

Abstract
Germinal‐center kinase‐like kinase (GLK, Map4k3), a GCK‐I family kinase, plays multiple roles in regulating apoptosis, amino acid sensing, and immune signaling. We describe here the crystal structure of an activation loop mutant of GLK kinase domain bound to an inhibitor. The structure reveals a weakly associated, activation‐loop swapped dimer with more than 20 amino acids of ordered density at the carboxy‐terminus. This C‐terminal PEST region binds intermolecularly to the hydrophobic groove of the N‐terminal domain of a neighboring molecule. Although the GLK activation loop mutant crystallized demonstrates reduced kinase activity, its structure demonstrates all the hallmarks of an “active” kinase, including the salt bridge between the C‐helix glutamate and the catalytic lysine. Our compound displacement data suggests that the effect of the Ser170Ala mutation in reducing kinase activity is likely due to its effect in reducing substrate peptide binding affinity rather than reducing ATP binding or ATP turnover. This report details the first structure of GLK; comparison of its activation loop sequence and P‐loop structure to that of Map4k4 suggests ideas for designing inhibitors that can distinguish between these family members to achieve selective pharmacological inhibitors.
Keywords: Map4k3, GLK, germinal‐center kinase‐like kinase, crystal structure, GCK‐I, PEST sequence, activation loop, aminopyrrolopyrimidine inhibitor
Short abstract
PDB Code(s): 5J5T
Abbreviations
- GLK
germinal‐center kinase‐like kinase
- PEST
region with high content of 4 amino acids: proline (P), glutamic acid (E), serine (S), and threonine (T), which is associated with proteins that have short intracellular half life, possibly due to degradation
Introduction
GLK (Map4k3) is a member of the GCK‐I family of STE kinases and has been implicated in several disparate cellular signaling networks. First, GLK relays immunomodulatory signals upon T‐cell receptor ligation, resulting in the phosphorylation of T538 of PKC‐theta1 and in subsequent downstream T‐cell activation. The enhanced expression of GLK correlates with severity of adult's onset Still's disease, an inflammatory disorder.2 GLK‐deficient mice show impaired immune responses and were resistant to experimental autoimmune encephalomyelitis.1 Second, GLK mediates aminoacid induced mTORC1 activation, which is dependent on phosphorylation of its activation loop residue Ser170.3 During aminoacid starvation conditions, the phosphatase subunit PR61 of PP2A dephosphorylates Ser170.4, 5 GLK activation of mTORC1 has also been demonstrated in Drosophila.6 Third, GLK is known as a proapoptotic kinase and has a proposed role in cancer progression. Recent studies have shown that GLK effects apoptosis by modulating BH3‐only proteins at the post‐transcriptional level, resulting in activation of the mitochondrial‐dependent intrinsic apoptosis pathway.7 Recently GLK upregulation has been found to be associated with recurrence of nonsmall cell lung cancer.8
The GCK‐I family includes GCK (Map4k2), GLK (Map4k3), HGK (Map4k4), KHS or GCKR (Map4k5), and HPK1 (Map4k1). The GCK‐I family of kinases have a similar architecture—the kinase domain located at its N‐terminus followed by modular domains that enable protein‐protein interactions—namely the central proline‐rich domain (including PEST sequences that could mediate degradation) and the C‐terminal SH3 binding regions and Citron‐homology domains.9 Upon stimulation through the SH2/SH3 binding region, the GCK‐I kinases translocate to membrane‐associated receptor complexes and activate stress‐activated protein kinases (SAPKs). The middle PEST region (amino acids 431–540) is necessary for GLK to interact with phosphatase PP2A which regulates its autophosphorylation.4 The Citron‐homology domain mediates binding of the GCK‐I kinases to the GTP‐bound forms of Rac and Rho, which activates the SAPKs.10
HPK1 is an example of a well‐studied GCK‐I family kinase which acts as a negative regulator of the T‐cell signalosome. Upon T‐cell receptor ligation, HPK1 is activated by phosphophorylation of tyrosine 379 by Lck and then it interacts with the adaptor SLP‐76 through its SH2 domain, which down‐modulates IL‐2 production.11 Like HPK1, GLK contains a tyrosine at a similar location within the PEST domain sequence (Y379), but activation of GLK kinase activity is thought to upregulate T‐cell signaling; thus the two kinases may have different downstream effectors though they may have similar regulatory mechanisms.
Kinase activity is often regulated by the phosphorylation of an activation loop serine or threonine.12, 13, 14, 15, 16, 17 Protein kinases have an activation segment that runs from the “DFG” motif to the “APE” motif. The activation loop is anchored by the DFG at the N‐terminus and the P + 1 loop at the C‐terminus.12 The phosphorylation of Ser170 to activate GLK for downstream regulation of mTOR is consistent with the common activation mechanism of Ser/Thr kinases. Other features of the active conformation of Ser/Thr kinases described in the literature include alignment of the regulatory spine (R‐spine),15 a salt bridge between the phosphorylated Ser/Thr and catalytic loop arginine of the “HRD” motif,12 and a salt bridge between the catalytic lysine and a glutamate of the α‐C helix.13
The structures of the GCK‐I family have been somewhat elusive. The structure of only one kinase domain of the GCK‐I family has been published to date–Map4k4 or HGK18(PDB IDs: 4U43, 4U44, 4U45,4OBO, 4OBP, and others). The HGK structure is an activation loop‐swapped dimer, similar to STE‐20 kinases such as MST4, SLK (LOSK), OSR1, CHK2, LOK, DAPK3, and SPAK.19, 20, 21, 22 The MST4 dimer forms an extended beta strand between the “FVGT” sequences of the swapped activation loops, similar to the extended β‐strand observed for Map4k4 “TFIGT” sequence. The literature suggests that activation loop swapping allows a single kinase molecule to catalyze phosphorylation to two different subsets of residues (the autophosphorylation sequence of the dimer versus the physiological consensus sequence of the substrate).20 We hypothesized that the structure of the GLK kinase domain could contribute to our understanding of the mechanism of activation of this kinase by Ser170 phosphorylation and shed light on its mechanism of regulation in response to amino acid starvation conditions.
