

ABSTRACT
The poliovirus (PV) is currently targeted for worldwide eradication and containment. Sanger-based sequencing of the viral protein 1 (VP1) capsid region is currently the standard method for PV surveillance. However, the whole-genome sequence is sometimes needed for higher resolution global surveillance. In this study, we optimized whole-genome sequencing protocols for poliovirus isolates and FTA cards using next-generation sequencing (NGS), aiming for high sequence coverage, efficiency, and throughput. We found that DNase treatment of poliovirus RNA followed by random reverse transcription (RT), amplification, and the use of the Nextera XT DNA library preparation kit produced significantly better results than other preparations. The average viral reads per total reads, a measurement of efficiency, was as high as 84.2% ± 15.6%. PV genomes covering >99 to 100% of the reference length were obtained and validated with Sanger sequencing. A total of 52 PV genomes were generated, multiplexing as many as 64 samples in a single Illumina MiSeq run. This high-throughput, sequence-independent NGS approach facilitated the detection of a diverse range of PVs, especially for those in vaccine-derived polioviruses (VDPV), circulating VDPV, or immunodeficiency-related VDPV. In contrast to results from previous studies on other viruses, our results showed that filtration and nuclease treatment did not discernibly increase the sequencing efficiency of PV isolates. However, DNase treatment after nucleic acid extraction to remove host DNA significantly improved the sequencing results. This NGS method has been successfully implemented to generate PV genomes for molecular epidemiology of the most recent PV isolates. Additionally, the ability to obtain full PV genomes from FTA cards will aid in facilitating global poliovirus surveillance.
KEYWORDS: FTA cards, cell culture, metagenomics, next-generation sequencing, picornavirus, poliovirus
INTRODUCTION
Poliovirus (PV) is a highly infectious agent that causes poliomyelitis, an irreversible paralysis (1). During the prevaccine era, PV caused more than 350,000 paralytic cases per year (1, 2). Since 1988, the efforts of the Global Polio Eradication Initiative (GPEI) have reduced global polio cases by >99.9% (3, 4). Wild PV type 2 is now extinct, and the genetic diversity of the remaining strains is markedly low, indicating a limited circulation (5). Still, PV persists in places with low vaccine coverage and poor sanitation. Thus, in addition to broad, national-level immunization campaigns, efforts to locate and eliminate remaining PV reservoirs have been accelerated.
Molecular detection and comparative genomic sequencing of polioviruses are major surveillance tools essential for eradication. Due to the rapid evolution of PV (∼1% per year) (6), molecular epidemiology provides information to link cases and identify persistent reservoirs. For example, viral strain evolution is analyzed to estimate the extent and duration of infections (4). Sequence comparisons can also determine the source of a PV infection and distinguish among viruses imported into a new area or country, endemic virus circulation, repeated reintroduction of PV to a population, and vaccine-derived poliovirus (VDPV) strains, all of which help to direct vaccination efforts (5).
Poliovirus is monitored through its detection in stool samples from acute flaccid paralysis (AFP) cases (5, 7), as well as through environmental surveillance of sewage samples. Traditionally, stool samples collected through the acute flaccid paralysis (AFP) surveillance system are cultured in L20B cells and then in rhabdomyosarcoma (RD) cells before performing PCR to identify the poliovirus and whether it is vaccine-like or wild-like (8). Mouse L20B cells express the human poliovirus receptor, enabling the propagation of poliovirus without extensive growth of other human enteroviruses. The isolates flagged as potential wild, VDPV, or indeterminate are referred for sequencing of the viral protein 1 (VP1) capsid region (9).
To facilitate the shipment of poliovirus nucleic acids in a noninfectious form, the WHO Global Polio Laboratory Network (GPLN) has adopted, in many circumstances, the use of FTA cards for international shipments (10). Heat-inactivated culture supernatants from virus isolates are spotted onto FTA cards and dried. Polioviruses are inactivated during the spotting, but their RNA genomes remain intact. This facilitates the transfer of PV RNA to a sequencing lab, though at the expense of some loss of sensitivity (11).
Traditional molecular surveillance of PV employs Sanger dideoxy sequencing to obtain the VP1 capsid sequence (1). In some cases, more comprehensive genome sequencing is needed for characterizing VDPVs or for higher resolution analysis of transmission. While sequencing of the short VP1 capsid region (906 nucleotides) is highly effective, sequencing of the whole PV genome (∼7,400 nucleotides) by the Sanger method is time consuming and difficult to scale up. However, the analysis of whole PV genomes has several advantages over VP1 analysis, as it enables the detection of recent recombination events and provides a higher resolution view of viral lineages. Next-generation sequencing (NGS) platforms feature the ability to rapidly obtain full-length genomes, and they dramatically increase throughput via multiplexing. Here, we describe the optimization of NGS of PV genomes from FTA cards and virus isolates.
RESULTS
Poliovirus RNA pretreatment comparisons.
