

Abstract
Preeclampsia is associated with chronic inflammation and an imbalance among T-helper cell subtypes with an increase in T-helper 17 (TH17) cells. The objective of this study was to determine a role for TH17s, from the reduced uterine perfusion pressure (RUPP) rat model of preeclampsia, in the etiology of hypertension and chronic inflammation during pregnancy. CD4+/CD25− T cells were isolated from rat spleens, cultured in TH17 media, and were verified as TH17s via flow cytometry. On day 12 of gestation, 1×106 TH17 cells from RUPP rats were adoptively transferred into NP rats, carotid catheters were inserted on day 18, and on day 19, mean arterial pressure (MAP) was recorded, serum and plasma were collected, and oxidative stress and production of agonistic autoantibodies to the ANG II type I receptor (AT1-AA) were analyzed. MAP increased from 100.3 ± 1.7 mmHg in normal pregnant (NP; n = 17) to 124.8 ± 2.1 mmHg in RUPP (n = 22; P < 0.0001) and to 110.8 ± 2.8 mmHg in NP+RUPP TH17 (n = 11). Pup weights in NP+RUPP TH17s were decreased to 1.92 ± 0.09 g from 2.39 ± 0.14 in NP rats (P < 0.01). AT1-AA significantly increased from 0.1 ± 0.2 beats/min in NP to 15.6 ± 0.7 beats/min in NP+RUPP TH17s. IL-6 was 22.3 ± 5.7 pg/ml in NP and increased to 60.45 ± 13.8 pg/ml in RUPP (P < 0.05) and 75.9 ± 6.8 pg/ml in NP+RUPP TH17 rats (P < 0.01). Placental and renal oxidative stress were 238 ± 27.5 and 411 ± 129.9 relative light units·min−1·mg−1 in NP and 339 ± 104.6 and 833 ± 331.1 relative light units·min−1·mg−1 in NP+RUPP TH17, respectively. In conclusion, RUPP TH17 cells induced intrauterine growth restriction and increased blood pressure, AT1-AA, IL-6, and tissue oxidative stress when transferred to NP rats, indicating a role for autoimmune associated TH17 cells, to cause much of the pathophysiology associated with preeclampsia.
Keywords: hypertension, pregnancy, T-helper 17, IL-17, oxidative stress, intrauterine growth restriction
preeclampsia (PE) is a pregnancy-associated multisystem disorder that is characterized by new-onset hypertension after 20-wk gestation, which is oftentimes accompanied by low birth weight of the fetus. One important characteristic of PE is immune activation. The imbalance among CD4+ T cell and natural killer cell subtypes, inflammatory cytokines, agonistic autoantibodies to the ANG II type 1 receptor (AT1-AA), and placental oxidative stress, are thought to contribute to the increase in blood pressures, proteinuria, and low birth weight in PE pregnancies. Although the exact pathophysiological mechanisms that lead to the development of PE are not fully elucidated, it is hypothesized that PE develops in two phases. The first phase is thought to involve the shallow invasion of trophoblasts during placentation and early alteration of immune cells, such as natural killer cells and T cells (7, 15, 21). The shallow trophoblast invasion is believed to lead to inefficient remodeling of the uterine spiral arteries and decreased vasculogenesis, which results in insufficient oxygen and nutrient delivery to the placenta. This causes the development of a hypoxic environment leading to placental ischemia (7). During phase two in the development of PE, oxidative stress and endothelial dysfunction in the maternal vasculature and chronic immune activation occur (7, 10, 15, 21). This is thought to be due to the placental ischemia of phase one.
Previous research suggests that an imbalance between two types of CD4+ T-helper cell populations, T regulatory (TReg) and T helper 17 (TH17), is involved in the pathophysiology of PE (2, 23, 26). We have previously shown that placental ischemia, in the reduced uterine perfusion pressure (RUPP) rat model of PE, stimulates this same CD4+ T-cell imbalance, and contributes to an increase in proinflammatory cytokine production, AT1-AA, oxidative stress, and a shift in angiogenic factors, leading to the development of hypertension during pregnancy (27). We have recently shown that adoptive transfer of the total CD4+ T-cell population from RUPP rats into normal pregnant (NP) rats caused elevations in blood pressure, AT1-AA, reactive oxygen species (ROS), and inflammatory cytokines (28), similar to levels seen in RUPP rats and PE patients.
CD4+ T-cell populations can be composed of several subsets of helper T cells, including Type 1 (TH1), Type 2 (TH2), regulatory (TReg), and TH17 cells. Type 1 CD4+ T cells produce a variety of inflammatory cytokines and are the major effectors of phagocyte-mediated host defense against intracellular pathogens (25). TH17 cells secrete the proinflammatory cytokine IL-17 and are proposed to be a pathogenic mechanism in autoimmune disease (22). Previous studies demonstrate an increase in the population of TH17 cells in preeclamptic women compared with women with normal pregnancies (2, 26). Moreover, we have shown that both IL-17 and TH17 cells are increased in the RUPP rat compared with NP rats (1, 27). We have also demonstrated that infusion of IL-17 into NP rats leads to an increase in TH17 cells, urinary isoprostanes, placental oxidative stress, AT1-AA, and hypertension compared with NP control rats. Importantly, we have also shown that blockade of IL-17 function with the soluble receptor, IL-17 RC, attenuated hypertension; improved fetal weight and uterine artery resistance; blunted TH17 cell increase; and decreased urinary isoprostane, placental oxidative stress, and AT1-AA production in response to placental ischemia in the RUPP rat (1). However, a specific role for TH17 cells isolated from RUPP rats to cause PE-like features, such as hypertension or reduction in fetal weight, has yet to be clearly demonstrated. To date, no other intervention studies of TH17 cells in pregnant rodent models have been performed. Although we have shown that NP recipients of RUPP CD4+ T cells exhibited hypertension and other characteristics of PE, there were no decreases in fetal weight. Previous adoptive transfer studies of CD4+ T cells contained ~20% TH17 cells in a mixture with TReg cells and other CD4+ T cell subsets (27). Moreover, blockade of IL-17 and TH17 cell proliferation in RUPP rats was associated with improved pup weight and blood pressure. Therefore, in this current study, we wanted to determine the effects of a purer population of RUPP TH17 cells on pregnancy outcomes. We hypothesized that RUPP TH17 cells cause hypertension and oxidative stress, induce inflammation, and AT1-AA, and contribute to reduced fetal weight during PE. To test this hypothesis, we performed adoptive transfer experiments in which CD4+/CD25− T cells were isolated from NP or RUPP rats, differentiated into TH17 cells in vitro, and transferred to NP rats, which were subsequently evaluated for PE-like features.
