

Abstract
The vacuolar H+ ATPase (V-ATPase) is a complex multi-subunit machine that regulates important cellular processes through controlling acidity of intracellular compartments in eukaryotes. Existing small-molecule modulators of V-ATPase either are restricted to targeting one membranous subunit of V-ATPase or have poorly understood mechanisms of action. Small molecules with novel and defined mechanisms of inhibition are thus needed to functionally characterize V-ATPase and to fully evaluate the therapeutic relevance of V-ATPase in human diseases. We have discovered electrophilic quinazolines that covalently modify a soluble catalytic subunit of V-ATPase with high potency and exquisite proteomic selectivity as revealed by fluorescence imaging and chemical proteomic activity-based profiling. The site of covalent modification was mapped to a cysteine residue located in a region of V-ATPase subunit A that is thought to regulate the dissociation of V-ATPase. We further demonstrate that a previously reported V-ATPase inhibitor, 3-bromopyruvate, also targets the same cysteine residue and that our electrophilic quinazolines modulate the function of V-ATPase in cells. With their well-defined mechanism of action and high proteomic specificity, the described quinazolines offer a powerful set of chemical probes to investigate the physiological and pathological roles of V-ATPase.
Graphical Abstract
The vacuolar H+ ATPase (V-ATPase) is a pivotal player in controlling intracellular pH to regulate various biological processes including membrane trafficking, endocytosis, protein degradation, bone resorption, and small-molecule uptake in eukaryotic cells.1 Dysregulation of V-ATPase has been implicated in a number of diseases, including renal disease (renal tubular acidosis)2, bone disease (osteoporosis)3 and tumor metastasis.4 V-ATPase is a large multi-subunit protein complex that functions as a rotary molecular motor and is organized into two domains, V0 and V1. V1 is located on the cytoplasmic side of the membrane and carries out ATP hydrolysis, whereas the V0 domain is a membrane-embedded complex that is responsible for driving proton translocation across the membrane.1,5 The V1 domain is composed of eight different subunits (A, B, C, D, E, F, G, H) while the V0 domain contains five different subunits (a, b, c, d and e) in mammals. The core of the V1 domain contains a hexamer of A and B subunits, which participates in ATP binding and hydrolysis.6–8 A so-called non-homologous region (NHR) in V-ATPase subunit A has been shown to be important for regulating the disassembly of the V1–V0 complex. This 90-amino acid insert in subunit A is highly-conserved among V-ATPase A subunits from numerous species but is absent from the homologous β subunit of the F-ATPase.9 Mutations in the NHR can alter in vivo dissociation of V-ATPase independent of its catalytic activity.10
A number of inhibitors of V-ATPase including both natural products and synthetic compounds have been identified and characterized previously.11 Among these, the macrolides such as bafilomycin, concanamycin and archazolid were all found to bind to the membranous subunit c and perturb rotation of the b/c-ring.12–14 While diverse synthetic V-ATPase inhibitors such as benzolactone enamide,15 indole derivatives,16 and 3-bromopyruvate17 have been reported recently, their binding site within V-ATPase and exact modes of action are often not known. Small molecules with new and defined modes of action against V-ATPase are needed to elucidate the working mechanism and validate the therapeutic relevance of V-ATPase. Herein we describe the discovery and characterization of novel quinazoline-based compounds that covalently target a cysteine (Cys) residue within the NHR of the A subunit with exquisite specificity across the human proteome and modulate the function of V-ATPase in cells.
Our lab has recently prepared a series of quinazolines featuring electrophiles at the 7 position, developed as covalent inhibitors of the receptor tyrosine kinase EphB3.18 In an effort to monitor the target engagement of these electrophilic quinazolines, we derivatized one electrophilic quinazoline with a terminal alkyne group at the 3′ position of the anilino group as a “clickable” tag (Figure 1A). Treatment of HEK293H (293) cells with the resulting small-molecule probe 1 followed by cellular lysis, CuAAC (Cu(I)-catalyzed [3+2] azide-alkyne cycloaddtion)19 with TAMRA-azide, SDS-PAGE, and ingel fluorescence scanning revealed one major fluorescent band of ~70 kDa across the tested concentration range for 1 (Figure 1B). A time-course experiment showed that this target protein (dubbed p70) was near-completely labeled by 1 after 5 min (Figure 1C). Taken together, these data suggest that probe 1 can potently and selectively engage a major target protein of unknown identity in the human proteome. The p70 target appears to be present in many human cell types as probe 1 exhibited a similar labeling pattern in other tested cell lines from different tissue origins (Figure S1).