We compared the effects of different sample pretreatments. DNase-treated viral RNA from culture supernatants (CS) produced significantly more overall reads than non-DNase-treated samples (P = 0.0314; Fig. 1B). DNase treatment also significantly increased the percentage of reads mapped to the targeted poliovirus genomes (62.7% ± 29.1%) compared with that from non-DNase treatment (49.2% ± 22.3%) (P < 0.001; Fig. 1A). Additionally, DNase treatment yielded slightly higher percentages of genome coverage (99.3% ± 1.5%) compared with those from non-DNase treatments (98.5% ± 5.2%) (P = 0.0326; Fig. 1C).
FIG 1.

Comparison of DNase and non-DNase treatments in FTA card and cell culture supernatant (CS) poliovirus isolate samples from experimental design 1 in Materials and Methods (n = 16). (A) Proportions of total reads mapping to poliovirus genome. (B) Total numbers of reads passing quality control (trimmed and deduplicated). (C) Percentages of poliovirus genome mapped or covered by reads. The top- and bottom-most dark lines in the box-and-whisker plots represent 1.5 times the interquartile range (IQR); the gray zones represent upper and lower quartiles; the lines between the two gray zones represent arithmetic means. *, P < 0.05; ***, P < 0.0001.
For FTA samples, DNase treatment produced significantly fewer overall reads than were obtained when samples were not treated with DNase (P < 0.0001; Fig. 1B), though they contained a higher proportion of poliovirus reads (36.3% ± 23.6%) compared with those from the nontreatment group (21.8% ± 9.0%) (P < 0.0001; Fig. 1A). This paucity of reads affected the percentage of genome coverage for FTA card samples that underwent DNase treatment (70.5% ± 31.7%) compared with those from the non-DNase-treated group (98.3% ± 5.5%) (P < 0.0001; Fig. 1C). We hypothesized that DNase-treated FTA samples would produce satisfactory results if they were not sequenced together with high-quality CS samples. To rule out the possible effect of read competition obtained from multiplexing FTA and CS samples in the same run, we performed an additional sequencing run using only FTA DNase-treated samples and found that the overall numbers of reads (mean FTA-only, 206,156 reads versus FTA multiplexed with CS, 6,851 reads) and percentages of viral reads mapping to target poliovirus genomes (mean FTA-only, 90% versus FTA multiplexed with CS, 70%) were improved even for FTA card samples subjected to DNase treatment (see Fig. S1 in the supplemental material).