MATERIALS AND METHODS
Pregnant Sprague-Dawley rats purchased from Harlan Sprague Dawley (Indianapolis, IN) were used in this study. Animals were housed in a temperature-controlled room (23°C) with a 12:12-h light-dark cycle. All experimental procedures executed in this study were in accordance with the National Institutes of Health's “Guidelines for the Care and Use of Laboratory Animals.” All protocols were approved by the Institutional Animal Care and Use Committee at the University of Mississippi Medical Center.
Reduced uterine perfusion pressure procedure.
On gestational day 14, under isoflurane anesthesia, NP rats underwent a RUPP procedure with the application of a constrictive silver clip (0.203 mm) to the aorta superior to the iliac bifurcation that was performed while ovarian collateral circulation to the uterus was reduced with restrictive clips (0.100 mm) to the bilateral uterine arcades at the ovarian end.
T-helper 17 cell isolation, culture, and differentiation.
At the time of harvest (gestational day 19), spleens were collected from NP or RUPP rats, and lymphocytes were isolated from spleen by centrifugation on a cushion of Ficoll-Hypaque (Lymphoprep, Accurate Chemical & Scientific, Westbury, NY), according to the manufacturer’s instructions. Anti-CD4 and anti-CD25 antibodies (BD Biosciences, San Jose, CA) were biotinylated using the DSB-X biotin protein-labeling kit (Life Technologies, Grand Island, NY), according to the manufacturer’s instructions. Isolated lymphocytes were incubated with biotinylated anti-CD4 antibody. The CD4-positive (CD4+) population was isolated using FlowComp Dynabeads (Invitrogen, Oslo, Norway), according to the manufacturer’s protocol. The CD4+ population of lymphocytes were then incubated with biotinylated anti-CD25 antibody, and the supernatant containing the unbound CD4+/CD25− population was collected again using FlowComp Dynabeads to sequester the CD4+/CD25+ population. Biotinylated antibodies and FlowComp Dynabeads were separated from cells according to manufacturer’s protocol, before culture and expansion. The CD4+/CD25− population of splenocytes were incubated on anti-CD3 and anti-CD28 magnetic beads in T-helper media (RPMI, 10% FBS, 5% PenStrep, 1% HEPES) in 96-well plates at 103 cells/well on day 0. On day 2, cells were removed from magnetic beads and cultured in T-helper-specific media (RPMI, 10% FBS, 5% Pen-Strep, 1% HEPES, 20 ng/ml IL-6, 3 ng/ml TGFβ-1, and 20 ng/ml IL-23) for 5 or 6 days. Differentiation of isolated cells into TH17 cells was verified via flow cytometry.
Determination of cultured T-helper TH17 lymphocyte population.
We used flow cytometry analysis to determine differentiation of the cultured T cells into the specific CD4+ TH17 cell population: CD4+CD25−RORγ+ (retinoic acid receptor-related organ receptor γ). After culture, 1 × 106 cells were incubated for 30 min at 4°C with antibodies against rat CD4 and rat CD25 (BD Biosciences, San Jose, CA). After washing, we labeled cells with secondary fluorescein isothiocyanate (FITC; Southern Biotech, Birmingham, AL) and phycoerythrin with cyanin-5 (PE-Cy5; Santa Cruz Biotechnology, Santa Cruz, CA) antibody for 30 min at 4°C. Cells were washed, permeabilized, and stained with rabbit polyclonal RORγ (Santa Cruz Biotechnology) for 30 min at 4°C. After washing, cells were labeled with secondary phycoerythrin (PE; Southern Biotech, Birmingham, AL) for 30 min at 4°C. As a negative control for each individual rat, cells were treated exactly as described above except they were incubated with anti-FITC, anti-PE-Cy5, and anti-PE secondary antibodies alone. Subsequently, cells were washed and resuspended in 500 µl of Rosswell Park Memorial Institute medium (RPMI) and analyzed for single and double staining on a Gallios flow cytometer (Beckman Coulter, Brea, CA). The lymphocyte population of cells was gated, and cells were examined for RORγ-positive staining. The RORγ-positive-stained cells were then examined for CD4 and CD25-positive staining. The percentage of positive-stained cells above the negative control was collected for 3 separate cultures or individual rats, and the mean values for each experimental group were calculated.
Adoptive transfer of T-helper 17 cells.
For consistency with previous adoptive transfer studies performed in our laboratory, 500 μl of TH17 cells, in sterile saline, were injected intraperitoneally into NP rats at 2 × 106 cells/ml on day 12 of gestation (GD12). This results in two groups of rats designated as NP recipients of RUPP TH17 cells (NP+RUPP TH17s) and NP recipients of NP TH17 cells (NP+NP TH17s). NP recipients of TH17 cells were compared with NP and RUPP controls.
Administration of IL-17 RC to NP recipients of RUPP T-helper 17 cells.
On gestational day 14, under isoflurane anesthesia, NP+ RUPP TH17s rats were intraperitoneally implanted with a miniosmotic pump (model 2002, Alzet Scientific) that infused recombinant mouse IL-17 receptor C (IL-17RC) (100 pg/day) (R&D Systems, Minneapolis, MN). IL-17RC was infused from day 14 through day 19 of in 5 NP+RUPP TH17 rats. Murine IL-17RC has 87% homology and 86% identity to rat IL-17 RC, indicating high biological similarity to the naturally occurring rat protein. The dose was determined on the basis of binding ability of the soluble receptor to IL-17 A–F, as performed by the manufacturer, as well as a previous publication by our laboratory.