Results
We wished to characterize the binding mode and selectivity of a tool compound that inhibits GLK. To characterize its selectivity, we compared the activity inhibition characteristics of an analogue of Crizotinib against a panel of kinases at Reaction Biology Corps. Compound 1 is a closely related analogue of Crizotinib [Fig. 1(A)] and demonstrates potent inhibition of GLK with an IC50 of 110 nM by radiometric transfer assay [Fig. 1(B)], but it also inhibits many of the other GCK‐I family of kinases [Fig. 1(C)]. Compound 1 inhibits all GCK‐I family kinases tested at approximately 1–60 nM IC50, with a slight preference for Map4k4 over other kinases in that family. In contrast to Crizotinib, this compound does not appear to potently inhibit c‐Met and Abl (May‐Dracka et al., paper in communication). Because this compound appears to have a preference for this subfamily of Map4 kinases we tried to characterize its binding mode to GLK through structural and functional studies.
Figure 1.
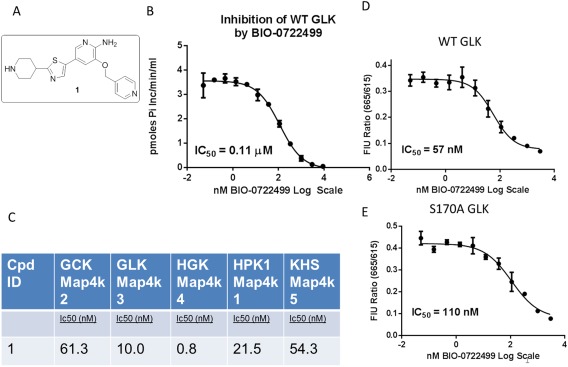
Inhibition activity and structure of compound 1. (A) Structure of compound 1. (B) Inhibition of wildtype GLK by compound 1. IC50 is 110 nM in our radioactive phosphor‐transfer activity assay. (C) Subpanel of Map4k tested for inhibition levels, measuring IC50 in nM by compound 1 as tested by Reaction Biology Corp. Compound 1 mainly inhibits other Map4k family kinases with similar affinities, with greater preference for Map4k4. The ATP‐binding site of GLK is not affected by the S170A mutation. Displacement of a probe by compound 1 demonstrates a similar IC50 for wildtype (C) and S170A (D), 57 and 110 nM, respectively. In contrast, the overall kinase activity of the mutant is 1–3% that of the wildtype kinase (Table I), suggesting it is impaired in a step other than ATP binding.
WT GLK versus GLK S170A protein purification and characterization
We expressed and purified wildtype GLK (WT GLK) in E. coli because it could not be overexpressed in baculovirus without causing toxicity. Wildtype GLK 1–384 (Tap195) was expressed in a customized E. coli host strain and was purified by Nickel affinity chromatography followed by S200 size exclusion chromatography. Fractions that had the most specific activity appeared as a broad peak, which was only about 50% pure. The protein was characterized by radiometric phosphotransfer activity to MBP with a specific activity of 167 nmol phosphate incorporated/min/mol protein or to PKC‐theta‐tide peptide with a specific activity of 61 nmol phosphate incorporated/min/mol protein (Table 1). Phosphomapping of the isolated protein band (MS Bioworks) demonstrated that several sites were phosphorylated, namely: Thr38, Thr145, Thr164, Ser170, Thr227, Ser280, Thr327, Ser329, Thr332, and Tyr379. Because GLK is a Ser/Thr kinase, we reasoned that the Tyr379 phosphorylation must have been the result of another kinase acting on GLK as a substrate. The phosphorylation at Ser170 is likely due to autophosphorylation.
Table 1.
Moles Phosphate Incorporated/Min/Mol WT Versus GLK S170A
| Substrate | WT GLK | GLK S170A |
|---|---|---|
| PKC theta‐tide (48 μM) | 61 ± 7 | 1.9 ± 0.5 (3% of WT) |
| MBP substrate (24 μM) | 167 ± 13 | 1.9 ± 0.2 (1% of WT) |
The WT GLK construct was dephosphorylated with lambda phosphatase and complete dephosphorylation at S170 was confirmed by Western [Fig. 2(A,B)] using a specific antibody directed to the phosphopeptide sequence. As a result of dephosphorylation, the activity of the protein in phosphotransfer to a PKC‐derived peptide was markedly reduced [Fig. 2(C)] at both concentrations of protein tested. This suggests that phosphorylation of one of the many sites (including Ser170) is critical for activity.
Figure 2.
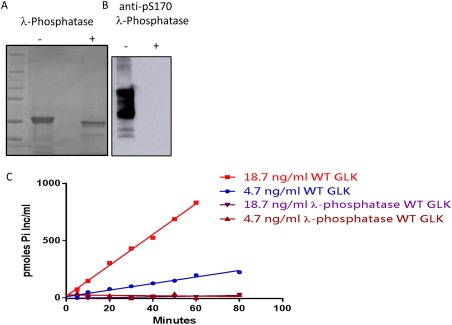
Phosphorylation of WT GLK impacts its activity. (A) Coomassie stained SDS‐PAGE of WT GLK before and after dephosphorylation with lambda‐phosphatase. (B) Phosphorylation state of WT GLK at S170A as measured by western blot with an anti‐S170 phospho antibody. (C) Phosphotransfer activity of increasing amounts of WT GLK over time (red and blue) compared to dephosphorylated WT GLK (brown and purple). The dephosphorylated protein has no measurable activity over the time range of 80 min.
The WT GLK preparation was not sufficiently chemically homogeneous to be used for crystallography so we employed a strategy of engineering a reduced‐activity mutant. This mutant form of GLK, in which Ser170 in the activation loop was mutated to alanine (GLK S170A), could be overexpressed in baculovirus without conferring toxicity. Using a His‐SNAP fusion at its amino‐terminus, the construct (13–380; S170A) was easily purified such that we could isolate ∼1 mg/L at 95% purity after a 3‐step purification process. Upon cleavage with thrombin, the protein forms a doublet which corresponds to 13–370/380 based on mass spectrometry.