Illumina MiSeq library construction kit comparisons.
The comparison of library kits showed that the Nextera and New England BioLabs (NEB) DNA libraries generated significantly more usable reads per sample than the NEB-RNA library (Fig. 2B). The percentage of poliovirus-specific reads was significantly higher using Nextera (84.2% ± 15.6%) than those with NEB-RNA (68.5% ± 17.6%) and NEB-DNA (51.9% ± 13.3%) libraries (Fig. 2A). Two samples failed due to a possible technical error when preparing the Nextera library, so only 11 samples were included in the final analysis from the Nextera preparation compared with 13 for each of the NEB-DNA and NEB-RNA kits. Overall, the Nextera kit performed the best considering the numbers of post-trimming reads and the percentages of those reads mapping to targeted poliovirus genomes. Despite these differences, we obtained poliovirus sequences with complete or near-complete genome coverage (99% to 100%), independent of the library construction method used (P = 0.99; Fig. 3). In cases where the final sequence comprised less than 100% of the genome, we observed that only a few bases (10 to 50 base pairs [bp]) of the termini were missing. For most applications, this missing sequence will not affect the analysis.
FIG 2.
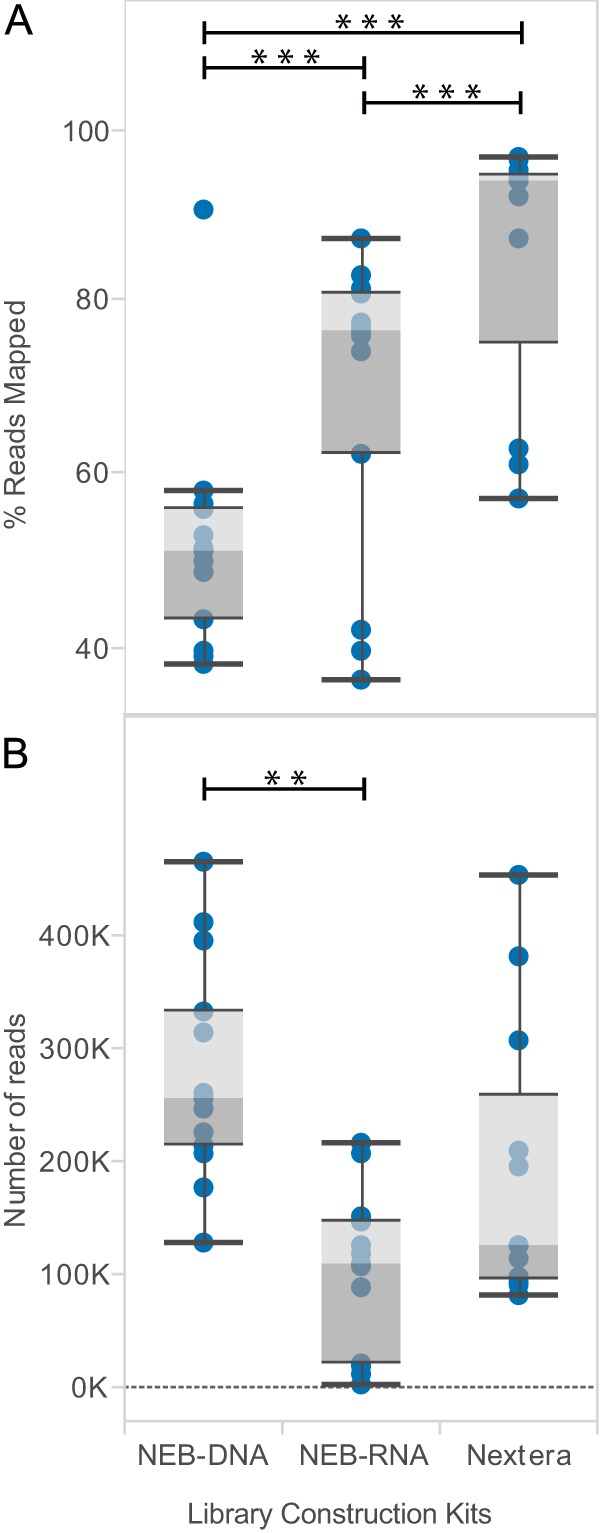
Comparison of three library preparation kits. (A) Percentages of reads mapping to poliovirus genome. (B) Total numbers of trimmed and filtered sequencing reads passing quality control (trimmed and deduplicated). Box-and-whisker plots are presented as described in Fig. 1. Nextera kit (n = 11), NEB kits (n = 13); **, P < 0.01; ***, P < 0.0001.
FIG 3.
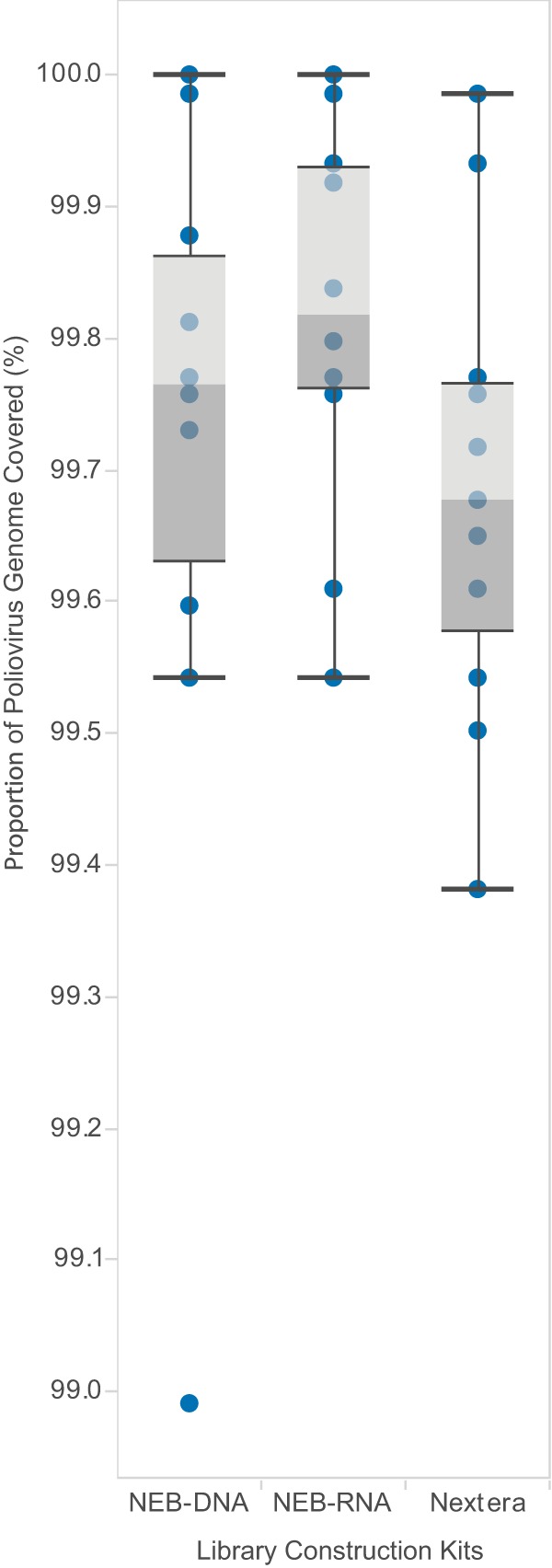
Comparison of the proportions of poliovirus genome covered by reads using three different library preparation kits. Box-and-whisker plots are presented as described in Fig. 1.
Poliovirus sample pretreatment prior to nucleic acid extraction.