Administration of an SOD mimetic and AT1 Receptor blockade.
To determine a role for oxidative stress in the blood pressure response to RUPP TH17 cells, NP recipient rats of RUPP TH17 cells were treated with tempol (30 mg·kg−1·day−1), a superoxide dismutase mimetic, via their drinking water, provided ad libitum, beginning on gestation day 12 (NP+RUPP TH17+Temp) (6). To determine the effect of AT1 receptor blockade and, thus, the role of the AT1-AA in the hypertensive response to RUPP TH17 cells, NP recipients of RUPP TH17 cells were treated with losartan (10 mg/day) via their drinking water, provided ad libitum, beginning on gestation day 12 (NP+RUPP TH17+Los) (16).
Sample collection and determination of mean arterial pressure.
Under isoflurane anesthesia, on day 18 of gestation, carotid arterial catheters were inserted for blood pressure measurements. The catheters inserted are V3 tubing (Scientific Commodities, Lake Havasu City, AZ), which is tunneled to the back of the neck and exteriorized. On day 19 of gestation, arterial blood pressure was analyzed after placing the rats in individual restraining cages. Arterial pressure was monitored with a pressure transducer (Cobe III Transducer CDX Sema) and recorded continuously for 1 h after a 1-h stabilization period. Subsequently, a blood and urine sample was collected; kidneys, placentas, and spleens were harvested; and litter size and pup weights were recorded under anesthesia.
Determination of placental and renal ROS.
Superoxide production in the placenta and renal cortex was measured by using the lucigenin technique, as we have previously described (19). Rat placentas and renal cortices from NP, RUPP, and NP+RUPP TH17s rats were snap frozen in liquid nitrogen directly after collection and stored at −80°C until further processing. Placentas and renal cortices were removed and homogenized in RIPA buffer (PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and a protease inhibitor cocktail; Santa Cruz, Santa Cruz, CA), as described previously (19). The samples were centrifuged at 16,000 g for 30 min, the supernatant was aspirated, and the remaining cellular debris was discarded. The supernatant was incubated with lucigenin at a final concentration of 5 μmol/l. The samples were allowed to equilibrate for 15 min in the dark, and luminescence was measured every second for 10 s with a luminometer (Berthold, Oak Ridge, TN). Luminescence was recorded as relative light units (RLU) per minute. An assay blank with no homogenate but containing lucigenin was subtracted from the reading before transformation of the data. Each sample was read 5 times, and the average was used for data transformation. The protein concentration was measured using a protein assay with BSA standards (Pierce, Rockford, IL). The data are expressed as RLU·min−1·mg protein−1.
Determination of circulating AT1-AA.
On day 19 of gestation, blood was collected, and immunoglobulin was isolated from 200 µl of serum by protein G Sepharose protein purification system (Knauer, Berlin, Germany). Chronotropic responses were measured as previously described (5, 29). The results express the difference between the basal beating rate of the cardiomyocytes and the beating rate measured after the addition of the AT1-AA (increase in number of beats/min or beats/min) (3, 4, 12, 17–19). AT1-AAs were assessed in NP, RUPP controls, and NP+RUPP TH17 rats.
Determination of cytokine production.
Plasma was collected from all pregnant rats, and circulating levels of IL-17, IL-6, and TNF-α were measured, according to the manufacturer’s protocol, using commercial ELISA kits available from R&D Systems (Quantikine). The minimal detectable dose for the IL-17 Quantikine ELISA was <5 pg/ml, with maximum being 800 pg/ml with an intra-assay/interassay precision of 5 and 5.1% coefficient of variation, respectively. The minimal detectable dose for the IL-6 Quantikine ELISA was <36 pg/ml, with maximum being 4,000 pg/ml with an intra-assay/interassay precision of 8.8 and 10% coefficient of variation, respectively. The minimal detectable dose for the TNF-α Quantikine ELISA was <12.5 pg/ml, with maximum being 800 pg/ml with an intra-assay/interassay precision of 3.1 and 9.4% coefficient of variation, respectively.
Statistical analysis.
All of the data are expressed as means ± SE. Comparisons of control with experimental groups were analyzed by ANOVA with a Tukey multiple-comparisons test performed as post hoc analysis. A value of P < 0.05 was considered statistically significant.
RESULTS
After T cells were isolated from NP and RUPP spleens and cultured as described above, flow cytometry was performed to verify differentiation of cells into TH17 population. Seventy-eight percent of the stained cells were double-positive for CD4 and RORγ with ~70% of cultured cells stained as CD4+, CD25−, and RORγ+, and were identified as TH17 cells. A representative flow cytometry dot plot of RUPP TH17 (CD4+/CD25−/RORγ+) T cells prepared for adoptive transfer is shown in Fig. 1.
Fig. 1.

TH17 differentiation. To examine cultured TH17 cells, a subset of cells from culture were stained for the external markers CD4 and CD25. Cells were permeabilized and fixed before staining for the intracellular transcription factor, RORγ. Cells were gated in the forward and side scatterplot (gated cells colored red). The percentage of RORγ + (red) cells were measured within the gate (selected population). TH17s are CD4+ and CD25− cells within the RORγ+ gate. Representative flow cytometry plot showing TH17 cells differentiated in culture.
Adoptive transfer of RUPP TH17 cells, but not NP TH17 cells, significantly increase blood pressure in NP rats.
Mean arterial pressure (MAP) was measured on day 19 of gestation in NP, RUPP, NP+NP TH17, and NP+RUPP TH17 cells. The MAP increased significantly from 100.3 ± 1.7 mmHg in NP rats (n = 17), to 124.8 ± 2.1 mmHg in RUPP rats (n = 22, P < 0.001; Fig. 2). RUPP TH17 cells also significantly increased blood pressure to 110.8 ± 2.8 mmHg in NP+RUPP TH17s (n = 11) compared with NP rats (P < 0.01; Fig. 2). However, blood pressure in response to adoptive transfer of NP TH17 cells was not significantly increased in NP animals [97.17 ± 2.8 mmHg, n = 6, not significant (n.s.) vs. NP].
Fig. 2.