To determine the boundaries of the kinase domain, we incubated GLK S170A with the protease clostripain (endoproteinase‐ArgC) which cleaves just after arginine residues. Using mass spectroscopy to define the size of the fragment, we found that GLK S170A has a proteolytically sensitive cleavage site just after R314, suggesting that that the region between the predicted end of the kinase domain (amino acid 294) and R314 was protected [Supporting Information Fig. S2(A)].
Compared to WT GLK, the GLK S170A mutant displayed reduced specific activity. Specifically, phosphoryltransfer to a PKCtide substrate, as measured by mole percent, was found to be 3% of that of the wildtype E. coli expressed protein (1.9 vs. 61 mol phosphates incorporated/min/mol protein) (Table 1). Thus, the importance of the Ser170 phosphorylation as a trigger for kinase activity has been demonstrated by dephosphorylation of the WT GLK and by the site directed mutation of Ser170 to alanine.
Using an ATP‐competitive fluorescent probe we wished to compare the ability of the mutant and WT proteins to bind compounds. We found that the GLK S170A binds an ATP‐competitive probe with similar affinities as the wildtype GLK—6 versus 10 nM respectively [Supporting Information Fig. S1(A,B)]. The probe is further displaced by compound 1 with similar IC50s as described in the biochemical phosphoryl transfer assay—57 nM for WT GLK and 110 nM for GLK S170A [Fig. 1(D,E)]. This suggests that the S170A mutation does not impact the binding affinity of ATP, or the binding of inhibitors that compete for ATP binding in the active site.
Structure of co‐crystal
We attempted to determine a co‐crystal structure of GLK S170A bound to compound 1 to understand the differences in activity between mutant and wildtype proteins. The GLK S170A was co‐crystallized with compound 1 in a high salt condition (using ammonium sulfate) and was solved by molecular replacement in a C2 spacegroup with 1 molecule/asymmetric unit. It was refined to an R free of 20.6% and R of 17.5% to 2.85 Å resolution (Table 2). The density for the compound was clearly distinct, as was in general the backbone density for residues between 13 and 314 (the site of Clostripain cleavage in solution), but the residues that follow were disordered.
Table 2.
Data Collection and Refinement Statistics
| Data collection | |
| Space group | C2 |
| Wavelength (Å) | 0.98 |
| Unit cell dimensions (a, b, c) (Å) | 92.70, 63.24, 64.91 |
| Unit cell dimensions (α, β, γ) (°) | 90, 116, 90 |
| Resolution range (Å)a | 50.0–2.85 (2.95–2.85) |
| Total number of reflections | 30,098 |
| Total number of unique reflections | 8004 |
| Number of molecules in asymmetric units | 1 |
| Average redundancy | 3.7 (3.7) |
| Completeness (%)a | 99.6 (100) |
| R merge(%)b | 11.4 (63.9) |
| I/Σi | 6.4 (1.1) |
| CC1/2 | 99.4 (75.1) |
| Refinement | |
| Resolution (Å) | 41.65–2.85 |
| R‐factor (%) | 17.50 |
| R free (%)c | 20.58 |
| RMS deviations from restraint values | |
| Bond length (Å) | 0.009 |
| Total no. of non‐H atoms in ASU | 2381 |
| No. of solvent molecules | 50 |
| Avg. protein B value (Å2) | 44.50 |
| Avg. solvent B value (Å2) | 25.1 |
| Ramachandran plot (%) | |
| Preferred | 96 |
| Generous | 4.0 |
| Disallowed | 0 |
| PDBID | 5J5T |
The highest resolution shell is shown in parentheses.
R merge , where I is the integrated intensity of a given reflection.
R free was calculated using 5% of data omitted from refinement.
The overall architecture of this kinase must be viewed through its crystallographic symmetry, which produces an activation loop swapped dimer [Fig. 3(A)]. Its symmetrical, activation‐loop swapped dimer with a long, structured C‐terminal trailing region is an important part of its higher order structure. The GLK kinase has a typical kinase fold, in which an N‐terminal β‐sheet containing domain and C‐terminal α−helical domain are connected by a hinge, which is the site of inhibitor binding [Fig. 3(C)].
Figure 3.
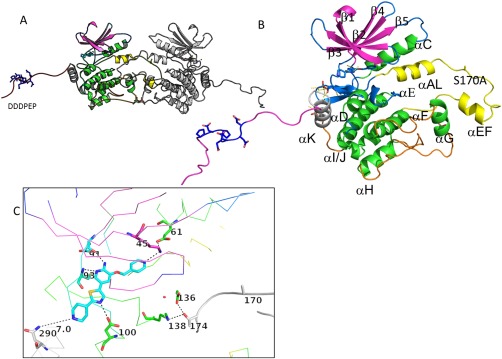
Overall structure of GLK S170A bound to compound 1. (A) Structure of the crystallographic dimer. (B) Close‐up of the GLK S170A monomer demonstrating the N‐terminal strands (purple) the C‐terminal lobe (mainly green helices), the last structured helix (gray; α−K), the ordered C‐terminus (red), and the activation loop (yellow). Ala170 is located between the α‐AL and the alpha EF region. The highly acidic region of the c‐terminal extension is highlighted in blue sticks. (C) Structure of the active site demonstrating residues in close proximity to compound 1. H‐bond networks that demonstrate the active conformation of this kinase are highlighted through sticks (K45‐E61) and in the catalytic loop (D136, K138—neighbor T174). An interactive view is available in the electronic version of the article.
One unique aspect of this structure is that the activation loop is ordered, containing an α‐AL and α‐EF helices [Fig. 3(B), yellow]. This region forms an intramolecular dimer interaction with a neighboring GLK molecule such that the activation loops are crossed [Figs. 3(A) and 4(B,C)]. One monomer's activation loop is bound by a symmetry‐mate such that the α‐EF helix and loop of one monomer is bound in a pocket between α‐G helix and the activation loop of its symmetry mate [Fig. 3(A,B)]. The kinase domain ends with a helix (α‐K) which comes close to the inhibitor binding site; a trailing C‐terminal ordered region makes interactions with a cleft in the neighboring N‐terminal GLK domain [Supporting Information Fig. S2(B)].