In previously published metagenomics protocols (12, 13), specimens containing encapsidated viral nucleic acids were filtered and/or treated with a nuclease cocktail before viral nucleic acids were extracted. These procedures were proposed to reduce the amount of nonviral material, such as host cellular debris and bacteria (filtration) and host nucleic acids (nuclease treatment). We performed a pilot study comparing the total numbers of reads, as well as the numbers of virus-specific reads, for two samples undergoing various pretreatment methods (see Fig. S2 and S3). The results from the specimens subjected to filtration and/or nuclease cocktail were similar to those from untreated samples, suggesting this step could be omitted when processing poliovirus isolates, provided that a DNase treatment was performed after extraction. By combining the results from both experiments, we concluded that an efficient protocol involves extraction, DNase, single-primer amplification (SISPA), and Nextera library preparation (see the methods in the supplemental material).
Comparison of NGS and Sanger sequences.
We compared the poliovirus genomes generated using NGS with available Sanger sequences of either the VP1 region (n = 14) or the entire genome (n = 25). A sequence comparison of the VP1 regions confirmed that almost all of the sequences matched in their entirety (Table 1), while two matched at 99.9% and 92.7%. Full-genome comparisons of the 25 samples showed that all except four samples shared 99.5% to 100% pairwise identities between NGS and Sanger sequences. In these highly matched samples (99.5% to 100%), the small sequence divergences reflected different variant base calls in Sanger sequencing for quasispecies mixtures. Sanger consensus might have reflected either major or minor bases in loci with single nucleotide polymorphisms (SNPs), whereas the NGS consensus always reflected the majority base calls in our analysis. Four samples showed higher discrepancies, with pairwise identities of 92.0% to 98.5%. It is likely that multiple coinfecting PVs in each of these samples were subcultured into two or more isolates, which were subsequently sequenced separately in Sanger or NGS.
TABLE 1.
GenBank accession numbers and pairwise identities of PV sequences obtained by NGS and Sanger methods
| GenBank accession no. | Strain name | Length (bp) |
Identical sites |
||
|---|---|---|---|---|---|
| NGS | Sanger | No. | % | ||
| KX162696 | NIE0611195 | 7414 | 7434 | 7411 | 99.96 |
| KX162697 | NIE0611432 | 7422 | 7440 | 7422 | 100.00 |
| KX162706 | NIE0611579 | 7244 | 7434 | 7132 | 98.50 |
| KX162702 | NIE0611523 | 7414 | 7434 | 7406 | 99.90 |
| KX162705 | NIE0611535 | 7414 | 7434 | 7414 | 100.00 |
| KX162698 | NIE0611449 | 7418 | 7439 | 7418 | 100.00 |
| KX162703 | NIE0611531 | 7422 | 7434 | 7417 | 99.90 |
| KX162700 | NIE0611517 | 7421 | 7440 | 7320 | 98.60 |
| KX162699 | NIE0611452 | 7422 | 7439 | 7421 | 99.99 |
| KX162701 | NIE0611522 | 7422 | 7439 | 7417 | 99.90 |
| KX162704 | NIE0611534 | 7417 | 7434 | 7416 | 99.99 |
| KX162708 | NIE1116622 | 7363 | 906a | 906 | 100.00 |
| KX162709 | NIE1116623 | 7076 | 906a | 906 | 100.00 |
| KX162682 | CAF2019340 | 7360 | 906a | 906 | 100.00 |
| KX162710 | NIE1119343 | 7331 | 903a | 903 | 100.00 |
| KX162711 | NIE1119344 | 7357 | 903a | 903 | 100.00 |
| KX162707 | NIE1116178 | 7399 | 903a | 903 | 100.00 |
| KX162683 | CHA1119341 | 7362 | 906a | 905 | 99.90 |
| KX162684 | CHA1218878 | 7541 | 906a | 906 | 100.00 |
| KX162685 | CHA1218985 | 7402 | 7441 | 7377 | 99.70 |
| KX162690 | CHA1219348 | 7377 | 7442 | 7352 | 99.70 |
| KX162686 | CHA1218986 | 7440 | 7442 | 6843 | 92.00 |
| KX162687 | CHA1218987 | 7368 | 7443 | 6704 | 91.00 |
| KX162688 | CHA1219345 | 7381 | 7373 | 7373 | 99.90 |
| KX162689 | CHA1219346 | 7334 | 7450 | 6746 | 92.00 |
| KX162678 | CHA1219347 | 7478 | 7403 | 7403 | 99.50 |
| KX162679 | CAE1419303 | 7363 | 7444 | 7362 | 99.99 |
| KX162680 | CAE1419304 | 7431 | 7444 | 7444 | 99.97 |
| KX162681 | CAE1419305 | 7430 | 7444 | 7424 | 99.90 |
| KX162692 | EQG1419328 | 7428 | 7442 | 7442 | 99.99 |
| KX162693 | EQG1419331 | 7418 | 7442 | 7418 | 100.00 |
| KX162695 | EQG1419333 | 7301 | 7320 | 7294 | 99.90 |
| KX162691 | EQG1418881 | 7424 | 7442 | 7422 | 99.97 |
| KX162713 | NIE1419323 | 7429 | 903a | 903 | 100.00 |
| KX162712 | NIE1419321 | 7421 | 903a | 837 | 92.70 |
| KX162714 | NIE1519322 | 7440 | 903a | 903 | 100.00 |
| KX162715 | NIE1519324 | 6707 | 903a | 903 | 100.00 |
| KX162716 | NIE1519325 | 7439 | 903a | 903 | 100.00 |
| KX162717 | NIE1519342 | 7398 | 903a | 903 | 100.00 |
VP1.
