

ABSTRACT
The interaction of Candida albicans with the innate immune system is the key determinant of the pathogen/commensal balance and has selected for adaptations that facilitate the utilization of nutrients commonly found within the host, including proteins and amino acids; many of the catabolic pathways needed to assimilate these compounds are required for persistence in the host. We have shown that C. albicans co-opts amino acid catabolism to generate and excrete ammonia, which raises the extracellular pH, both in vitro and in vivo and induces hyphal morphogenesis. Mutants defective in the uptake or utilization of amino acids, such as those lacking STP2, a transcription factor that regulates the expression of amino acid permeases, are impaired in multiple aspects of fungus-macrophage interactions resulting from an inability to neutralize the phagosome. Here we identified a novel role in amino acid utilization for Ahr1p, a transcription factor previously implicated in regulation of adherence and hyphal morphogenesis. Mutants lacking AHR1 were defective in growth, alkalinization, and ammonia release on amino acid-rich media, similar to stp2Δ and ahr1Δ stp2Δ cells, and occupied more acidic phagosomes. Notably, ahr1Δ and stp2Δ strains did not induce pyroptosis, as measured by caspase-1-dependent interleukin-1β release, though this phenotype could be suppressed by pharmacological neutralization of the phagosome. Altogether, we show that C. albicans-driven neutralization of the phagosome promotes hyphal morphogenesis, sufficient for induction of caspase-1-mediated macrophage lysis.
KEYWORDS: Ahr1, Candida albicans, alkalinization, macrophages, pyroptosis
INTRODUCTION
The fungus Candida albicans is a common human commensal which frequently causes superficial and invasive opportunistic infections, such as oral thrush, vaginitis, and disseminated candidiasis. Phagocytes, particularly macrophages and neutrophils, are the first line of defense against many microbial pathogens, including C. albicans. The macrophage phagolysosome exposes the pathogens to multiple challenges, including oxidative, nitrosative, and nutritional stresses. Upon phagocytosis, C. albicans adapts to the nutrient conditions by inducing alternative metabolic pathways, such as the glyoxylate cycle, gluconeogenesis, and β-oxidation of fatty acids (1–5). C. albicans metabolism plays a pivotal role in many other host niches: it governs fungal susceptibility to stress conditions and antifungal drugs, the expression of key virulence factors, and fungal vulnerability to innate immune defenses. Furthermore, some metabolic pathways are required for full virulence in animal models of candidiasis (2–4, 6–8), highlighting the importance of nutrient acquisition and assimilation to the success of C. albicans as a commensal and infectious agent.
We and others have previously shown that upon utilization of alternative carbon sources, such as amino acids, organic acids, and N-acetylglucosamine, C. albicans can actively neutralize acidic environments (9–11). The metabolism of these nutrients is also essential for neutralization of the macrophage phagosome, a process that contributes to hyphal growth and fungal escape from the phagocyte (8, 9). When C. albicans utilizes amino acids as the sole carbon source, it generates excess ammonia that can raise the extracellular pH from pH 4 to pH 7 to 8 in a matter of hours; some key factors in this phenomenon have been identified via genetic and genomic approaches (8, 11). These are enriched in functions related to amino acid uptake and catabolism and include the SPS plasma membrane amino acid sensor; its regulatory target, Stp2p; a transcription factor that controls expression of amino acid permeases; and a family of putative ammonia transporters, at least two of which (Ato1 and Ato5) are required for the neutralization of acidic environments (11–13). The stp2Δ mutant completely fails to alkalinize in vitro and does not autoinduce hyphal morphogenesis (11). The ability of C. albicans to utilize amino acids and modulate the phagosomal pH was also critical for escape from the immune cells, since mutants defective in environmental alkalinization remain in acidic phagosomes, fail to damage the host cells, and do not switch to the hyphal form, a process restored upon pharmacological neutralization of the phagosomes (8, 13). C. albicans cells lacking STP2 are also attenuated in virulence in a mouse model of disseminated candidiasis, demonstrating that alteration of phagosomal pH is an important virulence adaptation in this species (8).
