fluorescence Microscopy
COS7 cells transfected with plasmids in 35-mm-diameter glass-bottom dishes were fixed with 3% paraformaldehyde in PBS for 10 min. After being rinsed with 50 mm NH4Cl in PBS, the cells were permeabilized with 0.1% Triton X-100 in PBS at 25 °C for 10 min or 0.002% digitonin in PBS containing 10 mm HEPES (pH 7.5), 300 mm sucrose, 100 mm KCl, 2.5 mm MgCl2, and 0.5% bovine serum albumin at 4 °C for 15 min. After being treated with ImmunoBlock (DS Pharma) for 30 min, the samples were incubated with specific antibodies, diluted 1:2000 with blocking buffer, at 4 °C overnight, followed by incubation with Alexa Fluor-conjugated secondary antibodies, diluted 1:1000 with blocking buffer, at 25 °C for 2 h. For microscopy, an Olympus FV10i confocal microscope equipped with a ×60 water objective was used with Olympus Fluoview version 3.1 Viewer.
Treatment with BFA
A 50 μm stock solution of BFA (Wako) was prepared in DMSO and diluted to 50 nm in cell culture medium for use in experiments. COS7 cells were plated into dishes and cultured for 9 h to allow attachment to the plate. The cells were treated with 50 nm BFA for 14 h, and the plasmid for SMS1-V5 or SMS2-V5 was transfected with Lipofectamine 2000. The cells were further cultured in cell culture medium containing BFA for 24 h. Then, the cells were harvested for BN-PAGE analysis or fixed for immunocytochemistry analysis as described above.
Chemical Cross-linking
BMH, BMB, and BMOE were from Thermo Scientific. Cross-linking was performed according to the instructions of the manufacturer. Briefly, COS7 cells transfected with plasmids, HEK293/SMS1-V5 cells, or HEK293/SMS2-V5 cells were washed twice in HEPES-KCl buffer consisting of 25 mm HEPES-NaOH (pH 7.5) and 125 mm KCl. Cross-linking reagents were dissolved at 25 mm in 100% DMSO and diluted to the indicated concentrations in HEPES-KCl buffer for use in experiments. After 15-min incubation at 25 °C, the cross-linking solution was removed, and the reaction was quenched with PBS containing 5 mm glycine and 5 mm cysteine for 10 min. The cells were detached with Cellstripper (Corning), collected by centrifugation at 500 × g for 1 min, and washed twice with PBS. The cells were lysed in buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1% Triton X-100, and CompleteTM protease inhibitor. Postnuclear supernatants were mixed with SDS sample buffer and subjected to SDS-PAGE and immunoblotting analysis.
Assay of SMS Activity in Vitro
SMS activity was assayed using C6-NBD-Cer as described previously (11).
Biotin Maleimide Labeling Assay
The assay was performed as described previously (19). Briefly, the pellet prepared as the cell membrane fraction was resuspended in buffer consisting of 100 μm Nα-(3-maleimidylpropionyl) biotin (biotin maleimide, Thermo Scientific), 10 mm HEPES-NaOH (pH 7.5), 10 mm KCl, 1.5 mm MgCl2, 5 mm EDTA, 250 mm sucrose, and 100 mm NaCl and incubated at 25 °C for 30 min. After quenching of the reaction with 10 mm β-mercaptoethanol, the cell membranes were collected by centrifugation. The pellet was resuspended in lysis buffer containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1.5% Nonidet P-40, 0.1% SDS, and CompleteTM protease inhibitor, and the extracts were incubated with NeutrAvidin agarose resins (Thermo Scientific) at 4 °C overnight. The agarose resins were washed four times with lysis buffer, and the proteins were eluted in SDS sample buffer by incubation at 65 °C for 4 min. The eluted proteins were subjected to SDS-PAGE and immunoblotting analysis.