DISCUSSION
Surveillance of poliovirus using whole-genome analysis provides an essential tool for the final stages of poliovirus eradication. It enables the investigation of new poliovirus outbreaks, VDPV emergence, and the characterization of VDPVs from immunocompromised chronic shedders (iVDPV). Here, we aimed to optimize a sequence-independent protocol for the whole-genome sequencing of poliovirus isolates from culture supernatants and FTA card specimens, which are the standard sample types analyzed for global poliovirus surveillance. The use of a sequence-independent metagenomics approach ensures that divergent or recombinant genomes are not missed, as PCR-based approaches might be limited by primer specificity. An additional benefit of the NGS approach is the ability to multiplex samples on the MiSeq, which, in turn, reduces time and cost per sample. We estimated that the total cost per genome per sample is only ∼$65 when 64 samples are multiplexed in a MiSeq run and ∼$60 when 96 samples are multiplexed (estimates include steps from extraction to MiSeq sequencing; data not shown). In our current experiments, we multiplexed up to 64 samples in a single MiSeq run, and we estimate that even multiplexing 96 samples would readily generate full genomes with adequate (>10-fold) coverage. However, for samples that contain multiple PV types, such as samples from immunocompromised chronic shedders, fewer samples should be multiplexed to allow adequate depth of coverage for analyzing potentially complex mixtures and genetic variants.
The genomes generated by NGS were validated by comparing them to available Sanger sequences from the same isolates. Even though the NGS approach uses random amplification and the Sanger method uses poliovirus-specific PCR, the majority of VP1 and whole-genome sequences generated by the two methods were identical or nearly identical in pairwise comparisons. In many cases, the results from NGS enable more in-depth analyses, as the approach offers an opportunity to examine multiple PV variants in each sample. In our experience, the NGS approach is especially beneficial when the sample contains diverse variants or when multiple PVs are coinfecting a sample. Therefore, since the NGS approach enables the detection of minor or unexpected variants, it might facilitate the analysis of VDPV and iVDPV.
We developed a highly efficient protocol for sequencing poliovirus isolates, where, on average, 84% ± 15.6% (n = 11) of the reads generated were the targeted poliovirus in our experiment with DNase-treated CS samples sequenced using the Nextera kit (Fig. 1A and 2A). DNase treatment of the extracted nucleic acids greatly improved the yield, as demonstrated by the increased percentages of reads that mapped to poliovirus compared with non-DNase treated (63% versus 49%, respectively; Fig. 1A and see Fig. S4 in the supplemental material) and by the slightly higher proportion of genome coverage (99% versus 98%, respectively; Fig. 1C). Previous studies found that RNA and DNA are eluted during Qiagen spin-column extractions (12, 14), thus necessitating the need for DNase treatment to reduce the amount of cellular DNA that is coextracted. In isolate samples, DNase treatment produced better results, likely by reducing the number of sequences from cellular DNA. Alternatively, to further simplify our protocol, DNase treatment can also be performed “on-column” during the final wash steps of the extraction procedure, and preliminary experiments have found this approach to be equally effective (data not shown). In conclusion, we recommend using a final DNase treatment step to improve sequencing results.
Compared with the sequences from virus isolates, those derived from FTA card samples exhibited lower proportions of PV-specific reads (Fig. 1A) and a lower percentage of genome mapped (Fig. 1C). FTA cards also yielded fewer total reads than the CS (Fig. 1B), possibly due to the lack of a cold chain and to inherently more manipulations. This reduction in yield likely resulted from the characteristically low quality and quantity of poliovirus RNA obtained from FTA cards. In many FTA samples, when a PV sequence was not obtained using the standard Sanger method, a transfection was performed to biologically amplify the PV before resequencing. This transfection approach requires a considerable amount of time and effort. Since we showed that NGS enables sequencing directly from FTA card RNA, it offers an attractive alternative to transfection. Of note, when multiplexing CS and FTA samples in the same NGS run, fewer reads were generated from the FTA samples. However, in an additional experiment where only FTA samples were included in the run, we consistently assembled complete poliovirus genomes. Genome coverage increased to 99% (from a mean of only 70%) when FTA samples were not sequenced in parallel with CS samples (see Fig. S1B). Therefore, FTA samples should be sequenced separately from CS samples for optimal results.
In numerous studies involving metagenomic analyses of clinical specimens, viral sequencing was improved by filtration and nuclease pretreatments (12, 13, 15). Most strikingly, PV isolates that were pretreated showed only slightly improved results (see Fig. S2 and S3), so these steps can be omitted to streamline the protocol. Notably, for direct sequencing of total RNA (Fig. 1A, CS DNase group), we observed that a large proportion (mean of 63%) of the total RNA was poliovirus RNA. One explanation for this unusually high proportion of viral RNA in the isolate is that poliovirus shuts down host transcription during viral replication (16), thus reducing the amount of host cell RNA in the sample. This may be a specific case for PV and other picornaviruses. By comparison, smaller proportions of viral reads were reported in isolates of many other viral families (17). Separate experiments are required for evaluating noncultured samples, such as stool and sewage samples.