It was long assumed that macrophage lethality resulted from physical disruption of the cell caused by C. albicans hyphal extension, yet recent findings demonstrate that C. albicans induces macrophage pyroptosis, a programmed cell death pathway, in a manner only partially dependent on hyphal morphogenesis (14–16). The mechanism remains unclear, but this is thought to be a two-step process in which C. albicans stimulates pattern recognition receptors on the surface of the immune cell, activating NF-κB and inducing expression of pro-interleukin-1β (pro-IL-1β) and/or pro-IL-18, which results in inflammasome priming. Following engulfment of the fungal cell, a second and yet unknown signal engages the NLRP3 inflammasome and the cysteine protease caspase-1, which mediates the processing and secretion of mature forms of IL-1β (14, 17, 18) and subsequent recruitment of additional immune cells. Caspase-1 activation also induces pyroptotic cell death of the macrophage, resulting in cell swelling, DNA fragmentation, and the lytic release of intracellular inflammatory contents. Evaluation of the intraphagosomal morphology of mutants inducing both low and normal levels of IL-1β revealed that two genes, UPC2 and AHR1, encoding transcription factors regulating ergosterol biosynthesis and expression of adhesins, respectively, are impaired in IL-1β release but not in hyphal morphogenesis. On the other hand, mutants with mutations in NDT80, encoding a transcription factor with multiple roles in C. albicans, were deficient in hypha formation but induced normal IL-1β release. Thus, there is some disconnect between morphology and pyroptosis, and the exact signals and sequence of steps that lead to induction of pyroptotic cell death by C. albicans remain unknown.
In this study, we investigated whether phagosomal neutralization plays a role in C. albicans-driven macrophage pyroptosis. We demonstrate that the transcription factor Ahr1p, involved in the expression of adhesins and induction of macrophage pyroptosis, is required for alkalinization in vitro and during macrophage phagocytosis. Cells lacking AHR1 have an alkalinization defect similar to that of the stp2Δ mutant, and the double ahr1Δ stp2Δ mutant is not further compromised, suggesting that the two transcription factors might act in the same pathway to drive alkalinization by C. albicans. Similar to the previously reported stp2Δ phenotype, macrophage phagocytosis of ahr1Δ and ahr1Δ stp2Δ mutant cells resulted in impaired hyphal morphogenesis and killing of the immune cells, which were restored in phagosomes neutralized with the vacuolar ATPase (vATPase) inhibitor bafilomycin A1 (BafA), demonstrating the importance of neutral phagosomes for C. albicans-induced damage of the host cells. Finally, C. albicans strains lacking genes involved in different steps of the alkalinization process displayed defects in macrophage lysis and IL-1β release upon interaction with the immune cells, a defect restored upon neutralization of the phagosome. Thus, our data indicate that the ability of C. albicans to interfere with phagosomal maturation, in particular, by maintaining a neutral pH in this organelle, is a key inducer of macrophage pyroptosis.
RESULTS
Ahr1p is required for environmental alkalinization by C. albicans.
A recent article by Askew et al. (19) revealed that Ahr1p, a transcription factor which controls adhesion, hyphal morphogenesis, and white-opaque switching in C. albicans (20), interacts with STP2 and other genes involved in amino acid transport and utilization. Thus, we were interested to understand in more detail the role of Ahr1p in carbon metabolism and pH modulation in vitro and within the macrophage phagosome. We tested growth and pH modulation in the ahr1Δ strain available in one of the C. albicans deletion libraries (21). The cells lacking AHR1 had a defect comparable to that of the stp2Δ library strain, revealing that Ahr1p plays an important role in the utilization of amino acids. To verify the library strains, we constructed independent mutants lacking Ahr1p (ahr1Δ) in the wild-type strain SC5314 background using the SAT1 flipper method (22). Since the ahr1Δ strain had no notable phenotypes during growth on rich medium (data not shown), we tested the newly developed strains for growth and pH modulation on acidic amino acid-rich medium (yeast nitrogen base [YNB], 1% Casamino Acids [CAA], pH 4.0). Cells lacking AHR1 were impaired in growth on amino acids compared to the growth of cells of the wild-type and the complemented strains (Fig. 1A). The poor metabolism of amino acids was also reflected in the impaired ability of the ahr1Δ mutants to neutralize the environment, reaching only pH 4.9 from a beginning culture at pH 4.0, a significant defect compared to the metabolism of wild-type and complemented ahr1Δ+AHR1 cells, in which the medium pH reached about 7.5 (Fig. 1B). Interestingly, the growth and alkalinization defects of the ahr1Δ strains were almost identical to those of cells lacking STP2. Thus, given the dramatic phenotype and the predicted role of Ahr1p in the transport and utilization of amino acids, we questioned if there is a connection between nutrient catabolic pathways and the pH modulation controlled by Ahr1p and Stp2p. To address this, we generated strains lacking both transcription factors (ahr1Δ stp2Δ) and assayed the strains for growth and alkalinization on amino acids (Fig. 1A and B). The phenotype of the double mutant was indistinguishable from that of cells lacking STP2.
FIG 1.

C. albicans AHR1 is important for growth on amino acids and for in vitro pH neutralization. C. albicans strains were grown in YNB plus 1% CAA, pH 4.0. (A) The growth of the cells was assessed by measuring the OD600 at the indicated time points. (B) The pH of the growing cultures was measured. The experiment was performed in triplicate. (C) Ammonia release from strains grown on YAC medium, pH 4.0, was assessed as described in Materials and Methods.