BiFC Assay by Flow Cytometry
The procedures were modified from those described by Ozalp et al. (23). COS7 cells transfected with plasmids were detached with Cellstripper, collected by centrifugation at 500 × g for 1 min, and washed twice with PBS. The cells were fixed with 3% paraformaldehyde for 10 min, washed twice with PBS, and permeabilized with 0.1% Triton X-100 in PBS at 25 °C for 10 min. Then the cells were washed twice with 1% FBS in PBS by centrifugation at 5000 × g for 1 min and treated with ImmunoBlock for 30 min. The samples were incubated with mouse IgG2a monoclonal anti-V5 antibody for 90 min and washed three times with 1% FBS in PBS. The cells were incubated with rat PerCP/Cy5.5-conjugated anti-mouse IgG2a antibody and mouse APC-conjugated IgG1 monoclonal anti-FLAG antibody for 60 min, washed three times with 1% FBS in PBS, and resuspended in 1 mm EDTA in PBS. Venus fluorescence intensity, detected by the FITC channel, was analyzed (5000 events) in double-positive cells (PerCP/Cy5.5 + APC) using the BD FACSCanto II (BD Biosciences). Data were manually compensated using singly stained cell controls.
Statistical Analysis
All analyses were performed with GraphPad Prism 6 (GraphPad Software). For comparisons, Student's t test or one-way analysis of variance followed by the Tukey-Kramer multiple comparisons test were used. p < 0.05 was considered significant.
Author Contributions
Y. H. and Y. N. S. designed the study, conducted experiments, and wrote the paper. N. M., T. T., S. O., and Y. T. performed the chemical cross-linking experiments. S. A. and I. W. performed the fluorescence microscopy experiments. T. S. and A. Y. designed the experiments and prepared the manuscript. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We acknowledge the assistance of T. Saeki, T. Kumasaka, and K. Asano (Teikyo University, Japan) in plasmid construction. We would like to thank Editage for English language editing.
This work was supported in part by Grants-in-Aid for Young Scientists (B) 15K18868 (to Y. H.) and for Scientific Research (C) 15K07946 (to A. Y.) from the Japan Society for the Promotion of Science. This work was also supported in part by the Supported Program for the Strategic Research Foundation at Private Universities from the Ministry of Education, Culture, Sports, and Technology. The authors declare that they have no conflicts of interest with the contents of this article.
- SM
- sphingomyelin
- SMS
- sphingomyelin synthase
- PC
- phosphatidylcholine
- Cer
- ceramide
- DAG
- diacylglycerol
- ER
- endoplasmic reticulum
- HIV-1
- HIV, type 1
- BN
- blue native
- BMH
- bismaleimidohexane
- BMB
- bismaleimidobutane
- BMOE
- bismaleimidoethane
- PDI
- protein disulfide isomerase
- BiFC
- bimolecular fluorescence complementation
- GlcT
- ceramide glucosyltransferase
- BFA
- brefeldin A
- OSER
- organized smooth endoplasmic reticulum
- COP
- coat protein complex
- TM
- transmembrane
- MBOAT
- membrane bound O-acyl transferase.