For virus isolates and FTA samples, we found that the most efficient protocol involved RNA extraction, DNase treatment, reverse transcription, and random amplification, followed by library construction. A robust and efficient protocol is important in outbreak situations where immediate results are critical. The processing time for our current protocol is 72 h from the time a typical set of 64 specimens is received to the time the final genome sequences are reported. Of the 72 h, 48 h were attributed to the MiSeq sequencing run. This time can be further shortened by substituting a shorter 150-bp single-end run for our current 250-base paired-end run, by processing fewer total samples simultaneously, or by using faster sequencing instruments as they become available.
In most laboratories, poliovirus genomes are obtained by specific, long PCR amplification of overlapping fragments, followed by Sanger sequencing. Although this Sanger methodology is well established for virus isolates (18), the quality and quantity of long PCR fragments (from 2 kbp to >7 kbp) vary and depend on virus titer, RNA quality, sequence similarity between primers and the viral RNA, and the presence of inhibitors. The efficiency of amplifying long PCR fragments from RNA eluted from FTA cards is reduced compared with that of RNA extracted from virus isolates, and thus, can sometimes lead to suboptimal results. For RNA of good quality, such as that from the oral poliovirus vaccine, long PCR fragments were sequenced with NGS for monitoring mutations in PV vaccine production (19). This paper describes an alternative protocol to the Sanger method involving random amplification and NGS, which enables a more automated workflow, higher throughput, and increased sequence depth for SNP and variant determinations.
MATERIALS AND METHODS
Polio RNA extraction.
Polioviruses were isolated and propagated in culture according to the WHO polio laboratory manual (10). Before nucleic acids were extracted, the culture supernatant (CS) was frozen and thawed three times and clarified at 15,294 × g for 10 min at 4°C. For some experiments, 160 μl of the clarified supernatant was then filtered through a sterile, 0.45-μm Ultrafree-MC filter (EMD Millipore, Darmstadt, Germany) at 8,000 × g for 5 min at 25°C to remove host cellular debris and bacteria. For FTA cards, a detailed, previously described card-processing procedure was used to extract PV RNA from the card (10, 20). Total nucleic acids were extracted from 140 μl of the filtered CS or from resuspensions from FTA cards using the QIAamp viral RNA minikit (Qiagen, Germantown, MD) according to the manufacturer's instructions. For certain experiments, DNase treatment and removal (rDNase I; Ambion of Thermo Fisher Scientific, Waltham, MA) were carried out using the extracted nucleic acids according to the manufacturer's instructions before reverse transcribing.
Experimental design 1: pretreatment comparisons.
First, we compared different sample pretreatments for their effects on sequencing outcomes for CS and FTA card specimens. Nucleic acids from a total of 32 samples were examined, including 16 CS and 16 FTA samples. Four treatment groups were tested, including (i) CS samples with DNase treatment, (ii) CS samples without DNase treatment, (iii) FTA samples with DNase treatment, and (iv) FTA samples without DNase treatment.
Experimental design 2: library construction kit comparisons.
In a second experiment, we compared the results from three library preparation kits from two different manufacturers that were compatible with the Illumina MiSeq platform: the Nextera XT DNA library preparation kit (Nextera) (Illumina, San Diego, CA) and the NEBNext Ultra DNA (NEB-DNA) and NEBNext Ultra RNA (NEB-RNA) kits (both from New England BioLabs, Ipswich, MA). A subset of 13 RNAs extracted from the original sample set of 32 used in experimental design 1 were included from CS (n = 10) and FTA card (n = 3) samples. All samples were subjected to DNase treatment prior to the library construction and consisted of RNA previously used in experimental design 1. For NEB-RNA, as RNA is used as a starting material, the DNase-treated viral nucleic acids were used as starting material. Viral RNA used in the NEBNext Ultra RNA kit was fragmented for 5 min at 94°C to generate products of approximately 200 bp in length. For Nextera and NEB-DNA kits, all sample RNA first underwent reverse transcription and random amplification using the protocol described below (12, 13). The resulting random amplicons were size selected using 1.8× volume Agencourt AMPure XP beads (Beckman Coulter, Brea, CA) before being used as input.
Reverse transcription and random amplification.
