

Abstract
Nonalcoholic fatty liver disease (NAFLD) is characterized by excess accumulation of fat in the liver. In some cases, NAFLD is also accompanied by insulin resistance, resulting in metabolic dysfunction. Dietary fat content probably influences both NAFLD and insulin resistance; however, the immediate effects of fat consumption have not been fully explored. In this issue of the JCI, Hernández et al. evaluated hepatic glucose and lipid metabolism in humans and mice following a single oral dose of saturated fat. This one bolus of fat resulted in a measurable increase in insulin resistance, hepatic triglycerides, and gluconeogenesis. In mice, the saturated fat bolus resulted in the induction of several NAFLD-associated genes. Together, the results of this study indicate that saturated fat intake has immediate effects on metabolic function.
Links between NAFLD and insulin resistance
Nonalcoholic fatty liver disease (NAFLD) covers a spectrum of disorders, ranging from simple steatosis to nonalcoholic steatohepatitis (NASH) and cirrhosis. Given the burgeoning national and global obesity epidemic, NAFLD is predicted to surpass all other forms of liver disease as a cause for liver transplantation (1). NAFLD is characterized by excess hepatic lipid accumulation; however, the added presence of insulin resistance (so-called “metabolic” NAFLD) results in features of metabolic syndrome and markedly increases the incidence of type 2 diabetes (T2D) and cardiovascular disease (CVD) (2). There is also a strong association between liver fat and peripheral insulin sensitivity in nondiabetic individuals, suggesting that intrahepatic triglyceride (IHTG) accumulation may have important systemic consequences that adversely affect insulin sensitivity in other tissues (3).
Some obese individuals remain metabolically healthy, and NAFLD is observed in the nonobese (2). In such individuals, NAFLD is linked to common genetic variants in genes, such as patatin-like phospholipase domain–containing 3 (PNPLA3) or transmembrane 6 superfamily member 2 (TM6SF2) (2, 4). PNPLA3 exhibits hydrolase activity against triglycerides (TGs), and variations in PNPLA3 can lead to TG accumulation in hepatocytes (5). TM6SF2 has been proposed to regulate liver fat metabolism and promote increases in cellular TG concentrations (6). Although variants in these genes impact IHTG and predict advanced liver disease, the variants are intriguingly not associated with insulin resistance or increased risk of T2D or CVD (2). Fatty acid and TG profiles in the liver of humans with genetic NAFLD markedly differ from profiles in individuals with metabolic NAFLD (7). It is not clear why some individuals, but not others, develop full-blown metabolic syndrome including NAFLD. Could interindividual differences in diet composition underlie the susceptibility to metabolic disorders?
Dietary intervention studies in human volunteers have shown that isocaloric substitution of carbohydrate with fat increases liver fat content (8). Moreover, the type of fat added to the diet matters. Diets rich in saturated fatty acids induce a greater increase in liver fat and insulin resistance compared with isocaloric diets enriched with monounsaturated or polyunsaturated fatty acids (7). The mechanisms whereby saturated fat modulates hepatic metabolism are incompletely understood; specifically, it is not entirely clear why glucose metabolism in the liver becomes insulin resistant in response to saturated fat feeding and how saturated fat increases liver fat content. Of note, various intrahepatic inflammatory processes and regulatory signaling pathways have been proposed to contribute to the pathogenesis of NAFLD. Inflammatory triggers, including LPS, promote TLR4-induced cytokine release by hepatic Kupffer cells, resulting in JNK-mediated insulin resistance (9). PPARs also play a role in modulating hepatic TG accumulation and hepatic fat oxidation (9). Since fatty acids can regulate the transcription of inflammatory and metabolically relevant genes (10), dietary fat might impact some or all of the above pathways.