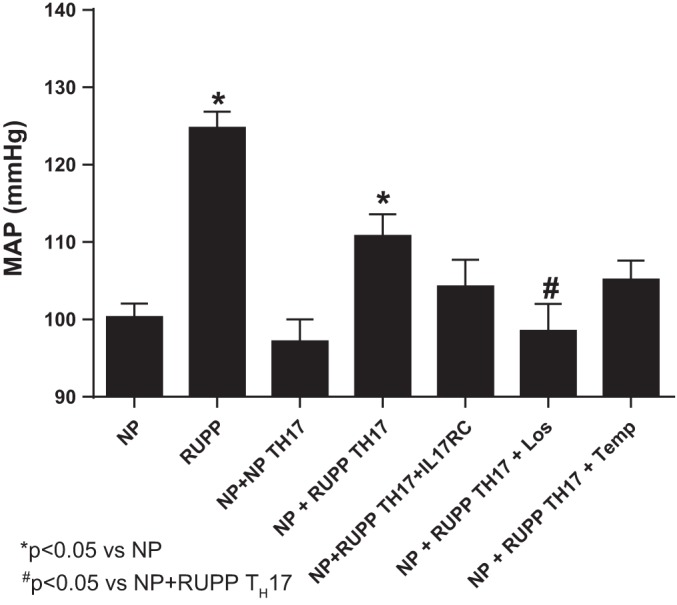
RUPP TH17 cells significantly increased blood pressure in NP rats. Mean arterial pressure (MAP) was measured on day 19 in NP (n = 17), RUPP (n = 22), NP+NP TH17s (n = 7), NP+RUPP TH17s (n = 11), NP+RUPP TH17s+ IL-17RC (n = 7), NP+TH17+Los (n = 7), and NP+TH17+Temp (n = 7) rats. NP, normal pregnant; RUPP, reduced uterine perfusion pressure; NP+ NP TH17s, NP recipients of TH17s from NP rats; NP+RUPP TH17s, NP recipients of TH17s from RUPP rats; NP+RUPP TH17s+IL-17RC, NP+RUPP TH17s treated with IL-17RC; NP+RUPP TH17s+Los, NP+RUPP TH17s treated with losartan; NP+RUPP TH17+Temp, NP+ RUPP TH17s treated with tempol. A Tukey test was performed as post hoc analysis to generate P values (*P < 0.05 vs. NP; #P < 0.05 vs. NP+RUPP TH17s).
To determine whether the blood pressure response to RUPP TH17 adoptive transfer was mediated by increased reactive oxygen species and/or AT1 receptor activation by AT1-AA, additional groups of rats were treated with either Tempol (Temp) or Losartan (Los) in their drinking water. MAP in NP+RUPP TH17+Temp was 105.1 ± 2.5 mmHg (n = 7), a nonsignificant decrease compared with NP+RUPP TH17 rats (110.8 ± 2.8 mmHg; P > 0.05; Fig. 2). However, MAP in NP+RUPP TH17+Los was significantly decreased to 98.5 ± 3.5 mmHg (n = 7; P < 0.05 vs. NP+RUPP TH17).
IL-17 is the hallmark proinflammatory cytokine secreted by TH17 cells. We have previously published that infusion of the proinflammatory cytokine IL-17 can induce hypertension and other PE-like characteristics in pregnant animals (6) and that blockade of IL-17 with the soluble receptor IL-17RC can attenuate these symptoms in RUPP rats, which are known to have an increased population of TH17 cells (1). Therefore, we set out to determine whether blockade of IL-17 signaling, with the soluble receptor IL-17RC, would inhibit the blood pressure increase after adoptive transfer of RUPP TH17 cells into NP rats. NP+RUPP TH17 rats were implanted with minipumps infusing IL-17RC on GD14. Infusion of the soluble receptor into NP recipient rats inhibited the increase in blood pressure induced by RUPP TH17 cells (104.3 ± 3 mmHg, n = 7, n.s. vs. NP).
Adoptive transfer of RUPP TH17 cells significantly decreased pup and placental weight in NP rats.
Pup weight of litters from RUPP rats (1.91 ± 0.06 g) was significantly lower than pup weight from NP rats (2.39 ± 0.14 g, P < 0.01). In previous studies, adoptive transfer of RUPP CD4+ T cells into NP recipient rats caused no changes in pup weight. In contrast, in Fig. 3A, adoptive transfer of RUPP TH17 cells into NP rats significantly decreased pup weight to 1.92 ± 0.09 (P < 0.05) to the same degree as pup weights observed in RUPP control rats. Importantly, these data identify an important role for TH17 cells stimulated during placental ischemia to cause fetal demise. Pup weight in NP+NP TH17 rats were not different from NP controls (2.22 ± 0.07 g), indicating that the population of TH17 cells stimulated by placental ischemia, specifically, have a role in mediating the intrauterine growth restriction (IUGR) aspect of preeclampsia pathophysiology. Treatment with Tempol yielded improvements in pup weights of NP recipients of RUPP TH17 cells. Pup weight was normalized with Tempol to 2.23 ± 0.03 g in NP+ RUPP TH17+Temp rats compared with NP+ RUPP TH17 (P < 0.05). However, pup weight showed minimal improvement to 1.99 ± 0.13 g in NP+RUPP TH17+Los, which was not significantly different from pup weight in NP+RUPP TH17 rats (P > 0.05). Additionally, inhibition of IL-17 signaling with IL-17RC completely attenuated IUGR induced by RUPP TH17 cells (2.5 ± 0.05 g, n.s. vs. NP), implicating IL-17 signaling in this component of preeclampsia pathology. Placental weights were likewise altered in each group. Placenta weights significantly decreased from 0.58 ± 0.01 g in NP rats to 0.51 ± 0.02 g in RUPP rats (P < 0.01). Moreover, adoptive transfer of RUPP TH17 cells into NP rats significantly decreased placental weight to 0.53 ± 0.01 g (P < 0.05), while NP TH17 cells did not change placental weights in NP rats (0.59 ± 0.02 g, n.s. vs. NP). Placental weight showed no improvement in NP+ RUPP TH17+Los (0.53 ± 0.04 g). However, treatment with Tempol significantly improved placental weights of NP recipients of RUPP TH17 cells. Placental weight was normalized with Tempol to 0.58 ± 0.02 g in NP+ RUPP TH17+Temp rats compared with NP+ RUPP TH17 (P < 0.05). Importantly, IL-17RC infusion also blocked the effects of RUPP TH17 cells on placental weight (0.57 ± 0.02 g, n.s. vs NP). These data further demonstrate that the observed IUGR stems from placental dysfunction induced by RUPP TH17 cell and IL-17-mediated oxidative stress, specifically.
