
Figure 3.
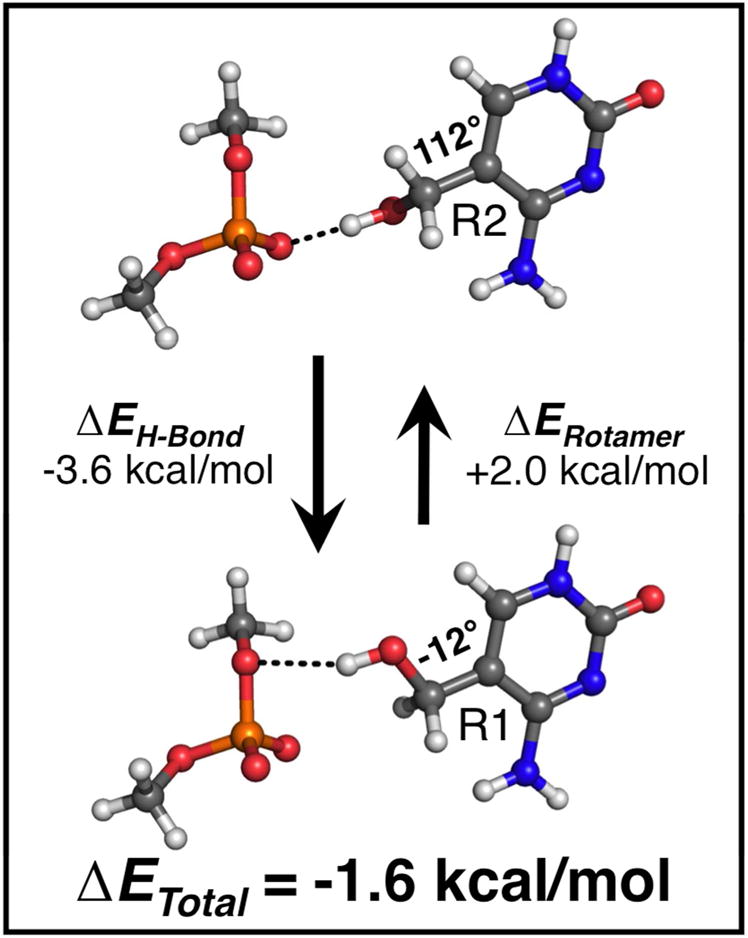
Comparison of H-bond energies (ΔEH-Bond) and rotamer energies (ΔERotamer) between the major (R1, bottom) and minor (R2, top) conformations of the hydroxymethyl substituent in the 5hmC structure. Quantum mechanical (QM) energies were calculated on small molecule models of the junction core (5hmC and dimethylphosphate), constructed from atomic coordinates taken from the crystal structure. The isolated 5hmC base has a 2.0 kcal/mol energy preference towards the R2 rotamer (112°) in the bond rotation energy. However, H-bonding interaction energy was calculated to favor the R1 rotamer (−12°) by −3.6 kcal/mol (signs of the energy terms are defined as the difference ER1 – ER2). In summation, the dominant R1 rotamer is favored by an overall energy (ΔETotal = ΔERotamer + ΔEH-Bond) of −1.6 kcal/mol.