Carefully controlled feeding studies that used multiple tracers have shown a measurable clearance of dietary lipid to the liver and the contribution of this clearance to intrahepatic IHTG (11–14). For example, Carpentier and colleagues used PET coupled with computerized axial tomography (CAT) technology to show the uptake of dietary fat in the liver over a six-hour period (15). Of note, it has long been established that dietary fat constitutes the smallest source of lipid that can enter IHTG pools, contributing roughly 10% to 20% of liver TG fatty acids compared with the plasma free fatty acid (FFA) pool, which can contribute 60% to 100% of liver TG fatty acids, and compared with de novo lipogenesis, which can contribute up to 30% to 40% of IHTG (13, 14, 16). However, a recent study described the development of gradient-enhanced heteronuclear single-quantum coherence (ge-HSQC) spectroscopy for the in vivo detection of hepatic 1H-[13C]-lipid signals. Using this technology, Lindeboom et al. showed incorporation of 13C-labeled fatty acids into IHTGs after a single high-fat meal, suggesting that a substantial amount of liver fat can originate directly from the storage of meal-derived fat (17), particularly given the increased insulin levels that would be expected following such a meal.
A single fat bolus packs a punch
The apparent discrepancy between these studies set the stage for the elegant and comprehensive human and mouse studies presented by Hernández et al. in this issue (18). Hernández and colleagues used state-of-the-art techniques to examine the short-term impact of oral saturated fat (palm oil) administration on in vivo metabolic fluxes and hepatic gene expression. Hepatic metabolism was studied in vivo with 13C/31P/1H and ex vivo with 2H magnetic resonance spectroscopy (MRS) combined with 2H2O ingestion before and during insulin clamps with isotope dilution. In humans, the fat bolus caused insulin resistance in muscle, liver, and adipose tissue, with increased hepatic TG and ATP content and enhanced gluconeogenesis. It is perhaps noteworthy that insulin levels increased substantially both from the fat bolus and during subsequent hyperinsulinemic clamps, which could potentiate the rise in IHTG. The relatively small change in IHTG following one oral fat bolus in these lean, nondiabetic subjects raises the question as to what extent this affected endogenous glucose production (EGP), or whether other factors such as increased TCA cycle activity, fat oxidation, or energy sensing in the liver might mediate these effects. Complementary rodent studies provided evidence to suggest that dietary saturated fat might promote NAFLD by activating LPS-induced genes, PPARs, and other known and suspected regulators of NAFLD, thus providing a potentially unified hypothesis whereby fat intake impacts both IHTG and insulin resistance (Figure 1). Intriguingly, in contrast to the human subjects, mice in this study did not exhibit peripheral insulin resistance or an increase in IHTG in response to the oral fat load. This discrepancy validates the observations that activation of various intrahepatic pathways mediates hepatic insulin resistance (9), while IHTG is more strongly linked with systemic insulin resistance (3).
Figure 1. Dietary fat influences both IHTG levels and insulin resistance.
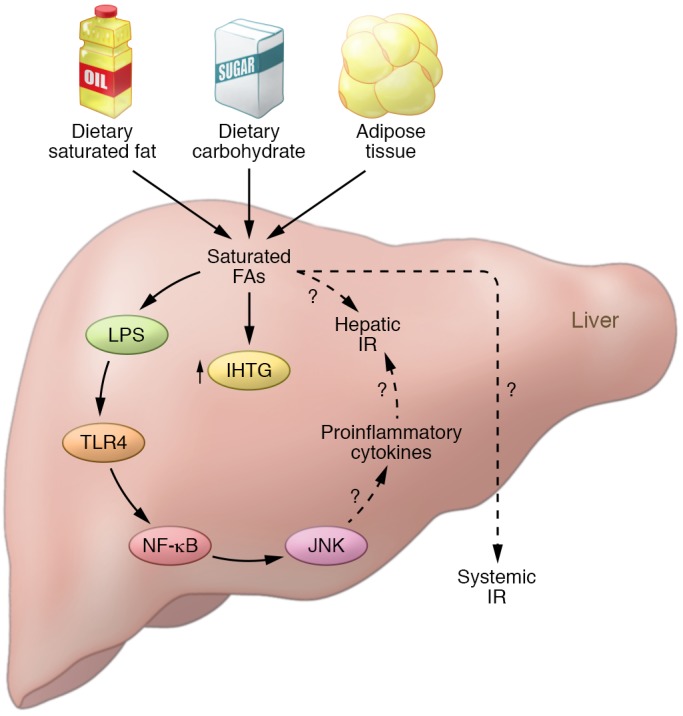
In this issue, Hernández et al. show that a single oral intake of saturated fat increases the level of IHTGs. In turn, saturated fatty acids from dietary fat and carbohydrate or from adipose tissue directly promote the upregulation of NAFLD-associated pathways and may promote both systemic and hepatic insulin resistance, as well as contribute to IHTG. FAs, fatty acids; IR, insulin resistance.