C. albicans cells grown on amino acids release increasing amounts of ammonia as a mechanism to neutralize the environment, a process abrogated in mutants that fail to modulate the pH (8, 11–13). Thus, we assayed ammonia release by the newly constructed strains following growth on amino acids. The ahr1Δ and ahr1Δ stp2Δ mutants failed to release ammonia after 72 h of growth on YNB medium with allantoin as the nitrogen source (0.17% yeast nitrogen base, 0.5% allantoin) and 1% Casamino Acids as the sole carbon source (YAC medium), pH 4.0, a phenotype comparable to that of the stp2Δ negative-control strain (Fig. 1C). In contrast, the wild type and the strain with restored AHR1 released an average of 200 ppm ammonia into the environment. Thus, Stp2p and Ahr1p seem to control in vitro alkalinization in a similar manner, suggesting a shared functionality.
Ahr1p is required for alkalinization of the macrophage phagosome.
Utilization of amino acids is required for efficient macrophage damage by C. albicans, which, at least in part, is linked to the ability to actively neutralize the macrophage phagosome (8). Ahr1p also seems to be important for macrophage lysis, since ahr1Δ cells show a significantly decreased release of lactate dehydrogenase (LDH) and IL-1β than the wild-type control (23), but the reasons for this were not known. To investigate whether this defect is related to alterations in phagosomal pH, we tested the ability of our strains to undergo hyphal morphogenesis within J774A.1 murine macrophages. All of the tested strains had comparable rates of phagocytosis (data not shown). After 1 h of coculture, the ahr1Δ, stp2Δ, and ahr1Δ stp2Δ mutants showed significant defects in hyphal morphogenesis, since the germination rates were 37%, 41%, and 32%, respectively (Fig. 2A), whereas they were 68% for the wild-type cells and 77% for the ahr1Δ+AHR1 cells. Further, the mutant cells that did initiate hyphal morphogenesis formed shorter hyphae than the control strains (Fig. 2C). To quantitate this, we calculated the length of the filaments by drawing a line from the middle of the cell to the tip of the hyphal projection (see Materials and Methods) using SlideBook (version 6) software. Indeed, image analysis revealed that the stp2Δ, ahr1Δ, and ahr1Δ stp2Δ mutants formed hyphae with average lengths of 10.5 μm, 10.9 μm, and 11.0 μm, respectively, slightly shorter than the average 12.6-μm hyphal length of the wild-type SC5314 strain. Interestingly, the complemented AHR1 strain formed longer hyphae, 14.9 μm on average, than the other strains. Hyphal morphogenesis of nonphagocytosed cells was robust in all strains tested (data not shown). Altogether, these results demonstrate that cells lacking AHR1 were impaired in hyphal morphogenesis.
FIG 2.

Cells lacking AHR1 are defective in hyphal morphogenesis during phagocytosis. FITC-stained C. albicans wild-type, ahr1Δ, ahr1Δ stp2Δ, ahr1Δ+AHR1, and stp2Δ strains were cocultured with J774A.1 macrophages for 60 min. Nonphagocytosed fungal cells were stained with calcofluor white, and then the cocultures were fixed and imaged. The percentage of phagocytosed hyphae (A, B) and hyphal length (C, D) were scored as described in Materials and Methods. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Since the stp2Δ and ahr1Δ mutants have similar in vitro alkalinization and macrophage morphogenesis phenotypes, we hypothesized that Ahr1p would also be required for alkalinization of the phagosome. We have demonstrated that pharmacological neutralization of the macrophage phagosome by bafilomycin A (BafA) restores normal hyphal morphogenesis of the stp2Δ cells (8), so we asked whether this treatment would allow ahr1Δ cells to form hyphae at a rate similar to that for wild-type cells. To test this, we pretreated LysoTracker red (LR)-loaded macrophages with 50 nM BafA 30 min prior to addition of fluorescein isothiocyanate (FITC)-stained fungal cells and visualized the cells after 1 h of coculture. The BafA treatment collapsed the pH gradient across the phagosomal membrane (Fig. 3). More importantly, neutralization of the macrophage phagosome fully restored the morphogenetic capacity of the ahr1Δ and ahr1Δ stp2Δ cells, since the rate of hyphal growth was equal to that of the control strains, which was in the range of 72.7 to 82.3%, on average (Fig. 2B). Addition of BafA to the medium enhanced hyphal growth in all strains, for which the average length was 13.2 to 14.9 mm, which is longer than that of the wild-type strain in untreated macrophages (Fig. 2D). Importantly, the pharmacological alkalinization of the phagosome suppressed the hyphal growth differences in the mutant strains relative to the control, indicating that the hyphal defects of the mutants are due to differences in phagosomal pH.
FIG 3.