Figure 1.
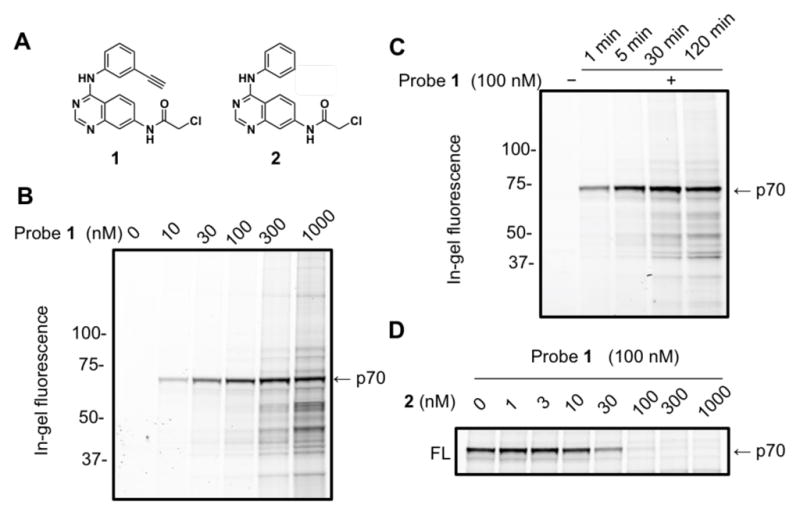
Potent, specific and rapid labeling of a cellular protein by the electrophilic probe 1 in 293 cells. (A) Structures of an electrophilic quinazoline 2 and its clickable analog (1). (B) Dose response of live cell labeling revealed potent engagement of probe 1 to an unknown cellular target of ~70 kDa (p70). (C) Time-course experiment showed rapid engagement of 1 to p70. (D) Dose-dependent blockade of probe labeling by pre-treatment of 2.
To evaluate the structure-activity relationship of the quinazoline-p70 interaction, we applied a competitive gel-based profiling strategy, screening our library of quinazolines (Figure S2) for blockade of p70 labeling by probe 1. Out of 8 quinazolines, only the chloroacetamide-containing compound 2 successfully blocked p70 target labeling by probe 1. By treating 293 cells with various concentrations of 2 for 30 min before addition of 100 nM of probe 1, we calculated an apparent IC50 value of ~30 nM for 2 labeling of p70 in situ (Figure 1D). Consistent with the in situ labeling, we also found that the p70 target was labeled by probe 1 in vitro in 293 cellular lysates and that pre-treatment with 2 blocked this labeling event with similar potency to that observed in live cells (Figure S3). Taken together, these results suggest that the p70 protein target potently and selectively reacts with chloroacetamide quinazolines both in vitro and in cells.
We next set out to identify the p70 protein by chemical proteomics. We treated 293 cells with 100 nM of probe 1 for 30 min, followed by lysis and CuAAC-mediated conjugation to a cleavable biotin-azo-azide tag. Biotin-linked target proteins were then captured by streptavidin agarose beads, eluted with sodium dithionite, and resolved by SDS-PAGE. Protein bands were excised, trypsinized, and subjected to LC-MS/MS analysis (Figure 2A).20 Using spectral counting to quantify the fold-enrichment for proteins derived from probe treated compared to DMSO-treated cells, we identified the vacuolar ATPase catalytic subunit A (ATP6V1A) as the likely target of 1 (Figure 2B). ATP6V1A has a predicted molecular size of 68.3 kDa, which matches the major fluorescent band observed in our proteomic labeling experiments. Western blot analysis, using an ATP6V1A antibody21 to label the proteins enriched during the biotin pulldown, revealed an intense band in the probe-treated sample but not in the mock-treated control, further confirming ATP6V1A as the target of probe 1 (Figure 2C). Finally, transfection of 293 cells with 3×FLAG-tagged human ATP6V1A followed by treatment with probe 1 led to the appearance of a new labeled band of slightly retarded mobility compared to the original band (Figure 3B, arrow). Thus, multiple lines of evidences converged to support ATP6V1A as the target of probe 1 in cells.