The greatest effects of the mechanisms underlying metabolic disease occur during the fed state as the result of entry of exogenous nutrients (a meal) against a backdrop of excess endogenous nutrient production. In this way, an elevated fasting concentration of glucose is made worse after a meal by both meal glucose absorption and the lack of suppression of endogenous EGP. The findings of Hernández et al. further advance this concept by highlighting the impact of one macronutrient (fat) on another (glucose) and by demonstrating the negative impact of fat consumption on subsequent whole-body and hepatic glucose metabolism, thereby pointing to mechanisms of nutrient toxicity. Indeed, a high-fat meal consumed the night before an oral glucose tolerance test (OGTT) was associated with higher glucose and FFA concentrations during the OGTT, despite insulin levels that were comparable to levels observed the morning after a high-carbohydrate meal (19). It is important to note that the higher FFA concentrations observed after fat consumption are not likely due to greater adipose FFA output but rather originate from spillover of fatty acids through chylomicron-TG hydrolysis (20, 21). The naturally extended time of absorption of such a large bolus of orally ingested fat results in a spillover of dietary fatty acids into the plasma FFA pool throughout the eight-hour postprandial period — the magnitude of which is proportional to the amount of fat in the meal (22). The ability of fatty acids to stimulate gluconeogenesis and EGP remains somewhat underappreciated (23), and the present study by Hernández et al. shows that such a “cross-macronutrient effect” may occur, even acutely after a single bolus of lipid.
Conclusions and future directions
Future avenues to extend the work of Hernández and colleagues include examining the relevance of these findings in women, as only male subjects were included in this study, and the uptake of fatty acids by the liver may be greater in men than in women (24). While Hernández et al. used water intake as the control to evaluate the effect of a fat bolus, future comparisons with an unsaturated fatty acid or a protein bolus will be helpful to distinguish the effects of saturated fatty acids per se under isocaloric conditions. The isolated impact of large quantities of saturated fat was examined with the intent of avoiding any confounding effects of other nutrients. Given the interaction between dietary lipid handling and glucose metabolism, it would be intriguing to use these sophisticated methodologies to investigate the impact of saturated fat present in mixed meals, with more physiologic fat content, to better understand cross-macronutrient effects. It will be particularly interesting to extend these studies to the physiology of sedentary, overweight individuals with fatty liver disease, as subjects with T2D acutely incorporated dietary fatty acids into IHTG to a greater extent than did lean individuals (25).
Acknowledgments
The authors wish to thank Emilce Carrasco (Albert Einstein College of Medicine) for her helpful intellectual contributions and creation of the figure and to acknowledge support from the NIH (DK-069861 and DK-48321, to MH).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2017;127(2):454–456. https://doi.org/10.1172/JCI92407.
See the related article at Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance.
Contributor Information
Elizabeth Parks, Email: parksej@missouri.edu.
Hannele Yki-Järvinen, Email: Hannele.Yki-Jarvinen@helsinki.fi.
Meredith Hawkins, Email: meredith.hawkins@einstein.yu.edu.