References
- 1. Huitema K., van den Dikkenberg J., Brouwers J. F., and Holthuis J. C. (2004) Identification of a family of animal sphingomyelin synthases. EMBO J. 23, 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamaoka S., Miyaji M., Kitano T., Umehara H., and Okazaki T. (2004) Expression cloning of a human cDNA restoring sphingomyelin synthesis and cell growth in sphingomyelin synthase-defective lymphoid cells. J. Biol. Chem. 279, 18688–18693 [DOI] [PubMed] [Google Scholar]
- 3. Hannun Y. A., and Obeid L. M. (2011) Many ceramides. J. Biol. Chem. 286, 27855–27862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Griner E. M., and Kazanietz M. G. (2007) Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 7, 281–294 [DOI] [PubMed] [Google Scholar]
- 5. Borochov H., Zahler P., Wilbrandt W., and Shinitzky M. (1977) The effect of phosphatidylcholine to sphingomyelin mole ratio on the dynamic properties of sheep erythrocyte membrane. Biochim. Biophys. Acta 470, 382–388 [DOI] [PubMed] [Google Scholar]
- 6. Marggraf W. D., and Kanfer J. N. (1984) The phosphorylcholine acceptor in the phosphatidylcholine:ceramide cholinephosphotransferase reaction. Biochim. Biophys. Acta 793, 346–353 [DOI] [PubMed] [Google Scholar]
- 7. Separovic D., Hanada K., Maitah M. Y., Nagy B., Hang I., Tainsky M. A., Kraniak J. M., and Bielawski J. (2007) Sphingomyelin synthase 1 suppresses ceramide production and apoptosis post-photodamage. Biochem. Biophys. Res. Commun. 358, 196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Asano S., Kitatani K., Taniguchi M., Hashimoto M., Zama K., Mitsutake S., Igarashi Y., Takeya H., Kigawa J., Hayashi A., Umehara H., and Okazaki T. (2012) Regulation of cell migration by sphingomyelin synthases: sphingomyelin in lipid rafts decreases responsiveness to signaling by the CXCL12/CXCR4 pathway. Mol. Cell Biol. 32, 3242–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Subathra M., Qureshi A., and Luberto C. (2011) Sphingomyelin synthase regulate protein trafficking and secretion. PLoS ONE 6, e23644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mitsutake S., Zama K., Yokota H., Yoshida T., Tanaka M., Mitsui M., Ikawa M., Okabe M., Tanaka Y., Yamashita T., Takemoto H., Okazaki T., Watanabe K., and Igarashi Y. (2011) Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. J. Biol. Chem. 286, 28544–28555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hayashi Y., Nemoto-Sasaki Y., Tanikawa T., Oka S., Tsuchiya K., Zama K., Mitsutake S., Sugiura T., and Yamashita A. (2014) Sphingomyelin synthase 2, but not sphingomyelin synthase 1, is involved in HIV-1 envelope-mediated membrane fusion. J. Biol. Chem. 289, 30842–30856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu Y., Belkina N. V., and Shaw S. (2009) HIV infection of T cells: actin-in and actin-out. Sci. Signal. 2, pe23. [DOI] [PubMed] [Google Scholar]
- 13. Heerklotz H., and Seelig J. (2000) Correlation of membrane/water partition coefficients of detergents with the critical micelle concentration. Biophys. J. 78, 2435–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nazari M., Kurdi M., and Heerklotz H. (2012) Classifying surfactants with respect to their effect on lipid membrane order. Biophys. J. 102, 498–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van Itallie C. M., Mitic L. L., and Anderson J. M. (2011) Claudin-2 forms homodimers and is a component of a high molecular weight protein complex. J. Biol. Chem. 286, 3442–3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crichton P. G., Harding M., Ruprecht J. J., Lee Y., and Kunji E. R. (2013) Lipid, detergent, and Coomassie Blue G-250 affect the migration of small membrane proteins in blue native gels: mitochondrial carriers migrate as monomers not dimers. J. Biol. Chem. 288, 22163–22173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walz T., Hirai T., Murata K., Heymann J. B., Mitsuoka K., Fujiyoshi Y., Smith B. L., Agre P., and Engel A. (1997) The three-dimensional structure of aquaporin-1. Nature 387, 624–627 [DOI] [PubMed] [Google Scholar]