Figure 4.
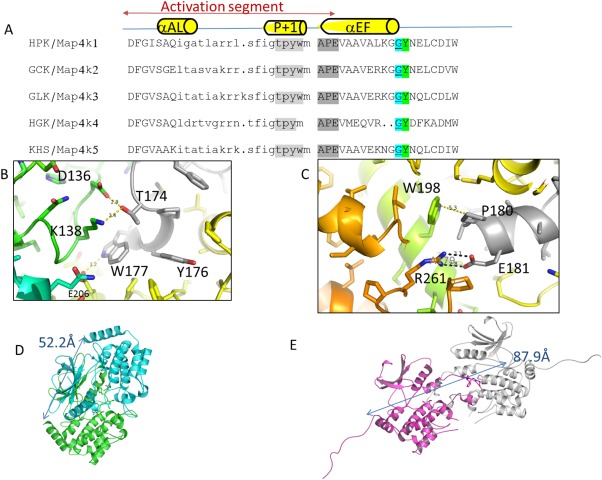
Comparison of sequence and structure of GCK‐1 family kinases.dimer viaactivation loop. (A) Comparison of the activation loop sequence of GCK‐I family kinases. Alignment based on conserved DFG and APE sequences shows that all subfamily members contain a glycine or proline (cyan) just before a conserved tyrosine (green) suggesting a cross‐swapped activation loop conformation as described previously by Taylor et al.17 Sequence is colored to highlight the P + 1 sequence (light gray) and the APE sequence (dark gray). (B) Closeup of the P + 1 region forming crossdimer interactions. (C) Closeup of the APE region of the activation loop forming crossdimer interactions. (D) Hydrodynamic radius of the Map4k4 dimer; blue and green (PDB 4OBP) as measured by distance from C‐terminal helix K of both monomers (52 Å) to (E) the Map4k3 S170A crystallographic dimer (magenta and white) distance from C‐terminal helix K of the two monomers (88 Å).
Compound 1 binds the active site hinge with two hydrogen bonds and additionally forms a direct H‐bond to Asp100. Its pyridine nitrogen is in hydrogen bond distance to the catalytic lysine (Lys45) [Fig. 3(C)]. The activation loop residues Ala170 and Thr174 from the “SFIGT” motif of a second GLK molecule approach the first molecule's active site, and Thr174 forms H‐bonds to Lys138 and Asp136 of the catalytic loop (HRD) [Fig. 3(C)]. However, in this mutant structure, the side chain of 170 does not appear to be close enough to the neighboring molecule's active site for phosphoryl transfer, as one might expect for autophosphorylation to occur at Ser170 in the wildtype GLK.
A second unique aspect of this structure is the ordered C‐terminal region that forms inter‐molecular interactions. The kinase has a C‐terminal α‐K helix (up to residue 294) that traverses from the C‐terminal domain up towards the hinge and then breaks into a trailing C‐terminal ordered region that binds back to a third molecule's N‐terminal domain. The highly negatively charged C‐terminal region extending away from the α‐K helix (PDHSTYHDFDDD‐PEPLVAVPHRI) binds the generally positively charged groove behind α‐C helix of a 3rd monomer's N‐terminal domain, and the side chains of Tyr297 and Phe300 are buried in a hydrophobic part of the groove [Supporting Information Fig. S2(B,C)].
Discussion
GLK is a highly desirable drug target for disease areas that include Still's disease (in which patients present with clinical symptoms similar to systemic lupus erythrmatosus),2 breast cancer,23 and possibly nonsmall cell lung cancer.8 We have observed that GLK, like Map4k4, has a glycine right before the Tyrosine in the activation loop and forms an activation loop‐swapped dimer [Fig. 4(A)]. This is consistent with the observation made by Taylor et al.21 that the presence of a glycine or proline right before the aligned tyrosine after the APE of the activation loop is the critical determinant of whether the STE kinase forms an activation loop swapped dimer or a monomer.
One of the most surprising aspects of this structure, which was not observed in the Map4k4 structures published prior, is that the last 20 amino acids of the C‐terminal end are ordered and binds to the N‐terminal lobe of a neighboring molecule. This behavior likely happens in solution as well, given that the region up to 314 is protected from proteolysis in solution. The intermolecular binding site for GLKs trailing C‐terminal PEST sequence (in a groove in the N‐terminal kinase domain) is reminiscent of the manner in which the PIFtide binds to the PIF pocket in AGC kinases. Superposition of the structure of the trans‐peptide C‐terminus of Map4k3 with the AGC kinase structure bound to its intramolecular‐PIF‐tide sequence [Supporting Information Fig. S2(B)] demonstrates that the aromatic residues (Tyr297 and Phe300) enter the PIF‐tide pocket in a similar manner as was observed in PDK24 and AKT2 (PDB ID: 2UW9) structures although the aromatic side chains do not superimpose. In both structures the α‐C helices superimpose well, suggesting that they are appropriately poised for catalysis. It is unclear whether this intermolecular interaction is physiologically relevant.
The phosphorylation of serine 170 may be critical for optimal substrate peptide binding. Consistent with this hypothesis, when we dephosphorylate the highly active WT GLK we observed a decrease in its phosphotransfer activity. The dephosphorylation at S170 was validated by Western blots using an anti‐phospho‐S170 specific antibody (Fig. 2). An important measured difference between the S170A mutant and WT proteins is that the overall specific activity of this mutant is 1–3% of that of the wildtype GLK (Table 1). We demonstrate that this is not due to differences in the structure of active site residues that bind ATP because an ATP‐competitive probe binds similarly to both mutant and WT enzymes. It follows that the other substrate of the reaction, the peptide, may bind differently to the WT versus the mutant GLK.