Figure 2.
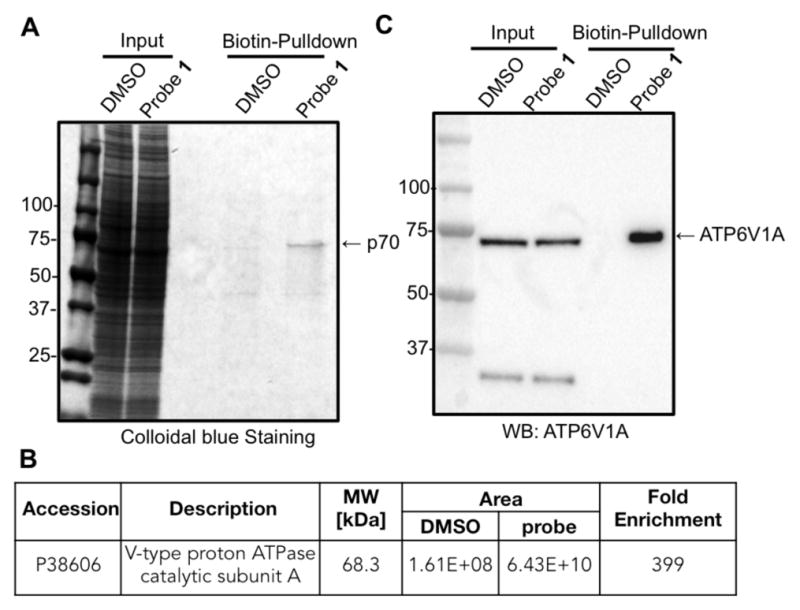
Identification of p70 as the vacuolar ATPase subunit A. (A) Streptavidin pull-down enriched p70. (B) LC-MS/MS analysis identified the target protein as ATP6V1A. (C) Immunoblot analysis using a house-made antibody confirmed the protein pulled down with probe 1 was ATP6V1A.
Figure 3.
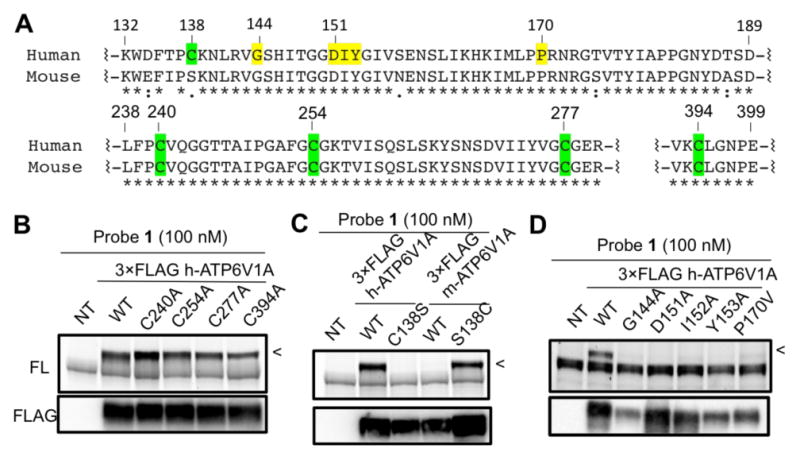
Identification of the probe-labeling site in ATP6V1A. (A) Sequence alignment of human and mouse ATP6V1A highlighting the mutated sites. (B) Mutation of four conserved Cys residues failed to significantly affect probe labeling of 3xFLAG-tagged ectopic ATP6V1A (<). (C) Mutation of C138 abolished probe labeling of human ATP6V1A while the converse mutation of S138C enabled labeling of the mouse protein. (D) Five mutations in distinct regions of NHR all diminished probe binding substantially. The top panels in B–D are in-gel fluorescence images (FL) and the bottom are western blots showing levels of FLAG-tagged ATP6V1A. NT: non-transfected.
