
Fig. 1.
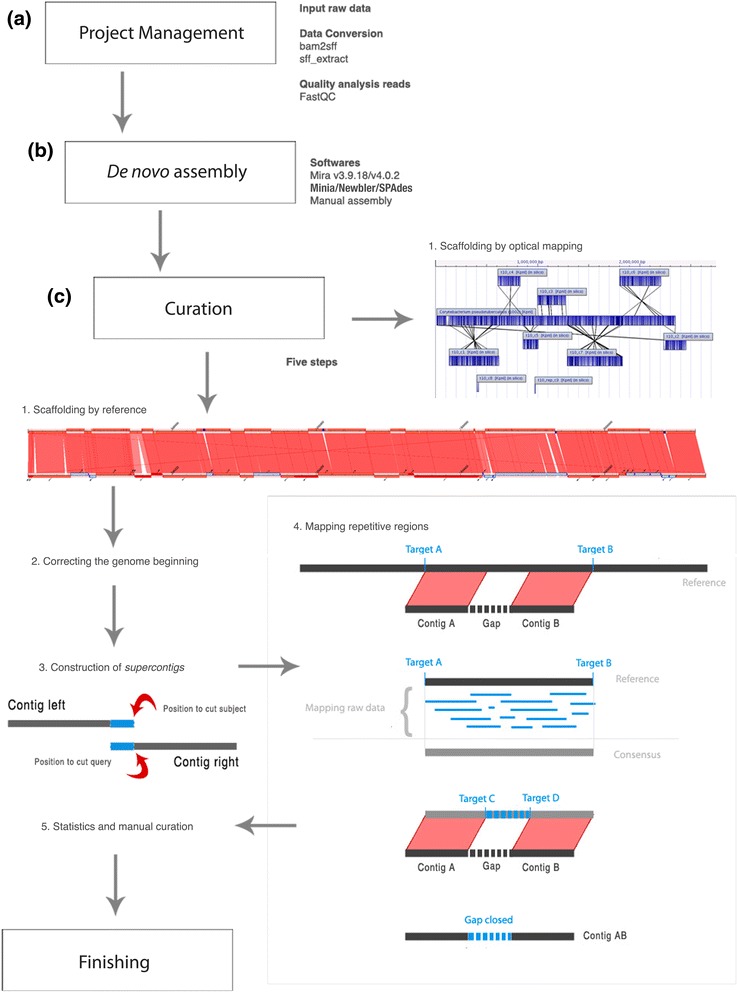
Workflow representing data fluxes in SIMBA’s process. a Projects management: creation and management of projects; and raw data conversion. b De novo assembly (Mira software – support to the assemblers Minia, Newbler and SPAdes). In this step the user can insert output files of de novo assembly yielded by external software. c Curation: This step has five subdivisions: 1) contigs orientation using CONTIGuator2 software by reference or by optical mapping report; 2) Start DNA correction based on reference genome; 3) merging neighbor contigs with overlap in flank regions using PHP parser and BLAST (blastn); 4) mapping 3,000 bp of flank regions of neighbor contigs (Contig A and Contig B) against reference genome; BLAST is used to determine the start and end position (targets A and B); the reference genome is trimmed at targets A and B positions; then the raw data is mapped using Mira4; the contigs A and B are mapped on consensus sequence obtained through raw data mapping; targets C and D are used to detect start and end position of specific GAP; the region is extracted, and the gap is closed. 5) Showing statistics about unknown nucleotides present in the genome and allow genome download for manual curation