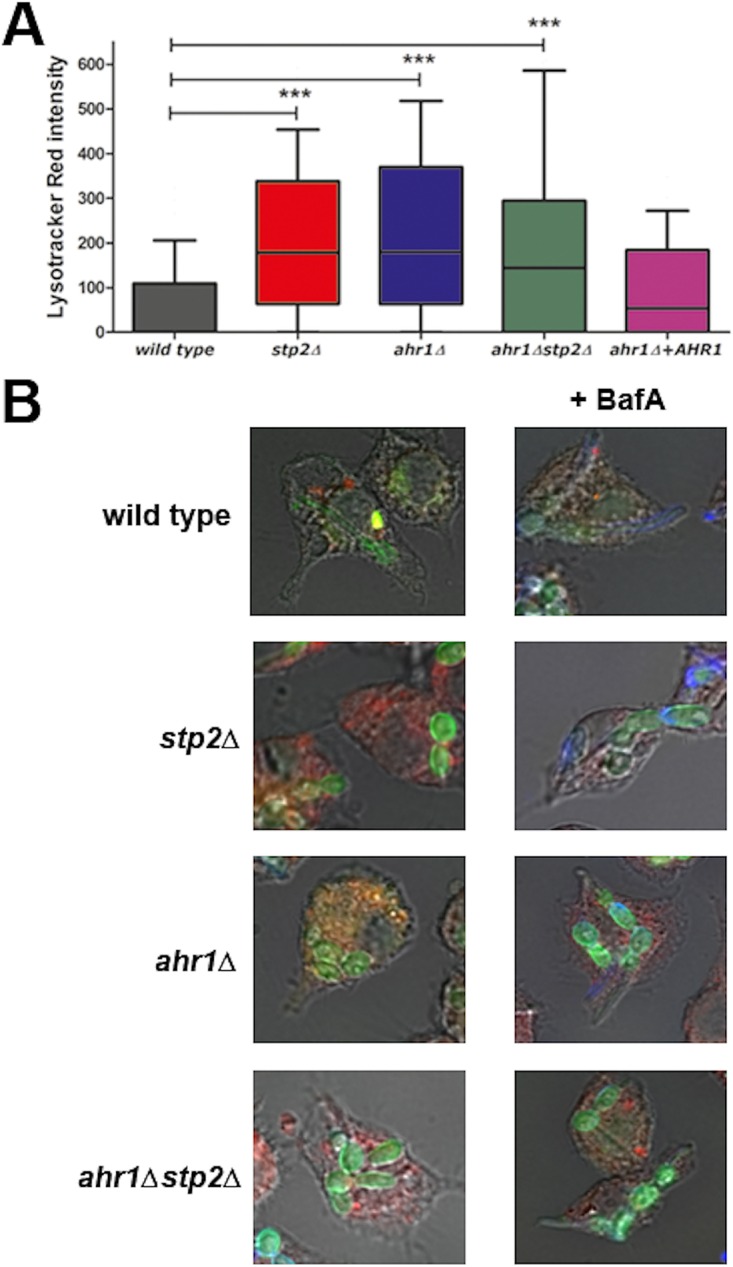
Ahr1p is required for alkalinization of the macrophage phagosome. FITC-stained C. albicans wild-type, ahr1Δ, ahr1Δ stp2Δ, ahr1Δ+AHR1, and stp2Δ strains were cocultured with LysoTracker red-preloaded J774A.1 macrophages for 60 min and imaged. (A) To estimate the phagosomal pH, we measured the LysoTracker red intensity as described in Materials and Methods. Results are reported as mean values ± SDs from triplicate experiments. ***, P < 0.001. (B) Representative images of cocultures incubated in the absence (left) or presence (right) of the vATPase inhibitor BafA.
To directly estimate phagosomal pH, we preloaded J774A.1 macrophages with the acidophilic dye LR for 2 h prior to coculture, followed by addition of FITC-labeled C. albicans cells. We used the intensity of the LR signal immediately next to the fungal cells as a measure of the phagosomal pH (12), since it is inversely proportional to the pH in our range of detection. As expected, the wild-type cells occupied a phagosome of neutral pH, whereas stp2Δ mutant cells were found in acidic phagosomes, as were the ahr1Δ and the ahr1Δ stp2Δ mutant cells (Fig. 3A). The ahr1Δ+AHR1 cells restored the ability to modify the intraphagosomal pH (Fig. 3A). Representative images are shown in Fig. 3B. Thus, Ahr1p is important for alkalinization of the environmental pH in vitro and within the macrophage phagosome. Thus, the neutralization of the macrophage phagosome significantly contributes to hyphal morphogenesis, and this effect is not driven solely by Stp2p.
Phagosomal neutralization is important for C. albicans-induced macrophage lysis.