We reasoned that the covalent interaction between ATP6V1A and probe 1 was likely mediated by a nucleophilic attack of a Cys thiol group in ATP6V1A at the chloroacetamide group of 1. To identify the residue being modified by the probe, we tested the probe-labeling sensitivity of 3×FLAG-tagged ATP6V1A alleles in which select Cys residues were individually mutated to serine (Ser) or alanine. Sequence analysis revealed that human ATP6V1A contained a total of seven Cys residues, out of which six are conserved in the mouse orthologue (Figure 3A). Excluding two predicted buried Cys residues, we first mutated the other four conserved Cys residues and examined their effects on probe labeling. However, mutation of these four Cys residues had little effect on probe labeling of the 3×FLAG-tagged ATP6V1A (Figure 3B). We then considered the possibility that the non-conserved Cys (C138) was the modification site (Figure 3A). Mutation of C138 to Ser, the corresponding residue in mouse ATP6V1A, indeed abolished probe labeling. The converse mutation (S138C) conferred the mouse ATP6V1A with a strong sensitivity to probe labeling (Figure 3C). It is interesting to note that the sequence surrounding position 138 is highly conserved between human and mouse ATP6V1A. We suspect that C138 might have been evolved to serve as a redox sensor in the human protein. C138 is located at the beginning of the ATP6V1A NHR, which has been shown to be important for regulation of complex assembly and dissociation.10 We introduced several mutations in this region and observed a consequent loss of probe labeling for five distinct residues (Figure 3D). Taken together these results imply that probe 1 binds to a pocket that is likely formed by the NHR and covalently targets C138. Consistent with the remoteness of NHR from the ATPase active site, up to millimolar concentrations of ATP failed to block ATP6V1A labeling by probe 1 in vitro (Figure S4).
The gel-based profiles of probe 1 reactivity suggested high specificity for ATP6V1A, but we recognized that additional, lower-abundance protein targets of 1 may also exist in human cells. We addressed this question by using the quantitative chemical proteomic method isoTOP-ABPP (isotopic tandem orthogonal proteolysis-activity-based protein profiling) following previously described protocols.22 MDA-MB-231 cell lysates were pre-treated with DMSO or 2, followed by treatment with the general Cys-reactive probe, iodoacetamide (IA)-alkyne, followed by quantitative isoTOP-ABPP analysis. C138 of ATP6V1A was fully protected from IA-alkyne labeling by 2 at 500 nM (competition R value of 20, reflecting 95% or greater protection); in contrast, none of the other > 2900 cysteines quantified in these experiments were substantially affected by 2 (competition R values < 4), including another Cys (C277) in ATP6V1A (Figure 4A, B). These chemical proteomic data indicate that 2 reacts with C138 of ATP6V1A with remarkable selectivity in human cells.
Figure 4.
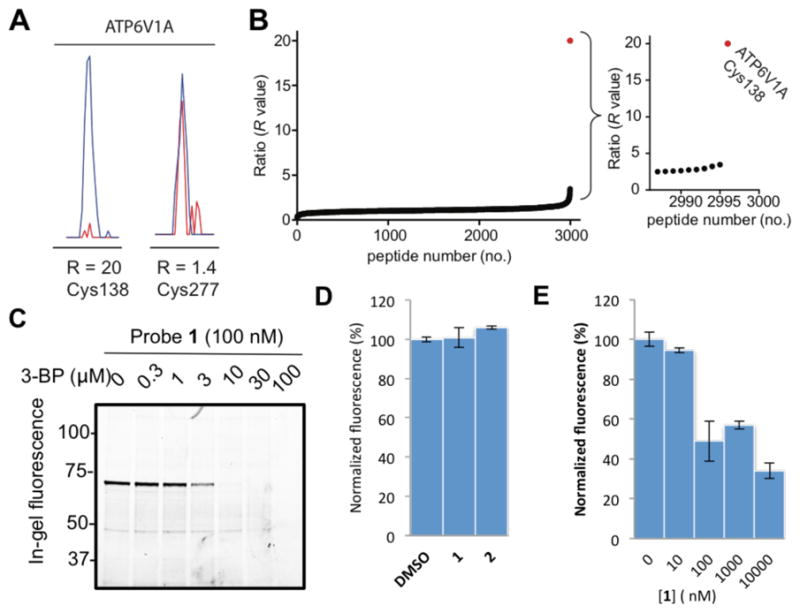
Chloroacetamide quinazolines exhibit exquisite proteomic selectivity, expose the mechanism of action of 3-bromopyruvate, and inhibit vesicle re-acidification. (A) Quantitative isoTOP-ABPP experiments22 revealed that pre-treatment with 2 in vitro fully protected C138 from iodoacetamide alkyne labeling while iodoacetamide alkyne labeling of a second Cys (C277) in ATP6V1A was insensitive to 2. Samples were treated with either 2 (Light-Red) or DMSO (Heavy-Blue) and the ratio of MS1 chromatographic peaks compared (R=20; >95% decrease in MS1 peak intensity). (B) C138 exhibits a maximal competition R value of 20 while all other Cys have R values less than 4. (C) 3-bromopyruvate (3-BP) caused dose-dependent blockade of ATP6V1A labeling by probe 1. (D) The two quinazolines at 10 μM had no significant effect on steady-state vesicle pH. (E) Probe 1 caused dose-dependent inhibition of vesicle re-acidification.