While the GLK S170A crystallographic dimer is stabilized by the activation loop swap, the activation loop is in the wrong conformation to enable peptide binding. Supporting this idea, by superimposing GLK S170A with the structure of active PKA bound to a substrate mimetic peptide (PDB ID: 1ATP)25 we note that the Ala170 is 8 Å from the site it would need to be for optimal (auto)phospho‐transfer and more than 10 Å from the site it would need to be to act as a platform for trans‐phosphorylation of a substrate peptide (data not shown). We did observe a high degree of phosphorylation at several sites in the WT GLK, not just at Ser170. As these sites are not in the activation loop, we discount their importance for the activity of this enzyme. Ser170 phosphorylation is the only site that when phosphorylated leads to downstream mTORC activation. Our S170A mutant protein is inactive for phosphotransfer to a peptide substrate, which is consistent with its importance in appropriately orienting the activation loop to act as a platform for peptide binding.
Other than this deficiency in forming the substrate‐binding platform, the kinase appears to be in a conformation that is catalytically competent. The structure reveals a highly active, primed conformation for this mutant Map4k3 kinase [Fig. 3(B)]. First, the side chains of the regulatory spine (Met64 of the C‐helix, Phe155 of the DFG motif, His134 of the HRD motif, and Ile137) are aligned, which is necessary to stabilize the active conformation of the kinase. The catalytic lysine (Lys45) forms a salt bridge to the glutamate (Glu61) of the C‐helix, which is also a hallmark of an active kinase conformation. Furthermore, the interactions that stabilize the HKD136 catalytic loop appear to be intact (H‐bonds between D136, R138, and Thr 174 of the 2nd monomer).
The relative orientation of the two kinase domains of the Map4k3 crystallographic dimer is very different from their orientation in the Map4k4 dimer. This may be due to the difference in the size of their activation loops, as measured between the DFG and APE conserved motifs. GLKs activation loop is longer than Map4k4 by two residues [Fig. 4(A)]. The Map4k4 dimer is a much more compact dimer (measuring 52.2 Å across), in which the “TFIGT” sequences within the two activation loops abut [Fig. 4(D)], similar to the architecture observed in Mst4 kinase and other STE20 kinases. In contrast, the GLK (Map4k3) dimer is more open (measuring 87.9 Å across), and the SFIGT sequences in the activation loops are not in close proximity [Fig. 4(E)]. The activation loop is stabilized by interactions between the P + 1 loop and APE region and the second monomer (Fig. 4). One cannot rule out the possibility that this interaction is due to crystallization packing. These differences between Map4K family members could affect the conformation of the P‐loop and thus the affinity for the compound.
Using this crystal structure, how would one design a compound with selectivity for Map4k3 over other Map4k family members? Compound 1 binds more tightly to HGK (Map4k4) than the other Map4k kinases; its binding may reflect an important difference in P‐loop conformation between GLK and HGK. The P‐loop of HGK bends over to bind small inhibitors and stacks with them using the Tyrosine at its tip; the P‐loop bent conformation is itself stabilized by the dimeric interface. In contrast, the P‐loop of GLK extends straight out and its tyrosine does not stack with the compound. In GLK there is no clear interaction between the P‐loop and the neighboring monomer's activation loop. This suggests that the affinity for compound could be tied to the nature of the dimer interface. A second region of sequence divergence near the active site is at the C‐terminal residues of α‐K helix. These surface residues of the α‐K helix could be exploited for enhancing inhibitor selectivity among the GCK‐I subfamily [Supporting Information Fig. S2(B)] even though the residues surrounding the ATP binding site are highly homologous. For example, hydrogen bond interactions with the N290 residue [Supporting Information Fig. S2(B)] could enable one to achieve inhibitor specificity for Map4k3 and Map4k5 within this family. Overall, the structure/function studies described here should be useful in understanding mechanisms of regulation of GLK and enable the design of selective compounds to treat Still's disease and other autoimmune diseases.
Materials and Methods
Materials
Clostripain was purchased from Hampton Research. The LanthaScreen Eu Kinase Binding Assay (Life Technologies, Carlsbad, CA). Compounds were submitted to Reaction Biology Corporation for inhibition profiling (Malvern, PA; www.reactionbiology.com).
Expression/purification of wildtype GLK kinase domain
An E. coli expression system (Tap440, which is a derivative of NEB2523 with a deletion of the Tsp protease) was used to produce wildtype kinase domain of human GLK (1–384) with a Tev‐cleavable N‐terminal His tag (Tap195). The strain was expressed in Luria broth with 50 μg/mL Kanamycin and in shake flasks grown at 37°C. Protein expression was induced by addition of IPTG to 1 mM overnight at 20°C.
All purification steps were performed at 4°C. The cell paste from 15 L was suspended in 400 mL cold lysis buffer (buffer A): 50 mM Tris pH 8.0, 0.5M NaCl, WS Protease inhibitors (benzamidine, bestatin, E‐64, leupeptin, aprotinin, pepstatin, PMSF), 20 mM Imidazole, 5% glycerol, 5 mM MgCl2, 5 mM ATP, 1% Triton X‐100, 1 mM DTT at a ratio of 4 mL of lysis buffer/g of cell paste and passed through a microfluidizer. Lysate was clarified by centrifugation. Supernatant was loaded on 5 mL NiNTA HiTrap column (GE Healthcare) and washed in 10 column volumes of lysis buffer without MgCl2, ATP, and Triton X‐100. Protein was eluted with buffer B (50 mM Tris pH 8.0, 0.5M NaCl, protease inhibitors, 250 mM imidazole, 5% glycerol. Protein was concentrated to 4 mg/mL and purified on a Superdex 200 16/600 column (120 mL) equilibrated in buffer C (50 mM Tris, 200 mM NaCl, 5% glycerol, 1 mM DTT, pH 8) and fractions were pooled according to their kinase activity.
Expression/purification of GLK S170A
The baculovirus vector expression system (BEVS) was used to produce an activation loop mutant of the kinase domain of human GLK S170A (VCID10799; residues 13 − 380 with the S170A mutation). Baculovirus was generated using the BestBac 2.0 baculovirus backbone (Expression Systems) in Sf9 cells and protein production was performed in High Five Cells (Life Technologies) using MOI = 1 and harvested 48 h post‐transfection using the BestBac 2.0 baculovirus backbone (Expression Systems). The protein was expressed as a cleavable fusion protein with 8XHis‐TEV‐SNAP tag‐thrombin attached to the N‐terminus. Constructs without the S170A mutation failed to express.