- 18. Yasuda S., Nishijima M., and Hanada K. (2003) Localization, topology, and function of the LCB1 subunit of serine palmitoyltransferase in mammalian cells. J. Biol. Chem. 278, 4176–4183 [DOI] [PubMed] [Google Scholar]
- 19. Feramisco J. D., Goldstein J. L., and Brown M. S. (2004) Membrane topology of human insig-1, a protein regulator of lipid synthesis. J. Biol. Chem. 279, 8487–8496 [DOI] [PubMed] [Google Scholar]
- 20. Zhu Q., and Casey J. R. (2007) Topology of transmembrane proteins by scanning cysteine accessibility mutagenesis methodology. Methods 41, 439–450 [DOI] [PubMed] [Google Scholar]
- 21. Hu C.-D., Chinenov Y., and Kerppola T. K. (2002) Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9, 789–798 [DOI] [PubMed] [Google Scholar]
- 22. Yang W., Qiu C., Biswas N., Jin J., Watkins S. C., Montelaro R. C., Coyne C. B., and Wang T. (2008) Correlation of the tight junction-like distribution of Claudin-1 to the cellular tropism of hepatitis C virus. J. Biol. Chem. 283, 8643–8653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ozalp C., Szczesna-Skorupa E., and Kemper B. (2005) Bimolecular fluorescence complementation analysis of cytochrome p450 2c2, 2e1, and NADPH-cytochrome p450 reductase molecular interactions in living cells. Drug Metab. Dispos. 33, 1382–1390 [DOI] [PubMed] [Google Scholar]
- 24. Romano C., Yang W.-L., and O'Malley K. L. (1996) Metabotropic glutamate receptor 5 is a disulfide-linked dimer. J. Biol. Chem. 271, 28612–28616 [DOI] [PubMed] [Google Scholar]
- 25. Tani M., and Kuge O. (2009) Sphingomyelin synthase 2 is palmitoylated at the COOH-terminal tail, which is involved in its localization in plasma membranes. Biochem. Biophys. Res. Commun. 381, 328–332 [DOI] [PubMed] [Google Scholar]
- 26. Gao X. D., and Dean N. (2000) Distinct protein domains of the yeast Golgi GDP-mannose transporter mediate oligomer assembly and export from the endoplasmic reticulum. J. Biol. Chem. 275, 17718–17727 [DOI] [PubMed] [Google Scholar]
- 27. Margeta-Mitrovic M., Jan Y. N., and Jan L. Y. (2000) A trafficking checkpoint controls GABA B receptor heterodimerization. Neuron 27, 97–106 [DOI] [PubMed] [Google Scholar]
- 28. Overton M. C., Chinault S. L., and Blumer K. J. (2003) Oligomerization, biogenesis, and signaling is promoted by a glycophorin A-like dimerization motif in transmembrane domain 1 of a yeast G protein-coupled receptor. J. Biol. Chem. 278, 49369–49377 [DOI] [PubMed] [Google Scholar]
- 29. Salahpour A., Angers S., Mercier J. F., Lagacé M., Marullo S., and Bouvier M. (2004) Homodimerization of the β2-adrenergic receptor as a prerequisite for cell surface targeting. J. Biol. Chem. 279, 33390–33397 [DOI] [PubMed] [Google Scholar]
- 30. Lopez-Gimenez J. F., Canals M., Pediani J. D., and Milligan G. (2007) The α1b-adrenoceptor exists as a higher-order oligomer: effective oligomerization is required for receptor maturation, surface delivery, and function. Mol. Pharmacol. 71, 1015–1029 [DOI] [PubMed] [Google Scholar]
- 31. Martzoukou O., Karachaliou M., Yalelis V., Leung J., Byrne B., Amillis S., and Diallinas G. (2015) Oligomerization of the UapA purine transporter is critical for ER-exit, plasma membrane localization and turnover. J. Mol. Biol. 427, 2679–2696 [DOI] [PubMed] [Google Scholar]
- 32. Snapp E. L., Hegde R. S., Francolini M., Lombardo F., Colombo S., Pedrazzini E., Borgese N., and Lippincott-Schwartz J. (2003) Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lenormand C., Spiegelhalter C., Cinquin B., Bardin S., Bausinger H., Angénieux C., Eckly A., Proamer F., Wall D., Lich B., Tourne S., Hanau D., Schwab Y., Salamero J., and de la Salle H. (2013) Birbeck granule-like “organized smooth endoplasmic reticulum” resulting from the expression of a cytoplasmic YFP-tagged langerin. PLoS ONE 8, e60813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rabouille C., and Klumperman J. (2005) The maturing role of COPI vesicles in intra-Golgi transport. Nat. Rev. Mol. Cell Biol. 6, 812–817 [DOI] [PubMed] [Google Scholar]