3-Bromopyruvate (3-BP) was previously found to affect intracellular pH by inhibiting V-ATPase activity putatively through modification of an unidentified cysteine.17 To test if 3-BP covalently targets C138 of ATP6V1A, we pretreated 293-cell lysate with various concentrations of 3-BP before labeling with probe 1. Pretreatment of 3-BP completely abolished ATP6V1A labeling by probe 1 (Figure 4C). These results support covalent modification of C138 in ATP6V1A as a plausible mechanism of 3-BP’s inhibition of V-ATPase.
To test if our covalent ATP6V1A probes affect V-ATPase function, we treated HeLa cells with probe 1 or compound 2 at 10 μM for 2 h, followed by labeling with the lysosomotropic red-fluorescent dye LysoTracker® Red DND-99 to report V-ATPase-driven intravesicular acidification as described before.23 Under these conditions, there was no significant difference between untreated cells and those treated with 1 or 2 (Figure 4D). We had previously developed a more sensitive assay to study V-ATPase inhibition in cells that involves pre-treatment with bafilomycin and measuring vesicle re-acidification after it has been washed off.23 Using this more sensitive assay, we determined the functional consequences of our electrophilic probes on V-ATPase-dependent vesicle acidification after exposing cells to bafilomycin (Figure S5). Immediately after bafilomycin washout, cells were treated with probe 1 at various concentrations, labeled with DND-99, followed by lysis and fluorescence measurement after 3 h. Probe 1 inhibited vesicle re-acidification with an apparent IC50 value of 30 nM (Figure 4E), which aligns with the potency of its target occupancy measured by in-gel fluorescence (Figure 1B). These results suggest that our electrophilic probes suppress the recovery of V-ATPase activity after its inhibition with bafilomycin.
In summary, we report the first covalent small-molecule modulators of V-ATPase. These quinazoline-based electrophilic probes specifically targeted a Cys residue in the catalytic subunit of V-ATPase, provided novel mechanistic insight into the actions of a previously reported inhibitor of V-ATPase, and modulated the function of V-ATPase in cells. With their well-defined mechanism of action and high proteomic specificity, the described quinazolines offer a powerful set of chemical probes to investigate the physiological and pathological roles of V-ATPase.
Supplementary Material
Acknowledgments
We thank Prof. Yi Tang (University of California, Los Angeles) for generous help with high-resolution mass spectrometry. This work was supported in part by National Science Foundation (CHE-1455306), American Cancer Society (IRG-58-007-51), University of Southern California and National Institute of Health (R37 DK042956 to D.B.).
Footnotes
The authors declare no competing financial interest.
Supporting Information. Supplementary figures, experimental protocol, synthetic scheme and compound characterization. This material is available free of charge on the ACS Publications website.