Fig. 3.

RUPP TH17 cells significantly decreased pup and placental weight in NP rats. At the time of harvest, the average weight of pups and placentas were calculated for all experimental groups. NP (n = 14), RUPP (n = 15), NP+NP TH17s (n = 7), NP+RUPP TH17s rat (n = 16), NP+RUPP TH17s+ IL-17RC (n = 7), NP+TH17+Los (n = 7) and NP+TH17+Temp (n = 7). A Tukey test was performed as post hoc analysis to generate P values (*P < 0.05 vs. NP; #P < 0.05 vs. NP+RUPP TH17s).
Adoptive Transfer of RUPP TH17 cells increased oxidative stress and AT1-AA in NP rats.
Figure 4A shows that placental reactive oxygen species (ROS) was significantly increased to 370.4 ± 81.6 RLU/min RUPP rats (n = 4) from 144.0 ± 20.1 RLU/min in NP rats (n = 6; P < 0.05). Adoptive transfer of RUPP TH17 cells into NP rats also significantly increased placental oxidative stress. Placental ROS was 431.5 ± 136.7 RLU/min in NP+ RUPP TH17 rats (n = 4; P < 0.05). Fig. 4B shows that renal ROS was also significantly increased in NP+ RUPP TH17 rats (1072 ± 324.2 RLU/min, n = 3) compared with NP rats (214.7 ± 18.5 RLU/min, n = 4; P < 0.05). Renal ROS was higher in RUPP rats, but not significantly higher than NP rats (682.5 ± 49.9 RLU/min, n = 4).
Fig. 4.

RUPP TH17 cells increase oxidative stress in NP rats. Oxidative stress is a hallmark of preeclampsia. Oxidative stress was measured in the placentas (A) and renal cortices (B) of rats in NP (n = 4–6), RUPP (n = 3–4), NP+RUPP TH17s (n = 4) groups using chemiluminescence methods. A Tukey test was performed as post hoc analysis to generate P values (*P < 0.05 vs. NP).
AT1-AA levels in NP rats were 0.11 ± 0.2 beats/min (n = 17). Autoantibody levels increased significantly to 17.27 ± 0.7 beats/min in RUPP rats (n = 21, P < 0.0001; Fig. 5). Likewise, adoptive transfer of RUPP TH17 cells into NP rats significantly increased production of the AT1-AA to 15.58 ± 0.97 compared with NP rats (n = 8, P < 0.0001; Fig. 5).
Fig. 5.
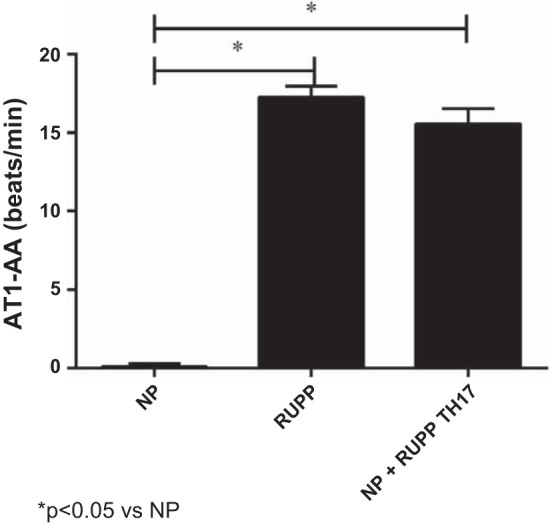
RUPP TH17 cells increase AT1-AA production in NP rats. AT1-AA levels from NP (n = 17), RUPP (n = 21), NP+RUPP TH17s (n = 8) rats were measured via chronotropic events in cardiomyocytes in culture (beats/min). A Tukey test was performed as post hoc analysis to generate P values (*P < 0.05 vs. NP).
Adoptive transfer of RUPP TH17 cells increased proinflammatory cytokines in NP rats.
Circulating levels of the proinflammatory cytokines IL-17, IL-6, and TNF-α were measured in each group of rats. IL-17 (Fig. 6A) was significantly increased from 0.40 ± 0.23 pg/ml in NP rats (n = 7) to 17.19 ± 7.42 pg/ml in RUPP rats (n = 7, P < 0.05). As expected, adoptive transfer of RUPP TH17 cells into NP rats also significantly increased circulating IL-17 to 22.61 ± 10.2 pg/ml (P < 0.05). Treatment with Losartan (n = 6) and Tempol (n = 6) normalized circulating IL-17 to 0.83 ± 0.25 pg/ml and 0.53 ± 0.06 pg/ml, respectively. Circulating levels of IL-6 (Fig. 6B) was significantly increased from 22.4 ± 5.7 pg/ml in NP (n = 9) to 60.5 ± 13.8 pg/ml in RUPP (n = 12; P > 0.05) and to 75.9 ± 6.8 pg/ml in NP+ RUPP TH17 rats (n = 22; P < 0.01). Losartan (n = 9) and Tempol (n = 9) both completely inhibited IL-6 production in NP+RUPP TH17 rats (0.75 ± 0.07 pg/ml and 0.53 ± 0.04 pg/ml, respectively). TNF-α (Fig. 6C) significantly increased from 14.9 ± 5 pg/ml in NP (n = 13) to 65.3 ± 12 pg/ml in RUPP (n = 15; P < 0.01). Likewise, adoptive transfer of RUPP TH17 cells into NP rats also significantly increased circulating IL-17 to 111.5 ± 26.4 pg/ml (n = 13; P < 0.001 vs. NP). Additionally, treatment with Losartan and Tempol also inhibited the increase in TNF-α. Circulating TNF-α was 9.8 ± 5 in NP+RUPP TH17+ Losartan (n = 5; P < 0.05 vs. NP+RUPP TH17) and 9.2 ± 8.1 in NP+RUPP TH17+ Tempol (n = 5, P < 0.05 vs. NP+RUPP TH17). These data demonstrate a modulatory role for RUPP-induced TH17 cells in the inflammatory response to placental ischemia.