Hyphal morphogenesis of C. albicans within the macrophage phagosome contributes to escape of the fungus from the immune cell (24). Since Ahr1p and Stp2p are required for macrophage damage (8, 23), we questioned whether neutralization of the phagosome plays a role in C. albicans-driven macrophage lysis. We measured the release of LDH from infected macrophages as a proxy for cell death. Mutants lacking AHR1 were significantly impaired in their ability to damage J774A.1 macrophages (18.7% of the macrophages were killed by mutants lacking AHR1, whereas 52.6% of the macrophages were killed by the wild-type strain), which was more pronounced than that in the cells lacking STP2 (which caused 31.5% macrophage damage). These differences were either completely (stp2Δ mutant) or partially (ahr1Δ mutant) suppressed in cultures pretreated with BafA (Fig. 4A). BafA treatment did not further increase the macrophage damage caused by the control wild type strains and strains complemented in AHR1 and STP2. The double ahr1Δ stp2Δ mutant strain phenocopied the ahr1Δ effect in terms of macrophage lysis in both the absence and presence of BafA (Fig. 4A). These findings were further validated in bone marrow-derived macrophages (data not shown). Thus, these results demonstrate that the ability of C. albicans to evade phagosomal acidification and induce hyphal growth is important for macrophage killing, and although both Ahr1p and Stp2p are required in this process, Ahr1p may have additional roles that contribute to macrophage damage.
FIG 4.

C. albicans strains defective in phagosomal neutralization fail to induce macrophage pyroptosis. C. albicans wild-type strain SC5314 and ahr1Δ, stp2Δ, and ahr1Δ stp2Δ mutant and complemented strains were pretreated or not with 50 nM BafA for 30 min. To calculate macrophage lysis, the C. albicans strains were cocultured with the immune cells for 5 h in the absence (no treatment) or presence of a caspase-1 inhibitor (z-YVAD-fmk). The extracellular release of lactate dehydrogenase (A, C) and IL-1β (B, D) was measured as described in Materials and Methods. The experiments were performed in triplicate. *, P < 0.05; ***, P < 0.001.
Phagosomal neutralization by C. albicans induces caspase-1-dependent IL-1β release.
The proinflammatory cytokine IL-1β is released as a result of C. albicans-driven NLPR3 inflammasome activation and has been used as a measurement of pyroptotic cell death in the immune cells in response to many microbial pathogens, including C. albicans (16, 23). Previous reports demonstrate that ahr1Δ mutant strains are defective in triggering IL-1β release from stimulated macrophages (14, 23). Thus, given that the ahr1Δ and stp2Δ strains behave similarly within macrophages, we decided to test the ability of cells lacking STP2 to induce caspase-1-dependent IL-1β release. For this we cocultured C. albicans strains with stimulated J774A.1 macrophages at a multiplicity of infection of 3:1 for 5 h in the presence or absence of the caspase-1 inhibitor z-YVAD-fmk (YVAD) and collected the medium to assess the IL-1β levels via enzyme-linked immunosorbent assay (ELISA). As expected, wild-type cells induced IL-1β release (3.2 ng/ml), whereas treatment with the YVAD caspase-1 inhibitor reduced the level of IL-1β production by all of the C. albicans strains tested to below 0.5 ng/ml (Fig. 4B). Further, our data agreed with the observations of Wellington et al. (14) that ahr1Δ is important for pyroptosis, since macrophages cocultured with the mutant strain elicited an ∼60% decrease in IL-1β release compared to the ahr1Δ+AHR1 strains (Fig. 4B). Interestingly, coincubation of J774A.1 macrophages with the stp2Δ strain reduced IL-1β release by an amount comparable to that achieved by coincubation with the ahr1Δ strain (Fig. 4B). The double ahr1Δ stp2Δ mutant phenocopied both single mutants, demonstrating that these transcription factors have similar functions in stimulating the release of the proinflammatory cytokine from macrophages.
The exact C. albicans factors triggering macrophage pyroptosis are currently unknown, yet recent investigations revealed that the level of hyphal formation and exposure of specific moieties on the fungal cell wall play a role in this process (14–16). Since neutral pH is a potent morphogenetic signal in C. albicans and cell wall composition depends on the available carbon sources (25, 26), we explored if the effects of the stp2Δ and ahr1Δ mutations on IL-1β release were related to the inability of the mutants to prevent phagosomal acidification. For this purpose, we pretreated the J774A.1 macrophages with the vATPase inhibitor BafA to generate neutral phagosomes, followed by addition of C. albicans strains. We noted that extended exposure to BafA increased the levels of spontaneous LDH release from macrophages even in the absence of C. albicans, so we adjusted our protocol to treat the cocultures with the vATPase inhibitor for 1 h and then replaced the medium and continued the incubation for 4 more hours. Addition of YVAD to the coculture completely inhibited LDH and cytokine release, an effect that was not rescued by BafA treatment (Fig. 4C and D). Thus, neutralization of the macrophage phagosome achieved by inhibition of the vATPase blocks the caspase-1-mediated cell death induced by C. albicans.