References
- 1.Forgac M. Nat Rev Mol Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- 2.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CWRJ, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Nat Genet. 1999;21:84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 3.Toyomura T, Murata Y, Yamamoto A, Oka T, Sun-Wada GH, Wada Y, Futai M. J Biol Chem. 2003;278:22023–22030. doi: 10.1074/jbc.M302436200. [DOI] [PubMed] [Google Scholar]
- 4.Sennoune SR, Bakunts K, Martinez GM, Chua-Tuan JL, Kebir Y, Attaya MN, Martinez-Zaguilan R. Am J Physiol Cell Physiol. 2004;286:C1443–C1452. doi: 10.1152/ajpcell.00407.2003. [DOI] [PubMed] [Google Scholar]
- 5.Maxson ME, Grinstein S. J Cell Sci. 2014;127:4987–4993. doi: 10.1242/jcs.158550. [DOI] [PubMed] [Google Scholar]
- 6.Liu Q, Kane PM, Newman PR, Forgac M. J Biol Chem. 1996;271:2018–2022. doi: 10.1074/jbc.271.4.2018. [DOI] [PubMed] [Google Scholar]
- 7.Liu Q, Leng XH, Newman PR, Vasilyeva E, Kane PM, Forgac M. J Biol Chem. 1997;272:11750–11756. doi: 10.1074/jbc.272.18.11750. [DOI] [PubMed] [Google Scholar]
- 8.MacLeod KJ, Vasilyeva E, Baleja JD, Forgac M. J Biol Chem. 1998;273:150–156. doi: 10.1074/jbc.273.1.150. [DOI] [PubMed] [Google Scholar]
- 9.Zimniak L, Dittrich P, Gogarten JP, Kibak H, Taiz L. J Biol Chem. 1988;263:9102–9112. [PubMed] [Google Scholar]
- 10.Shao E, Nishi T, Kawasaki-Nishi S, Forgac M. J Biol Chem. 2003;278:12985–12991. doi: 10.1074/jbc.M212096200. [DOI] [PubMed] [Google Scholar]
- 11.Huss M, Wieczorek H. J Exp Biol. 2009;212:341–346. doi: 10.1242/jeb.024067. [DOI] [PubMed] [Google Scholar]
- 12.Bowman BJ, Bowman EJ. J Biol Chem. 2002;277:3965–3972. doi: 10.1074/jbc.M109756200. [DOI] [PubMed] [Google Scholar]
- 13.Huss M, Ingenhorst G, Konig S, Gassel M, Drose S, Zeeck A, Altendorf K, Wieczorek H. J Biol Chem. 2002;277:40544–40548. doi: 10.1074/jbc.M207345200. [DOI] [PubMed] [Google Scholar]
- 14.Huss M, Sasse F, Kunze B, Jansen R, Steinmetz H, Ingenhorst G, Zeeck A, Wieczorek H. BMC Biochem. 2005;6:13. doi: 10.1186/1471-2091-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG, Xie XJ, White MA. Nature. 2007;446:815–819. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 16.Petrangolini G, Supino R, Pratesi G, Dal Bo L, Tortoreto M, Croce AC, Misiano P, Belfiore P, Farina C, Zunino F. J Pharmacol Exp Ther. 2006;318:939–946. doi: 10.1124/jpet.106.103481. [DOI] [PubMed] [Google Scholar]
- 17.Dell’Antone P. Life Sci. 2006;79:2049–2055. doi: 10.1016/j.lfs.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 18.Kung A, Chen YC, Schimpl M, Ni F, Zhu J, Turner M, Molina H, Overman R, Zhang C. J Am Chem Soc. 2016;138:10554–10560. doi: 10.1021/jacs.6b05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 20.Chen YC, Zhang C. Genes Cancer. 2016;7:148–153. doi: 10.18632/genesandcancer.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada GH, Bourgoin S, Casanova J, Wildeman A, Bechoua S, Ausiello DA, Brown D, Marshansky V. Nat Cell Biol. 2006;8:124–136. doi: 10.1038/ncb1348. [DOI] [PubMed] [Google Scholar]
- 22.Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF. Nature. 2016;534:570–574. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merkulova M, Paunescu TG, Azroyan A, Marshansky V, Breton S, Brown D. Sci Rep. 2015;5:14827. doi: 10.1038/srep14827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.