All purification steps were performed at 4°C. The cell paste was suspended in cold lysis buffer (buffer A): 50 mM Tris pH 8.0, 0.3M NaCl, 1 mM TCEP, protease inhibitors (benzamidine, bestatin, E‐64, leupeptin, aprotinin, pepstatin, PMSF), 20 mM imidazole, 5% glycerol at a volume of 4 mL of lysis buffer/g of cell paste and passed through a microfluidizer. Lysate was clarified by centrifugation. Supernatant was mixed with 20 mL Ni resin (Qiagen) and protein was bound by batch method over a period of 16 h. After overnight mixing, the protein was eluted with buffer B (50 mM Tris pH 8.0, 0.3M NaCl, 1 mM TCEP, protease inhibitors, 300 mM imidazole, 5% glycerol. Buffer exchange using Zeba spin columns pre‐equilibrated in buffer C (50 mM Tris pH 8, 0.3M NaCl, 1 mM TCEP, 5% glycerol, 2.5 mM CaCl2) was performed to remove protease inhibitors and then thrombin (Sigma, T6894) was added at a ratio of 25 μg of VCID107799/1 U of thrombin. The cleaved GLK was passed over a 5 mL Ni resin column and the FT and two column volume washes were pooled. The GLK S170A protein was further purified on a Superdex 200 size exclusion column (GE Healthcare) using buffer D (50 mM Tris pH 8, 0.3M NaCl, 1 mM TCEP, 5% glycerol, 1 mM EGTA). The protein was evaluated for purity by gel filtration and mass spectrometry revealed that the protein was partially cleaved at residue 370 (60%) versus residue 380 (30%). The resulting yield was 0.75 mg/1 L cell paste.
WT GLK (1–184) expressed in E. coli (TAP195) was dephosphorylated using GST‐tagged lambda protein phosphatase (Millipore EMD cat. #14‐598). A 300 μg sample of GLK was treated with 3 ug lambda phosphatase in buffer provided by manufacturer (50 mM Hepes pH 7.5, 0.1 mM EDTA, 2 mM MnCl2, 5 mM DTT) and incubated at room temperature for 1.5 h. Following dephosphorylation, lambda phosphatase was removed by adding 50 μL glutathione sepharose (GE Healthcare cat. #17‐0756‐01) to the reaction mixture and gently rocking for 15 min. Glutathione sepharose was removed using a Spin‐X centrifuge tube filter (Costar #8163).
The activity of the protein was assessed before and after lambda phosphatase treatment using a radioactive phosphotransfer assay Western analysis using an anti‐pS170 antibody was used to demonstrate that that site was completely dephosphorylated upon phosphatase treatment. The anti‐pS170 antibody was raised in rabbit using peptide sequence TIAKRKSFIGTPYC (hGLK‐164‐176aa) and affinity purified (New England peptide, MA). The activity of the WT and dephosphorylated WT proteins was carried out at 30°C, in 27 mM Mops, pH 7.2, 54 mM NaCl, 10 mM MgCl2, 0.02% Brij‐35, 10 μg/mL BSA, and 1 mM DTT, with 50 mM ATP (with [γ33P]ATP) and 40 mM peptide containing residues 529–547 derived from PKC‐theta:
PKC theta‐tide (SKNMLGDAKTNTFCGTPDYIAKRRRRR)
Protease digestion of GLK S170A to determine minimal domain
GLK S170A at 5 mg/mL was treated with clostripain at 1.25 μg/1 mg of GLK for 6 h at 4°C. The reactions were quenched using 1× protease inhibitor cocktail (Roche) + 1 mM EDTA and subjected to mass spectrometry analysis. To assess the mass of the fragment, proteolyzed GLK VCID10779 was reduced with 50 mM dithiothreitol in Tris‐buffered saline, pH 8.0, containing 4M urea and 5 mM EDTA. The sample was then analyzed on a LC–MS system comprised of a UPLC (ACQUITY, Waters Corp.), a TUV dual‐wavelength UV detector (Waters Corp.), and a ZQ mass spectrometer (Waters Corp.). A Vydac C4 cartridge was used for desalting. Molecular masses were obtained by deconvoluting the raw mass spectra using the MaxLynx 4.1 software (Waters Corp.). The resulting spectrum demonstrated that predominant species (86%) was residues 13–314 whose detected mass (34,396 Da) matched the predicted mass (34,394 Da).
Synthesis and characterization of pyrrolo‐pyridine compound 1
The synthesis of compound 1 was completed as depicted in Scheme 1. Alkylation of 2‐amino‐5‐bromopyridin‐3‐ol with 4‐(bromomethyl)pyridine followed by bis‐Boc protection of 3 delivers bispyridine 4. Installation of a pinacolboryl group followed by Suzuki crosscoupling with thiazole 6 affords compound 7. Acid mediated Boc removal affords compound 1.
Scheme 1.
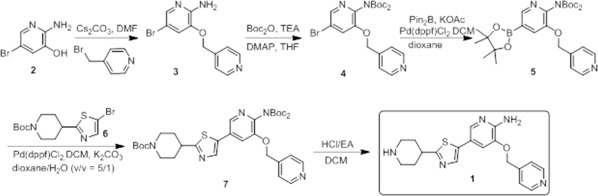
Synthesis of compound 1.