- 35. Lord C., Ferro-Novick S., and Miller E. A. (2013) The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the Golgi. Cold Spring Harb. Perspect. Biol. 5, a013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nishimura N., and Balch W. E. (1997) A di-acidic signal required for selective export from the endoplasmic reticulum. Science 277, 556–558 [DOI] [PubMed] [Google Scholar]
- 37. Yamashita A., Hayashi Y., Nemoto-Sasaki Y., Ito M., Oka S., Tanikawa T., Waku K., and Sugiura T. (2014) Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog. Lipid. Res. 53, 18–81 [DOI] [PubMed] [Google Scholar]
- 38. Zhao Y., Chen Y. Q., Bonacci T. M., Bredt D. S., Li S., Bensch W. R., Moller D. E., Kowala M., Konrad R. J., and Cao G. (2008) Identification and characterization of a major liver lysophosphatidylcholine acyltransferase. J. Biol. Chem. 283, 8258–8265 [DOI] [PubMed] [Google Scholar]
- 39. Yeang C., Ding T., Chirico W. J., and Jiang X. C. (2011) Subcellular targeting domains of sphingomyelin synthase 1 and 2. Nutr. Metab. (Lond) 8, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li E., Wimley W. C., and Hristova K. (2012) Transmembrane helix dimerization: beyond the search for sequence motifs. Biochim. Biophys. Acta 1818, 183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Korkhov V. M., and Zuber B. (2009) Direct observation of molecular arrays in the organized smooth endoplasmic reticulum. BMC Cell Biol. 10, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fasana E., Fossati M., Ruggiano A., Brambillasca S., Hoogenraad C. C., Navone F., Francolini M., and Borgese N. (2010) A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419–1430 [DOI] [PubMed] [Google Scholar]
- 43. Volkova E. G., Abramchuk S. S., and Sheval E. V. (2012) The overexpression of nuclear envelope protein Lap2β induces endoplasmic reticulum reorganisation via membrane stacking. Biol. Open 1, 802–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boulaflous A., Saint-Jore-Dupas C., Herranz-Gordo M. C., Pagny-Salehabadi S., Plasson C., Garidou F., Kiefer-Meyer M. C., Ritzenthaler C., Faye L., and Gomord V. (2009) Cytosolic N-terminal arginine-based signals together with a luminal signal target a type II membrane protein to the plant ER. BMC Plant Biol. 9, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cranfill P. J., Sell B. R., Baird M. A., Allen J. R., Lavagnino Z., de Gruiter H. M., Kremers G. J., Davidson M. W., Ustione A., and Piston D. W. (2016) Quantitative assessment of fluorescent proteins. Nat. Methods 13, 557–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Skube S. B., Chaverri J. M., and Goodson H. V. (2010) Effect of GFP tags on the localization of EB1 and EB1 fragments in vivo. Cytoskeleton 67, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Springer S., Malkus P., Borchert B., Wellbrock U., Duden R., and Schekman R. (2014) Regulated oligomerization induces uptake of a membrane protein into COPII vesicles independent of its cytosolic tail. Traffic 15, 531–545 [DOI] [PubMed] [Google Scholar]
- 48. Yeang C., Varshney S., Wang R., Zhang Y., Ye D., and Jiang X. C. (2008) The domain responsible for sphingomyelin synthase (SMS) activity. Biochim. Biophys. Acta 1781, 610–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wurch T., Lestienne F., and Pauwels P. J. (1998) A modified overlap extension PCR method to create chimeric genes in the absence of restriction enzymes. Biotechnology Techniques 12, 653–657 [Google Scholar]
- 50. Schägger H., and von Jagow G. (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199, 223–231 [DOI] [PubMed] [Google Scholar]