Fig. 6.

RUPP TH17 cells are proinflammatory in NP rats. Circulating levels of the proinflammatory cytokines IL-17 (A), IL-6 (B), and TNF-α (C) were measured in all animal groups. NP (n = 7–13), RUPP (n = 7–15), NP+ RUPP TH17s rat (n = 11–14), NP+TH17+Los (n = 7), and NP+TH17+Temp (n = 7). A Tukey test was performed as post hoc analysis to generate P values (*P < 0.05 vs. NP; #P < 0.05 vs. NP+ RUPP TH17s).
DISCUSSION
TH17 cells are implicated in autoimmune disorders, such as irritable bowel syndrome, Crohn’s disease, multiple sclerosis, rheumatoid arthritis, and others (30). Characteristics of PE that are similar to other autoimmune disease include inflammation, production of autoantibodies, and increased circulating IL-17 and TH17 cells (2, 13, 26, 27). PE is clinically recognized by hypertension and sometimes is associated with abnormal Doppler sonography identifying uterine artery resistance index, which is predictive of complications with placental or fetal growth. RUPP in pregnant rats induces an increase in blood pressure, oxidative stress, and circulating TH17 cells, while causing a decrease in fetal weight similar to what is seen in PE patients (8, 9, 24, 27). An imbalance in the ratio between two CD4+ T-helper populations, TReg, and TH17 cells is proposed to have a role in the pathophysiology of PE. We have previously established a role for IL-17 in the pathophysiology of PE by showing that infusion of IL-17 results in increased blood pressure, circulating TH17 cells, placental ROS, and AT1-AA (6). More recently, we have shown that blockade of IL-17 signaling, using a soluble receptor, significantly decreased blood pressure, AT1-AA production, and ROS in the placenta, while increasing pup and placenta weight and improving umbilical artery resitance index in the RUPP rat model of PE (1). However, a role specifically for TH17 cells to mediate the pathophysiology associated with PE or RUPP during pregnancy had never been examined. Therefore, in this study, we sought to determine whether TH17 cells stimulated in response to RUPP in pregnant rats would cause inflammation, oxidative stress, low birth weight, and hypertension during pregnancy.
We demonstrate that adoptive transfer of RUPP-induced TH17 cells into normal pregnant rats significantly increases blood pressure, inflammation, oxidative stress, and AT1-AA production, and causes reduced fetal and placental weight. We also show that these effects are specific to the RUPP-induced TH17 cell population, as TH17 cells from NP rats failed to elicit a PE-like response in NP recipient rats. These data, along with our previously published studies (1, 6), clearly establish a role for TH17 cells in mediating pathophysiology of PE. The introduction of RUPP TH17 cells in later gestation is consistent with the time at which the increased population of TH17 cells is observed during PE (2, 23). Earlier introduction of TH17 cells during pregnancy may result in total fetal demise, as these cell types are also implicated in recurrent miscarriage (14). Preeclampsia with fetal growth restriction may be a pregnancy that survived miscarriage but nevertheless endured many complications with its progression. Here, we show that adoptive transfer of RUPP TH17 cells into NP rats caused a significant decrease in pup and placental weight (Fig. 3). In our previous study, late-gestation blockade of IL-17 with a soluble receptor inhibited low birth weight and resulted in increased placental weight and improved uterine artery resistance in the RUPP rat model (1). In this study, we demonstrate this same effect of IL-17RC to attenuate the decreases in fetal and placental weights in response to RUPP TH17 cells. Further studies should be conducted to examine the levels of TH17 cells early in pregnancy to determine a timeline for which the altered immune response is initiated in PE. Importantly, neither pup nor placental weight were altered in our previous studies of adoptive transfer of CD4+ RUPP T cells into NP rats, where the CD4+ T-cell population comprised only 20% TH17 cells (27). In contrast, in this current study, the population of cells transferred into NP rats was 70% TH17 cells; thus, the greater population of RUPP-induced TH17 cells used in this current study is likely to be the cause for the increased circulating IL-17, and, as our data suggest, responsible for the decrease in fetal and placental weights in the NP recipients of RUPP TH17 cells. Furthermore, administration of IL-17RC to inhibit IL-17 signaling, in this study, resulted in normalization of pup weight, thereby, suggesting the importance of IL-17 to cause IUGR during pregnancy.
Furthermore, in this study, we demonstrated a significant increase in AT1-AA production after adoptive transfer of RUPP TH17 cells. The hypertension in response to RUPP TH17 cells was attenuated by administration of the AT1 receptor antagonist, losartan, suggesting that activation of the AT1 receptor, likely by the AT1-AA, is an important mechanism mediating hypertension when TH17 cells are increased during pregnancy. Importantly the increase in AT1-AA illustrates the importance of TH17 cells to act as facilitators for this antibody production. Although maternal blood pressure was improved with Losartan, pup weight did not increase, thereby, indicating the significance of other factors to cause fetal demise in response to RUPP TH17 cells.
To address this question, we included a group of recipient rats treated with Tempol, a reactive oxygen species scavenger, in the study. In this group, maternal blood pressures were not significantly improved; however, intrauterine growth restriction was attenuated as demonstrated by normalization of pup and placental weights. Our previous studies have shown that scavenging ROS with Tempol attenuates placental oxidative stress in response to chronic infusion of IL-17 (6). It is possible that this mechanism allowed for improved perfusion to the placenta and delivery of more nutrients to the fetus. Importantly, these two treatment arms of the study demonstrate a role for TH17 cells that are stimulated in response to placental ischemia to mediate activation of multiple mechanisms associated with the pathophysiology observed during PE.