Viral nucleic acids were amplified using a previously published sequence-independent, single-primer amplification (SISPA) protocol (12, 13). Briefly, viral RNA was reverse transcribed using SuperScript III reverse transcriptase (Invitrogen of Thermo Fisher Scientific, Waltham, MA) with a 28-base primer whose 3′ end consisted of eight random nucleotides (N1_8N, CCTTGAAGGCGGACTGTGAGNNNNNNNN). A complementary strand was synthesized using the Klenow fragment of DNA polymerase I (3′ to 5′ exo-; New England BioLabs). cDNA was then randomly amplified using 2 μM of the overhang primer (N1, CCTTGAAGGCGGACTGTGAG), 1.85 units AmpliTaq Gold polymerase (Thermo Fisher Scientific), 4 mM MgCl2, 0.25 mM deoxynucleoside triphosphates [dNTPs], and 1× PCR Gold buffer in a 25-μl reaction volume. PCR was performed in a thermocycler under the following conditions: 1 cycle of 95°C for 5 min, 5 cycles of 95°C for 1 min, 59°C for 1 min, and 72°C for 90 s, followed by either 20 or 30 cycles of 95°C for 30 s, 59°C for 30 s, and 72°C for 90 s with increments of 2 s per cycle. The amplicons were visualized by electrophoresis on an agarose gel or on a TapeStation (Agilent Technologies, Santa Clara, CA) for selecting appropriate quantities of DNA for downstream applications. After visualizing, the random amplicons were further purified to remove excess dNTPs and primers using a 1.8× volume of Agencourt AMPure XP beads. The purified amplicons were used as the input for Nextera XT and NEBNext Ultra DNA library preparation kits.
Sequencing.
Paired-end libraries were prepared according to the manufacturer's protocols with the following exceptions: (i) during NEB-DNA library construction, shearing and size selection of library input DNA was omitted as the SISPA protocol produced amplified products in the desired size range of 150 bp to 600 bp; (ii) the bead normalization step was omitted for the Nextera library, as all libraries were normalized and pooled based on qPCR results obtained using the Kapa library quantification kit (Kapa Biosystems, Wilmington, MA).
Prior to sequencing, the library size was visualized using the TapeStation instrument. Each uniquely barcoded sample was quantified using qPCR. Normalized samples were pooled and sequenced using 500-cycle (2 × 250-bp paired-end) MiSeq reagent kits (v2; Illumina, San Diego, CA). The pooled samples, including 13 barcodes from Nextera, 13 from NEB-DNA, and 13 from NEB-RNA, were then sequenced separately using the MiSeq platform.
Data analysis.
Raw sequencing reads were filtered to remove host DNA sequences, were trimmed to remove primers and adapters, and also were filtered by length. The remaining reads were then assembled de novo to produce sequence contigs. To facilitate the assembly of viral genomes, host genome sequences were first filtered from the data set using Bowtie2 version 2.1.0 (21), which removed any contaminating human sequences using the h19 human reference genome (22). Cutadapt version 1.8 (23) was used to trim specified primers and adapters and to filter out reads below Phred quality scores of 20. Duplicate reads were removed using the Python program Dedup.py (24) to prevent biased coverage of genomic regions. The remaining FASTQ reads were then assembled into contigs using SPAdes version 3.6.2 (25). The contigs were then classified into taxonomic groups using a BLAST search against a local GenBank database and a curated viral sequence database. First, the NCBI BLAST+ version 2.2.30 (26) was used to align the contigs against a viral database that contains a curated set of GenBank viral sequences. Next, a BLAST alignment of the contigs to the GenBank NT database was conducted. NT alignment scores were used to categorize alignments that had a greater score for nonviral pathogens than for viral hits. The outcome of this process is the assembly and identification of full-length and near full-length viral genomes.
Additional manual analysis was performed with the contigs, trimmed reads, and poliovirus reference genomes using Geneious version 8.1.6 (Biomatters, Auckland, NZ) for visualizing and quantifying the mapping of processed reads to the viral reference genomes.
Statistical analysis.
To compare sequencing results from different pretreatments for the two specimen categories (CS and FTA card), the percentages of genome covered, the read counts, and the percentages of reads mapped were analyzed statistically as three metrics for each sample. A generalized linear model with Tukey's comparisons was implemented to account for the multiple comparisons across different conditions using SAS 9.3 (SAS, Cary, NC). In cases where the observation was a percentage (fraction of genome covered and reads mapped), logit transformation was performed using the logit link function in SAS (Proc GENMOD), coupled with binomial distribution to guarantee that the variance of the distribution approached zero as the mean approached either 0 or 1 (27). For cases where data observations were counts, negative binomial regression was applied to adjust for overdispersion (28).
Accession numbers.
The sequences described here were submitted to the GenBank database under the accession numbers KX162678 to KX162693 and KX162695 to KX162717 as listed in Table 1.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Advanced Molecular Detection (AMD) program and the Polio Eradication line items at the CDC.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. The use of trade names is for identification only and does not imply endorsement by CDC or the U.S. government.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JCM.02121-16.