With these data, we have correlated defects in phagosomal acidification to the release of IL-1β in macrophages cocultured with C. albicans. While both Stp2 and Ahr1 regulate the ability of the fungus to neutralize environments, including the phagosome, they are also transcription factors with potentially pleiotropic effects. We have previously identified a number of mutations that alter alkalinization in vitro and in macrophages (8, 11–13). To establish a broader linkage between fungus-driven changes in phagosomal pH and IL-1β release, we measured macrophage lysis and extracellular cytokine levels from J774A.1 macrophages cocultured with some of these alkalinization-defective mutants in the presence or absence of the caspase-1 inhibitor YVAD. We tested mutants with mutations in genes involved in different steps of the alkalinization process: Ssy1p, an amino acid sensor; Ach1p, an acetyl coenzyme A hydrolase involved in gluconeogenesis; Dur1,2p, an urea amidolyase that converts amino groups into ammonia; and the putative ammonium transporter Ato5p. Overall, the strains defective in alkalinization in vitro showed a reduced ability to trigger macrophage lysis and IL-1β release compared to the strains complemented in the corresponding gene or the wild-type SC5314 cells (Fig. 5 and S1). The only exception was the ach1Δ mutant, which behaved similarly to the complemented ach1Δ+ACH1 strain (data not shown). For example, the levels of LDH and IL-1β released from macrophages cocultured with the dur1,2Δ strain were reduced from ∼50% to 60% relative to the levels released from macrophages cocultured with the wild-type control (Fig. 5A and B). As previously noted, pharmacological neutralization of the phagosomes resulted in very low levels of IL-1β release from all of the tested strains, and addition of YVAD to the macrophages led to almost undetectable cytokine concentrations in the supernatant (Fig. 5B). Since cells lacking DUR1,2 and ATO5, responsible for the generation and release of ammonia from the cells, respectively, had pronounced defects in inducing caspase-1-mediated cell death, we conclude that the ability of C. albicans cells to generate ammonia is important for inflammasome activation and macrophage cell death.
FIG 5.
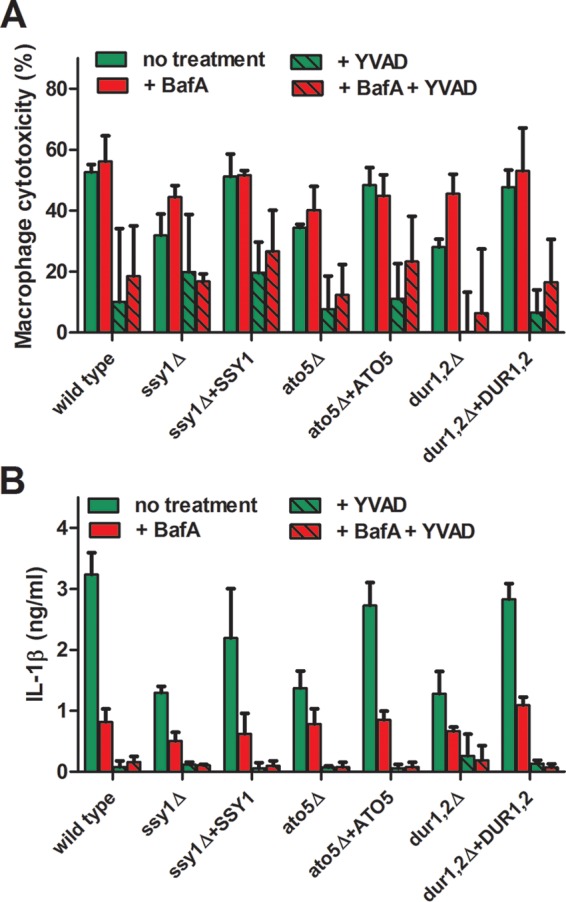
Alkalinization of the phagosome by C. albicans is required for macrophage killing and induction of pyroptotic cell death. C. albicans SC5314 and the ssy1Δ, dur1,2Δ, and ato5Δ mutants and their complemented strains were cocultured with J774A.1 macrophages for 5 h in a 96-well plate in the presence or absence of the caspase-1 inhibitor z-YVAD-fmk. Some of the wells were pretreated with 50 nM BafA for 30 min prior to coculture. The vATPase inhibitor was removed after 1 h of coculture, and all of the cultures were incubated for an additional 4 h. Macrophage lysis (A) and the release of IL-1β (B) were assessed as described in Materials and Methods. The experiments were performed in triplicate.