In this study, adoptive transfer of placental ischemia-induced TH17 into normal pregnant rats also causes a significant increase in circulating levels of the other proinflammatory cytokines; both TNF-α and IL-6 are known to be stimulated by TH17 cells and IL-17. In our previously published study, adoptive transfer of RUPP CD4+ T cells into NP rats did not significantly increase plasma IL-6 or IL-17 (27). TH17 cells are known to induce production of IL-6 through their secretion of IL-27, and IL-6 is required for differentiation and maintenance of TH17 cells. Therefore, the increased production of IL-6 may act as a positive feedback mechanism for the maintenance of the TH17 cell population. Additionally, IL-6 may influence progression of PE by activation of the vascular endothelium to cause dysfunction (20). Previous studies have shown that infusion of IL-6 into pregnant rats increased arterial pressure and decreased renal plasma flow and glomerular filtration rate (11). Furthermore, IL-6-treated pregnant rats show increased AT1-AA (11). We have previously published studies demonstrating that IL-17-induced hypertension is associated with AT1-AA and oxidative stress (1, 6), and this study established TH17 cells as a source of the IL-17 and a mediator of AT1-AA and oxidative stress during pregnancy. It is possible that communication between TH17, IL-17, and IL-6 could facilitate B-cell maturation and AT1-AA production. However, this is speculative, and follow-up studies should be performed to determine whether the route of AT1-AA production is dependent on increased B-cell activation or whether greater antigen presentation occurred more readily in NP recipients of TH17 cells. Increased understanding of these mechanisms could identify potential drug targets and lead to the development of novel treatment strategies for this disease. Newer strategies need to be developed to prolong pregnancy and yield beneficial outcomes for both mother and baby, and we believe these and other studies from our laboratory indicate targeting the TH17 pathway should be further investigated. In conclusion, this study demonstrates a role for TH17 cells stimulated in response to placental ischemia to be important mediators of many characteristics of PE, thus indicating the importance of research to better understand the pathophysiology behind PE and, thus, identify specific targets and strategies for treatment of PE.
Perspectives and Significance
This study demonstrates an important role for RUPP-induced TH17 cells to mediate the pathophysiology of hypertension and intrauterine growth restriction during pregnancy. The data presented here identify TH17 cell inhibition as a therapeutic target for the treatment of PE in women. The downregulation of this cell population could normalize function of the maternal vasculature, decrease oxidative stress, and improve placental and fetal growth even in the face of placental ischemia. These proposed outcomes could result in improved maternal and fetal health and lower the morbidity and mortality associated with preeclampsia.
GRANTS
This work was supported by National Institutes of Health Grants HL-126301, HL-105324 HD-067541 and American Heart Association Grant 16SDG27520000.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.C.C., L.M.A., N.C., A.J.T., J.S., F.H., G.W., and R.D. performed experiments; D.C.C., K.W., N.C., A.J.T., J.S., F.H., G.W., and B.D.L. analyzed data; D.C.C., K.W., and B.D.L. interpreted results of experiments; D.C.C. prepared figures; D.C.C. drafted manuscript; D.C.C., L.M.A., K.W., F.H., R.D., and B.D.L. edited and revised manuscript; D.C.C., L.M.A., K.W., N.C., A.J.T., J.S., F.H., G.W., R.D., and B.D.L. approved final version of manuscript.
REFERENCES
- 1.Cornelius DC, Hogg JP, Scott J, Wallace K, Herse F, Moseley J, Wallukat G, Dechend R, LaMarca B. Administration of interleukin-17 soluble receptor C suppresses TH17 cells, oxidative stress, and hypertension in response to placental ischemia during pregnancy. Hypertension 62: 1068–1073, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Darmochwal-Kolarz D, Kludka-Sternik M, Tabarkiewicz J, Kolarz B, Rolinski J, Leszczynska-Gorzelak B, Oleszczuk J. The predominance of Th17 lymphocytes and decreased number and function of Treg cells in preeclampsia. J Reprod Immunol 93: 75–81, 2012. doi: 10.1016/j.jri.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Dechend R, Homuth V, Wallukat G, Kreuzer J, Park JK, Theuer J, Juepner A, Gulba DC, Mackman N, Haller H, Luft FC. AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation 101: 2382–2387, 2000. doi: 10.1161/01.CIR.101.20.2382. [DOI] [PubMed] [Google Scholar]
- 4.Dechend R, Homuth V, Wallukat G, Müller DN, Krause M, Dudenhausen J, Haller H, Luft FC. Agonistic antibodies directed at the angiotensin II, AT1 receptor in preeclampsia. J Soc Gynecol Investig 13: 79–86, 2006. doi: 10.1016/j.jsgi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Dechend R, Müller DN, Wallukat G, Homuth V, Krause M, Dudenhausen J, Luft FC. Activating auto-antibodies against the AT1 receptor in preeclampsia. Autoimmun Rev 4: 61–65, 2005. doi: 10.1016/j.autrev.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Dhillion P, Wallace K, Herse F, Scott J, Wallukat G, Heath J, Mosely J, Martin JN Jr, Dechend R, LaMarca B. IL-17-mediated oxidative stress is an important stimulator of AT1-AA and hypertension during pregnancy. Am J Physiol Regul Integr Comp Physiol 303: R353–R358, 2012. doi: 10.1152/ajpregu.00051.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.George EM, Granger JP. Recent insights into the pathophysiology of preeclampsia. Expert Rev Obstet Gynecol 5: 557–566, 2010. doi: 10.1586/eog.10.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert J, Dukes M, LaMarca B, Cockrell K, Babcock S, Granger J. Effects of reduced uterine perfusion pressure on blood pressure and metabolic factors in pregnant rats. Am J Hypertens 20: 686–691, 2007. doi: 10.1016/j.amjhyper.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 9.Granger JP, LaMarca BB, Cockrell K, Sedeek M, Balzi C, Chandler D, Bennett W. Reduced uterine perfusion pressure (RUPP) model for studying cardiovascular-renal dysfunction in response to placental ischemia. Methods Mol Med 122: 383–392, 2006. [DOI] [PubMed] [Google Scholar]