REFERENCES
- 1.Pallansch MA, Oberste MS, Whitton JL. 2013. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. In Knipe DM, Howley P (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Kew O. 2012. Reaching the last one per cent: progress and challenges in global polio eradication. Curr Opin Virol 2:188–198. doi: 10.1016/j.coviro.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Asghar H, Diop OM, Weldegebriel G, Malik F, Shetty S, El Bassioni L, Akande AO, Al Maamoun E, Zaidi S, Adeniji AJ, Burns CC, Deshpande J, Oberste MS, Lowther SA. 2014. Environmental surveillance for polioviruses in the Global Polio Eradication Initiative. J Infect Dis 210 (Suppl 1):S294–S303. doi: 10.1093/infdis/jiu384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathanson N, Kew OM. 2010. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol 172:1213–1229. doi: 10.1093/aje/kwq320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hovi T, Shulman LM, van der Avoort H, Deshpande J, Roivainen M, de Gourville EM. 2012. Role of environmental poliovirus surveillance in global polio eradication and beyond. Epidemiol Infect 140:1–13. doi: 10.1017/S095026881000316X. [DOI] [PubMed] [Google Scholar]
- 6.Jorba J, Campagnoli R, De L, Kew O. 2008. Calibration of multiple poliovirus molecular clocks covering an extended evolutionary range. J Virol 82:4429–4440. doi: 10.1128/JVI.02354-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esteves-Jaramillo A, Estívariz CF, Peñaranda S, Richardson VL, Reyna J, Coronel DL, Carrión V, Landaverde JM, Wassilak SGF, Pérez-Sánchez EE, López-Martínez I, Burns CC, Pallansch MA. 2014. Detection of vaccine-derived polioviruses in Mexico using environmental surveillance. J Infect Dis 210 (Suppl 1):S315–S323. doi: 10.1093/infdis/jiu183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wood DJ, Hull B. 1999. L20B cells simplify culture of polioviruses from clinical samples. J Med Virol 58:188–192. [PubMed] [Google Scholar]
- 9.Kilpatrick DR, Yang C-F, Ching K, Vincent A, Iber J, Campagnoli R, Mandelbaum M, De L, Yang S-J, Nix A, Kew OM. 2009. Rapid group-, serotype-, and vaccine strain-specific identification of poliovirus isolates by real-time reverse transcription-PCR using degenerate primers and probes containing deoxyinosine residues. J Clin Microbiol 47:1939–1941. doi: 10.1128/JCM.00702-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.World Health Organization. 25 to 26 June 2015. The 21st informal consultation on the global polio laboratory network, final report of recommendations. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 11.World Health Organization. 24 to 26 October 2011. Report on the fifteenth intercountry meeting of directors of poliovirus laboratories in the eastern Mediterranean region. Kuwait City, Kuwait. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 12.Ng TFF, Kondov NO, Deng X, Van Eenennaam A, Neibergs HL, Delwart E. 2015. A metagenomics and case-control study to identify viruses associated with bovine respiratory disease. J Virol 89:5340–5349. doi: 10.1128/JVI.00064-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng TFF, Marine R, Wang C, Simmonds P, Kapusinszky B, Bodhidatta L, Oderinde BS, Wommack KE, Delwart E. 2012. High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage. J Virol 86:12161–12175. doi: 10.1128/JVI.00869-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng TFF, Chen L-F, Zhou Y, Shapiro B, Stiller M, Heintzman PD, Varsani A, Kondov NO, Wong W, Deng X, Andrews TD, Moorman BJ, Meulendyk T, MacKay G, Gilbertson RL, Delwart E. 2014. Preservation of viral genomes in 700-y-old caribou feces from a subarctic ice patch. Proc Natl Acad Sci U S A 111:16842–16847. doi: 10.1073/pnas.1410429111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allander T, Emerson SU, Engle RE, Purcell RH, Bukh J. 2001. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proc Natl Acad Sci U S A 98:11609–11614. doi: 10.1073/pnas.211424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chase AJ, Semler BL. 2012. Viral subversion of host functions for picornavirus translation and RNA replication. Future Virol 7:179–191. doi: 10.2217/fvl.12.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, Deng X, Mee ET, Collot-Teixeira S, Anderson R, Schepelmann S, Minor PD, Delwart E. 2015. Comparing viral metagenomics methods using a highly multiplexed human viral pathogens reagent. J Virol Methods 213:139–146. doi: 10.1016/j.jviromet.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns CC, Shaw J, Campagnoli R, Jorba J, Vincent A, Quay J, Kew O. 2006. Modulation of poliovirus replicative fitness in HeLa cells by deoptimization of synonymous codon usage in the capsid region. J Virol 80:3259–3272. doi: 10.1128/JVI.80.7.3259-3272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neverov A, Chumakov K. 2010. Massively parallel sequencing for monitoring genetic consistency and quality control of live viral vaccines. Proc Natl Acad Sci U S A 107:20063–20068. doi: 10.1073/pnas.1012537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Centers for Disease Control and Prevention. Processing FTA cards. https://cdc.train.org/DesktopModules/eLearning/CourseDetails/CourseDetailsForm.aspx?tabid=96&courseid=1048222.
- 21.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, et al. . 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 23.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 24.Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, Delwart EL. 2015. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res 43:e46. doi: 10.1093/nar/gkv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McLean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baum CF. 2008. Stata tip 63: modeling proportions. Stata J 8:299–303. [Google Scholar]
- 28.Coxe S, West SG, Aiken LS. 2009. The analysis of count data: a gentle introduction to poisson regression and its alternatives. J Pers Assess 91:121–136. doi: 10.1080/00223890802634175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.