5‐(2‐(piperidin‐4‐yl)thiazol‐5‐yl)−3‐(pyridin‐4‐ylmethoxy)pyridin‐2‐amine (1): 1HNMR (400 MHz, Methanol‐d4) δ: 8.55 (d, J = 5.2 Hz, 2H), 7.78 (s, 1H), 7.75 (s, 1H), 7.58 (d, J = 5.2 Hz, 2H), 7.33 (s, 1H), 5.31 (s, 2H), 3.19–3.15 (m, 3H), 2.78 (t, J = 12.0 Hz, 2H), 2.10 (d, J = 13.2 Hz, 2H), 1.81–1.71 (m, 2H). LCMS (ESI) m/z 367.9 [M + H]+
Evaluation of kinase specific activity
To measure the specific activities of wildtype GLK construct (TAP195) and the GLK S170A (13 − 370/380 S170A GLK;VCID10799) we employed a radioactive phosphotransfer assay. Variable amounts of GLK were incubated for 60 min at 30°C in 27 mM Mops, pH 7.2, 54 mM NaCl, 10 mM MgCl2, 10 μg/mL BSA, 0.016% Tween 20, 1 mM DTT, with 50 μM ATP (200 μCi/μM ATP[γ33P]) and 24 μM myelin basic protein or 48 μM PKCtheta peptide (529–547; SKNMLGDAKTNTFCGTPDYIAKRRRRRR), prior to quenching with 2% phosphoric acid. The quenched reaction mixtures were vacuum filtered on a Multiscreen phosphocellulose plate. The plate was washed with 1% phosphoric acid, OmniFluor scintillation cocktail was added to the wells, and the 33P cpm/well counted in a Microbeta LumiJet Plate Counter. Aliquots of the ATP[γ 33P]) substrate were added to empty wells to calculate moles of phosphate incorporated. The molar concentration of GLK was estimated from the concentration of total protein, purity, and molecular weight of each construct.
Determining compound IC50s
Using the radiometric assay described above, the IC50 of compound 1 with GLK was measured as follows. The pmol inorganic phosphate incorporated/min/mL was measured to assess phosphorylation of the PKC‐theta‐tide using 0.3 nM WT GLK. Reactions were run at 30°C in 25 mM MOPS pH 7.2, 50 mM NaCl, 10 mM MgCl2, 0.016% Tween‐20, 1 mM DTT, 3.8% DMSO, 20 μM ATP and 48 μM PKCtide, and 12 concentrations of compound 1.
Binding of active‐site probe and displacement assay
LanthaScreen Eu Kinase Binding Assay (Life Technologies, Carlsbad, CA) was used to characterize binding of a tracer and compound 1 displacement of the tracer to WT GLK and GLK S170A. All reagents were purchased from Life Technologies and assay was carried out according to manufacturer's directions. Briefly, final assay conditions consisted of 5 nM GLK, 2 nM Eu‐Anti‐His Antibody, and 1× kinase buffer A. First, K D for Kinase Tracer 236 was determined for WT GLK and GLK S170A by titrating the tracer in the presence of constant amounts of enzyme. For subsequent assays measuring compound binding, Kinase Tracer 236 was used at concentration = Kd. Kinase, compound, Eu‐Anti‐His Antibody, and Tracer 236 were incubated together in a final volume of 16 μL in 384‐well white low volume plates for 1 h at room temperature and then read on an Envision plate reader (Perkin Elmer, Waltham, MA). Each compound concentration was measured in quadruplicate and data was fit using nonlinear regression in GraphPad Prism 6 (Hearn Scientific Software).
Kinase selectivity
Inhibition of a kinase activity by compound 1 was tested in a radiometric substrate phosphorylation assay or a phosphotransfer assay. The % inhibition of kinase activity at 0.5 mM compound 1 was compared across a panel of kinases. The compound demonstrated reasonably good differential specificity, mainly targeting other GCK‐I subfamily members when tested at 0.5 μM concentration at Reaction Biology Corporation (Malvern, PA). IC50 values of compound 1 for the GCK‐I family of kinases were performed in the presence of 10 μM ATP using a 10 point curve in which compound was diluted 1:3 for each point starting at 1 μM; the Hill slopes were between −0.5 and −0.6 for each kinase [Fig. 1(c)].
Crystallization, data collection, structure solution, and refinement of inhibitor‐bound GLK S170A
The baculovirus expression construct (VCID10799) encoding 13–370/380 S170A GLK binds to compound 1 as measured by thermal shift, in which the compound at 10 μM increased the melting temperature +2.5°C from 42.5 to 45°C. The thrombin cleaved protein was concentrated to 5 mg/mL, mixed with 1 mM compound 1 and was crystallized by vapor diffusion. Crystals appeared in 0.8M Ammonium Sulfate, 0.1M bisTris pH 5.5 at room temperature and were harvested in mother liquor containing 25% glycerol as a cryoprotectant. The crystals diffracted to 2.85 Å resolution at LRL‐CAT (APS beamline ID31) and belonged to the C2 space group with 1 molecule in the asymmetric unit.
Diffraction data was measured in 1 degree rotation frames on a MAR‐CCD detector at APS beamline 31‐ID‐D frozen at −180°C. The data was integrated and scaled using MOSFLM.26 The complex structure was solved by molecular replacement with MOLREP27 using a monomer from the Map4k4 structure (PDB ID: 40B0) as a search model. Several rounds of restrained refinement in REFMAC528 and chain replacement in Buccaneer29 led to a good starting model.
Refinement in PHENIX30 with 6 TLS groups led to an R free of 20.6% and R‐factor of 17.5% at 2.85 Å with good geometry and 50 waters added and small molecule inhibitor compound 1 (Table 2). Residues 12–314 are ordered and residues 316–370/380 are disordered. Residues 167–169 and 292–295 are also disordered and threonine 145 is phosphorylated. A Ramachandran plot shows that all residues are within the acceptable range.
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
ACKNOWLEDGMENTS
The authors would like to thank Hairuo Peng, Jason Fontenot and Linda Burkley for project team leadership. We would like to thank Jay Chodaparambil for excellent crystallography advice and help in the last stages of protein refinement.
PDB deposition: The GLK S170A/Cpd1 co‐crystal structure has been deposited at the PDB with ID: 5J5T.