DISCUSSION
In this study, we describe that Ahr1p, a transcription factor involved in adhesion and hyphal morphogenesis, has a role in amino acid utilization and environmental alkalinization very similar to that previously described for Stp2, a transcription factor that regulates amino acid permeases. Further, mutants lacking these two transcription factors have almost identical phenotypes, including roles in amino acid metabolism, interaction with macrophages, and neutralization of amino acid-rich medium in vitro and within the macrophage phagosome. Double ahr1Δ stp2Δ mutant cells have phenotypes similar to those of the single mutant cells, suggesting that these two transcription factors function in the same pathway. Both proteins are required for pH-dependent hyphal morphogenesis during phagocytosis, since fewer cells formed hyphae in acidic phagosomes than neutralized phagosomes. Ahr1p is required for inflammasome-driven macrophage cell death, and other C. albicans mutants defective in alkalinization in vitro are also impaired in IL-1β release from infected macrophages (Fig. 6). Thus, our results suggest that neutralization of the phagosome by C. albicans is an important signal in activating the macrophage NLRP3 inflammasome.
FIG 6.

Phagosomal neutralization triggers C. albicans-driven macrophage pyroptosis. Upon interaction with macrophages, C. albicans cells trigger activation of the NLRP3 inflammasome and expression of pro-IL-1β. Within the phagosome, the fungal cell takes up amino acids (AA), metabolizes them, and expels NH4+ into the phagosomal milieu, a process that is dependent on the transcription factors Stp2p and Ahr1p. This results in neutralization of the phagosome and hyphal morphogenesis, with the latter contributing to membrane distention and the loss of immune cell integrity. LDH and the proinflammatory cytokine IL-1β are released from the cell and serve as a danger signal to uninfected macrophages to react to infection. Most of the phagocytosed cells defective in pH modulation, such as cells of the ahr1Δ stp2Δ mutant, reside in acidic phagosomes in yeast form, fail to activate the inflammasome, and are cleared more readily by the macrophages. These cells restore the ability to form hyphae and to damage the immune cell upon pharmacological neutralization of the phagosome with the vATPase inhibitor bafilomycin A.
Many host niches are glucose-limited environments that contain complex mixtures of alternative carbon sources. Amino acids are abundant in certain host niches, such as the oral cavity and the macrophage phagosome, and C. albicans has an expanded family of secreted aspartic proteases, amino acid permeases, and oligopeptide transporters that participate in the acquisition of this nutrient (27–31). Genomic evidence had previously suggested a role for Ahr1p in amino acid utilization (19). Here we demonstrate that cells lacking AHR1 grow poorly on amino acids as the sole carbon source and fail to neutralize the medium upon growth on this nutrient. We speculate that Ahr1p regulates the expression of Stp2p, based on the localization of Ahr1p on the STP2 promoter and the very similar phenotypes of the single and double mutants; however, Stp2 is also posttranslationally regulated via a proteolytic cleavage event directed by the SPS amino acid sensor (13, 32). In our future work, we will focus on understanding whether the role of Ahr1p in amino acid metabolism is by direct interaction with the genes required for their assimilation or it is mediated by other factors.
Nutrient limitation within the host is a common strategy to reduce pathogen growth. Transcriptomics data have established that certain host niches, including but not limited to phagocytic cells, are glucose poor and suggest that alternative carbon sources, including proteins, amino acids, carboxylic acids (e.g., lactate), and fatty acids, are present (33–35). Metabolic pathways that are essential for the assimilation of these nutrients, such as gluconeogenesis and the glyoxylate cycle, are required for full virulence (1–3, 8, 33). Some of these same mutations compromise the ability of C. albicans to escape from the immune cells (8, 36, 37). Indeed, we have shown that strains defective in the utilization of amino acids cannot neutralize the phagosome and germinate less readily, which are reflected by the improved clearance by the macrophages (8, 12, 13). Thus, the ability of C. albicans to adapt these pathways to alter the extracellular environment and undergo hyphal morphogenesis in certain host niches might directly contribute to survival in vivo.
It was long believed that macrophage lysis was caused by physical disruption by C. albicans filaments, since both killed or inactivated yeast cells and mutant strains locked in the yeast form trigger minimal levels of macrophage lysis (14, 38–40). However, recent investigations have begun to disconnect macrophage death from hyphal growth and have provided evidence that C. albicans-containing macrophages initiate pyroptotic cell death via the NLRP3 inflammasome, leading to release of the proinflammatory cytokines, such as IL-1β. Several studies have identified key factors, including Ahr1p, that play a role in this process. Here we link IL-1β release, phagosomal pH, and nutrient utilization via Stp2 and Ahr1, as well as several other proteins known to mediate extracellular alkalization. Thus, here for the first time we show that phagosomal neutralization can trigger programmed cell death mechanisms. It may be that the macrophage responds to aberrant phagosomal pH homeostasis by activating the NLRP3 inflammasome, leading to pyroptosis; alternatively, inflammasome activation may result from the hyphal growth induced by the neutral pH environment. Recent work from O'Meara et al. has suggested that hyphal growth is not required for macrophage lysis (16), so we favor a model in which pH is the driving factor, but further studies will be needed to test this hypothesis.