References
- 1.Pais R, et al. NAFLD and liver transplantation: Current burden and expected challenges. J Hepatol. 2016;65(6):1245–1257. doi: 10.1016/j.jhep.2016.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yki-Järvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014;2(11):901–910. doi: 10.1016/S2213-8587(14)70032-4. [DOI] [PubMed] [Google Scholar]
- 3.Hwang JH, et al. Increased intrahepatic triglyceride is associated peripheral insulin resistance: in vivo MR imaging and spectroscopy studies. Am J Physiol Endocrinol Metab. 2007;293(6):E1663–E1669. doi: 10.1152/ajpendo.00590.2006. [DOI] [PubMed] [Google Scholar]
- 4.Kozlitina J, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic with fatty liver disease. Nat Genet. 2014;46(4):352–356. doi: 10.1038/ng.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang Y, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci U S A. 2010;107(17):7892–7897. doi: 10.1073/pnas.1003585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahdessian H, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A. 2014;111(24):8913–8918. doi: 10.1073/pnas.1323785111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luukkonen PK, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol. 2016;64(5):1167–1175. doi: 10.1016/j.jhep.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Green CJ, Hodson L. The influence of dietary fat on liver fat accumulation. Nutrients. 2014;6(11):5018–5033. doi: 10.3390/nu6115018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol. 2016;11:451–496. doi: 10.1146/annurev-pathol-012615-044224. [DOI] [PubMed] [Google Scholar]
- 10.Kishore P, et al. Adipocyte-derived factors potentiate nutrient-induced production of plasminogen activator inhibitor-1 by macrophages. Sci Transl Med. 2010;2(20):20ra15. doi: 10.1126/scitranslmed.3000292. [DOI] [PubMed] [Google Scholar]
- 11.Barrows BR, Parks EJ. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J Clin Endocrinol Metab. 2006;91(4):1446–1452. doi: 10.1210/jc.2005-1709. [DOI] [PubMed] [Google Scholar]
- 12.Hodson L, et al. The contribution of splanchnic fat to vldl triglyceride is greater in insulin-resistant than insulin-sensitive men and women: studies in the postprandial state. Diabetes. 2007;56(10):2433–2441. doi: 10.2337/db07-0654. [DOI] [PubMed] [Google Scholar]
- 13.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vedala A, Wang W, Neese RA, Christiansen MP, Hellerstein MK. Delayed secretory pathway contributions to VLDL-triglycerides from plasma NEFA, diet, and de novo lipogenesis in humans. J Lipid Res. 2006;47(11):2562–2574. doi: 10.1194/jlr.M600200-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Labbé SM, et al. Organ-specific dietary fatty acid uptake in humans using positron emission tomography coupled to computed tomography. Am J Physiol Endocrinol Metab. 2011;300(3):E445–E453. doi: 10.1152/ajpendo.00579.2010. [DOI] [PubMed] [Google Scholar]
- 16.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinctive characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindeboom L, et al. Quantum coherence spectroscopy to measure dietary fat retention in the liver. JCI Insight. 2016;1(13):e84671. doi: 10.1172/jci.insight.84671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernández EÁ, et al. Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance. J Clin Invest. 2017;127(2):695–708. doi: 10.1172/JCI89444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robertson MD, Henderson RA, Vist GE, Rumsey RD. Extended effects of evening meal carbohydrate-to-fat ratio on fasting and postprandial substrate metabolism. Am J Clin Nutr. 2002;75(3):505–510. doi: 10.1093/ajcn/75.3.505. [DOI] [PubMed] [Google Scholar]
- 20.Barrows BR, Timlin MT, Parks EJ. Spillover of dietary fatty acids and use of serum nonesterified fatty acids for the synthesis of VLDL-triacylglycerol under two different feeding regimens. Diabetes. 2005;54(9):2668–2673. doi: 10.2337/diabetes.54.9.2668. [DOI] [PubMed] [Google Scholar]
- 21.Miles JM, Nelson RH. Contribution of triglyceride-rich lipoproteins to plasma free fatty acids. Horm Metab Res. 2007;39(10):726–729. doi: 10.1055/s-2007-990273. [DOI] [PubMed] [Google Scholar]
- 22.Puga GM, Meyer C, Mandarino LJ, Katsanos CS. Postprandial spillover of dietary lipid into plasma is increased with moderate amounts of ingested fat and is inversely related to adiposity in healthy older men. J Nutr. 2012;142(10):1806–1811. doi: 10.3945/jn.112.162008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kehlenbrink S, Tonelli J, Koppaka S, Chandramouli V, Hawkins M, Kishore P. Inhibiting gluconeogenesis prevents fatty acid-induced increases in endogenous glucose production. Am J Physiol Endocrinol Metab. 2009;297(1):E165–E173. doi: 10.1152/ajpendo.00001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Browning JD, Baxter J, Satapati S, Burgess S. The effect of short-term fasting on liver and skeletal muscle lipid, glucose, and energy metabolism in healthy women and men. J Lipid Res. 2012;53(3):577–586. doi: 10.1194/jlr.P020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ravikumar B, et al. Real-time assessment of postprandial fat storage in liver and skeletal muscle in health and type 2 diabetes. Am J Physiol Endocrinol Metab. 2005;288(4):E789–E797. doi: 10.1152/ajpendo.00557.2004. [DOI] [PubMed] [Google Scholar]