Disclosure: The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- 1. Chuang H‐C, Lan J‐L, Chen D‐Y, Yang C‐Y, Chen Y‐M, Li J‐P, Huang C‐Y, Liu P‐E, Wang X, Tan T‐H (2011) The kinase GLK controls autoimmunity and NF‐κB signaling by activating the kinase PKC‐θ in T cells. Nat Immunol 12:1113–1118. [DOI] [PubMed] [Google Scholar]
- 2. Chen D‐Y, Chuang H‐C, Lan J‐L, Chen Y‐M, Hung W‐T, Lai K‐L, Tan T‐H (2012) Germinal center kinase‐like kinase (GLK/MAP4K3) expression is increased in adult‐onset Still's disease and may act as an activity marker. BMC Med 10:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Duan Y, Li F, Tan K, Liu H, Li Y, Liu Y, Kong X, Tang Y, Wu G, Yin Y (2015) Key mediators of intracellular amino acids signaling to mTORC1 activation. Amino Acids 47:857–867. [DOI] [PubMed] [Google Scholar]
- 4. Yan L, Mieulet V, Burgess D, Findlay GM, Sully K, Procter J, Goris J, Janssens V, Morrice NA, Lamb RF (2010) PP2AT61ɛ is an inhibitor of MAP4K3 in nutrient signaling to mTOR. Mol Cell 37:633–642. [DOI] [PubMed] [Google Scholar]
- 5. Suzuki T, Inoki K (2011) Spatial regulation of the mTORC1 system in amino acids sensing pathway. Acta Biochim Biophys Sin 43:671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Resnik‐Docampo M, de Celis JF (2011) MAP4K3 is a component of the TORC1 signalling complex that modulates cell growth and viability in Drosophila melanogaster . PLoS One 6:e14528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lam D, Dickens D, Reid EB, Loh SHY, Moisoi N, Martins LM (2009) MAP4K3 modulates cell death via the post‐transcriptional regulation of BH3‐only proteins. Proc Natl Acad Sci USA 106:11978–11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hsu CP, Chuang HC, Lee MC, Tsou HH, Lee LW, Li J‐, Tan TH (2016) GLK/MAP4K3 overexpression associates with recurrence risk for non‐small cell lung cancer. Oncotarget 7:41748–41757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kyriakis JM (1999) Signaling by the germinal center kinase family of protein kinases. J Biol Chem 274:5259–5262. [DOI] [PubMed] [Google Scholar]
- 10. Madaule P, Furuyashiki T, Reid T, Ishizaki T, Watanabe G, Morii N, Narumiya S (1995) A novel partner for the GTP‐bound forms of rho and rac. FEBS Lett 377:243–248. [DOI] [PubMed] [Google Scholar]
- 11. Di Bartolo V, Montagne B, Salek M, Jungwirth B, Carrette F, Fourtane J, Sol‐Foulon N, Michel F, Schwartz O, Lehmann WD, Acuto O (2007) A novel pathway down‐modulating T cell activation involves HPK‐1‐dependent recruitment of 14‐3‐3 proteins on SLP‐76. J Exp Med 204:681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nolen B, Taylor S, Ghosh G (2004) Regulation of protein kinases: controlling activity through activation segment conformation. Mol Cell 15:661–675. [DOI] [PubMed] [Google Scholar]
- 13. Taylor SS, Kornev AP (2011) Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci 36:65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roskoski R (2016) Classification of small molecule protein kinase inhibitors based upon the structures of their drug–enzyme complexes. Pharmacol Res 103:26–48. [DOI] [PubMed] [Google Scholar]
- 15. Meng Y, Roux B (2014) Locking the active conformation of c‐Src kinase through the phosphorylation of the activation loop. J Mol Biol 426:423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson LN, Noble ME, Owen DJ (1996) Active and inactive protein kinases: structural basis for regulation. Cell 85:149–158. [DOI] [PubMed] [Google Scholar]
- 17. Kornev AP, Haste NM, Taylor SS, Eyck LF Ten (2006) Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci USA 103:17783–17788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang L, Stanley M, Boggs JW, Crawford TD, Bravo BJ, Giannetti AM, Harris SF, Magnuson SR, Nonomiya J, Schmidt S, Wu P, Ye W, Gould SE, Murray LJ, Ndubaku CO, Chen H (2014) Fragment‐based identification and optimization of a class of potent pyrrolo[2,1‐f][1,2,4]triazine MAP4K4 inhibitors. Bioorg Med Chem Lett 24:4546–4552. [DOI] [PubMed] [Google Scholar]
- 19. Pike ACW, Rellos P, Niesen FH, Turnbull A, Oliver AW, Parker SA, Turk BE, Pearl LH, Knapp S (2008) Activation segment dimerization: a mechanism for kinase autophosphorylation of non‐consensus sites. EMBO J 27:704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Record CJ, Chaikuad A, Rellos P, Das S, Pike ACW, Fedorov O, Marsden BD, Knapp S, Lee WH (2010) Structural comparison of human mammalian ste20‐like kinases. PLoS One 5:e11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taylor CA, Juang YC, Earnest S, Sengupta S, Goldsmith EJ, Cobb MH (2015) Domain‐swapping switch point in Ste20 protein kinase SPAK. Biochemistry 54:5063–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee SJ, Cobb MH, Goldsmith EJ (2009) Crystal structure of domain‐swapped STE20 OSR1 kinase domain. Protein Sci 18:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Park J, Lee J, Choi C (2015) Evaluation of drug‐targetable genes by defining modes of abnormality in gene expression. Sci Rep 5:13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR (2001) The PIF‐binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J 20:4380–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng J, Trafny EA, Knighton DR, Xuong NH, Taylor SS, Ten Eyck LF, Sowadski JM (1993) 2.2 Angstrom refined crystal structure of the catalytic subunit of cAMP‐dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr D49:362–365. [DOI] [PubMed] [Google Scholar]
- 26. Leslie AGW, Powell HR (2007) In: Sussman J, Read RL, Eds. Evolving methods for macromolecular crystallography. New York: Springer, pp 41–51. [Google Scholar]
- 27. Vagin AA, Teplyakov VA (1997) MOLREP: an automated program for molecular replacement. J Appl Crystallogr 30:1022–1025. [Google Scholar]
- 28. Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011) REFMAC 5 for the refinement of macromolecular crystal structures. Acta Crystallogr D67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cowtan K (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D62:1002–1011. [DOI] [PubMed] [Google Scholar]
- 30. Urzhumtsev A, Afonine PV, Van Benschoten AH, Fraser JS, Adams PD (2015) From deep TLS validation to ensembles of atomic models built from elemental motions. Acta Crystallogr D71:1668–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.