- 10.LaMarca B. The role of immune activation in contributing to vascular dysfunction and the pathophysiology of hypertension during preeclampsia. Minerva Ginecol 62: 105–120, 2010. [PMC free article] [PubMed] [Google Scholar]
- 11.LaMarca B, Brewer J, Wallace K. IL-6-induced pathophysiology during pre-eclampsia: potential therapeutic role for magnesium sulfate? Int J Interferon Cytokine Mediat Res 2011: 59–64, 2011. doi: 10.2147/IJICMR.S16320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaMarca B, Parrish M, Ray LF, Murphy SR, Roberts L, Glover P, Wallukat G, Wenzel K, Cockrell K, Martin JN Jr, Ryan MJ, Dechend R. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1. Hypertension 54: 905–909, 2009. doi: 10.1161/HYPERTENSIONAHA.109.137935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laresgoiti-Servitje E. A leading role for the immune system in the pathophysiology of preeclampsia. J Leukoc Biol 94: 247–257, 2013. doi: 10.1189/jlb.1112603. [DOI] [PubMed] [Google Scholar]
- 14.Lee SK, Kim JY, Lee M, Gilman-Sachs A, Kwak-Kim J. Th17 and regulatory T cells in women with recurrent pregnancy loss. Am J Reprod Immunol 67: 311–318, 2012. doi: 10.1111/j.1600-0897.2012.01116.x. [DOI] [PubMed] [Google Scholar]
- 15.Lindheimer MD, Romero R. Emerging roles of antiangiogenic and angiogenic proteins in pathogenesis and prediction of preeclampsia. Hypertension 50: 35–36, 2007. doi: 10.1161/HYPERTENSIONAHA.107.089045. [DOI] [PubMed] [Google Scholar]
- 16.Novotny SR, Wallace K, Heath J, Moseley J, Dhillon P, Weimer A, Wallukat G, Herse F, Wenzel K, Martin JN Jr, Dechend R, Lamarca B. Activating autoantibodies to the angiotensin II type I receptor play an important role in mediating hypertension in response to adoptive transfer of CD4+ T lymphocytes from placental ischemic rats. Am J Physiol Regul Integr Comp Physiol 302: R1197–R1201, 2012. doi: 10.1152/ajpregu.00623.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parrish MR, Murphy SR, Rutland S, Wallace K, Wenzel K, Wallukat G, Keiser S, Ray LF, Dechend R, Martin JN, Granger JP, LaMarca B. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am J Hypertens 23: 911–916, 2010. doi: 10.1038/ajh.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parrish MR, Ryan MJ, Glover P, Brewer J, Ray L, Dechend R, Martin JN Jr, Lamarca BB. Angiotensin II type 1 autoantibody induced hypertension during pregnancy is associated with renal endothelial dysfunction. Gend Med 8: 184–188, 2011. doi: 10.1016/j.genm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parrish MR, Wallace K, Tam Tam KB, Herse F, Weimer A, Wenzel K, Wallukat G, Ray LF, Arany M, Cockrell K, Martin JN, Dechend R, LaMarca B. Hypertension in response to AT1-AA: role of reactive oxygen species in pregnancy-induced hypertension. Am J Hypertens 24: 835–840, 2011. doi: 10.1038/ajh.2011.62. [DOI] [PubMed] [Google Scholar]
- 20.Prins JR, Gomez-Lopez N, Robertson SA. Interleukin-6 in pregnancy and gestational disorders. J Reprod Immunol 95: 1–14, 2012. doi: 10.1016/j.jri.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 20a.Richards S, Wallace K, Weimer A, Dhillion P, Wallukat G, Wenzel K, Martin J, Dechend R, Lamarca B.. T Lymphocyte induced AT1-AAs cause hypertension in response to placental ischemia. Am J Obstet Gynecol 204: S100, 2011. [Google Scholar]
- 21.Roberts JM, Lain KY. Recent Insights into the pathogenesis of pre-eclampsia. Placenta 23: 359–372, 2002. doi: 10.1053/plac.2002.0819. [DOI] [PubMed] [Google Scholar]
- 22.Saito S, Nakashima A, Shima T, Ito M. Th1/Th2/Th17 and regulatory T-cell paradigm in pregnancy. Am J Reprod Immunol 63: 601–610, 2010. doi: 10.1111/j.1600-0897.2010.00852.x. [DOI] [PubMed] [Google Scholar]
- 23.Santner-Nanan B, Peek MJ, Khanam R, Richarts L, Zhu E, Fazekas de St Groth B, Nanan R. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J Immunol 183: 7023–7030, 2009. doi: 10.4049/jimmunol.0901154. [DOI] [PubMed] [Google Scholar]
- 24.Sedeek M, Gilbert JS, LaMarca BB, Sholook M, Chandler DL, Wang Y, Granger JP. Role of reactive oxygen species in hypertension produced by reduced uterine perfusion in pregnant rats. Am J Hypertens 21: 1152–1156, 2008. doi: 10.1038/ajh.2008.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sykes L, MacIntyre DA, Yap XJ, Teoh TG, Bennett PR. The Th1:th2 dichotomy of pregnancy and preterm labour. Mediators Inflamm 2012: 967629, 2012. doi: 10.1155/2012/967629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toldi G, Rigó J Jr, Stenczer B, Vásárhelyi B, Molvarec A. Increased prevalence of IL-17-producing peripheral blood lymphocytes in pre-eclampsia. Am J Reprod Immunol 66: 223–229, 2011. doi: 10.1111/j.1600-0897.2011.00987.x. [DOI] [PubMed] [Google Scholar]
- 27.Wallace K, Richards S, Dhillon P, Weimer A, Edholm ES, Bengten E, Wilson M, Martin JN Jr, LaMarca B. CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension 57: 949–955, 2011. doi: 10.1161/HYPERTENSIONAHA.110.168344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jüpner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest 103: 945–952, 1999. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilke CM, Bishop K, Fox D, Zou W. Deciphering the role of Th17 cells in human disease. Trends Immunol 32: 603–611, 2011. doi: 10.1016/j.it.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]