Previous work has linked aberrations in pH homeostasis to immune cell death: extracellular acidosis triggers the pH-dependent secretion of mature IL-1β from cultured human macrophages via a mechanism involving cellular potassium depletion and inflammasome activation, while an alkaline external pH strongly inhibits either the NLRP3 inflammasome assembly or an upstream event (41). Phagosomal pH also appears to be important for IL-1β production in response to phagocytosed pathogens: blockage of the lysosomal H+ ATPase system or inhibition of cathepsin B resulted in lower levels of IL-1β release in response to Listeria monocytogenes challenge, suggesting the importance of phagosome maturation and integrity. Other reports demonstrate that macrophages treated with agents that prevent acidification of the phagosome, including BafA, induce the release of IL-1β from affected cells and this improves the clearance of the fungal pathogen Cryptococcus neoformans (42). Caspase-1 also functions to reduce the pH of phagosomes containing Gram-positive bacteria, such as Staphylococcus aureus, as a part of the host defense (43). Thus, pH plays an important role in host inflammatory responses, including pyroptosis.
Here we demonstrate that BafA blocks IL-1β secretion by macrophages challenged with C. albicans, but it is not clear whether the observed effect is due to inhibited inflammasome activation, blocked phagosome maturation, or other factors. The results suggest that BafA effects are upstream of caspase-1, since addition of the vATPase blocks pyroptotic cell death in macrophages cocultured with the fungus and caspase-1 inhibitors have no further effect. We speculate that pharmacological neutralization of the phagosome disrupts the maturation of phagosomes containing the fungus, inflammasome activation driven by C. albicans, or both. Indeed, BafA treatment shifts C. albicans-induced death to other mechanisms, such as physical rupture following hyphal growth, lysosome-mediated cell death, or apoptosis. Neutralization of the macrophage by the fungal pathogen, on the other hand, occurs in mature phagolysosomes and results in hyphal morphogenesis-triggered IL-1β release. Whether the NLRP3 inflammasome is activated in response to pH-sensitive factors is yet to be determined.
Pyroptosis is a lytic cell death pathway in which the infected macrophages deprive intracellular pathogens of a potentially protective niche, including intracellular nutrients, while recruiting additional immune effectors (44), and many bacterial pathogens have developed mechanisms to modulate inflammasome activation. For example, Mycobacterium tuberculosis inhibits inflammasome activation by secreting the zinc metalloprotease Zmp1 (45), while the facultative zoonotic intracellular pathogen Francisella tularensis suppresses this process via the lipid/polysaccharide transporter protein MviN (46, 47). Virulent C. albicans strains, on the other hand, have a completely different strategy since they trigger pyroptotic cell death upon phagocytosis. This could be due to the transient interaction of the fungal pathogen with the macrophages, which results in rupture of the immune cell and release of the intracellular contents, an event that inevitably triggers activation of immune responses. Therefore, the strategy of C. albicans might be to trigger various cell death mechanisms, including pyroptosis, in order to expedite host cell destruction. How and if this contributes to survival in the host are yet to be determined.
MATERIALS AND METHODS
Strains and growth media.
C. albicans strains were propagated under standard conditions in yeast extract-peptone-glucose (YPD) medium (1% yeast extract, 2% peptone, 2% glucose) and are described in Table S1 in the supplemental material. For growth on plates, 2% agar was added to the medium. To select for nourseothricin (Nou)-resistant (Nour) transformants, 200 μg/ml of nourseothricin (Werner Bioagents, Jena, Germany) was added to the YPD agar plates (22). Alkalinization experiments were performed in minimal yeast nitrogen base (YNB) medium (0.17% yeast nitrogen base, 0.5% ammonium sulfate) prepared without glucose and supplemented with 1% Casamino Acids as the sole carbon source. Alkalinization on solid medium was performed in YNB medium with allantoin as the nitrogen source (0.17% yeast nitrogen base, 0.5% allantoin) and 1% Casamino Acids as the sole carbon source (YAC medium) plus 2% agar. J774A.1 macrophages were maintained in RPMI medium with glutamine and HEPES supplemented with penicillin-streptomycin and 10% inactivated fetal bovine serum. Measurement of intraphagosomal pH was performed in J774A.1 macrophages, which were maintained in phenol red-free RPMI medium with glutamine and HEPES supplemented with 10% inactivated fetal bovine serum. Assessment of macrophage lysis and release of IL-1β was performed entirely in phenol red-free RPMI medium with glutamine and HEPES, stimulated overnight with 100 mg/ml lipopolysaccharide (LPS).
Strain construction.
C. albicans strains lacking AHR1 were generated using the SAT1 flipper method (22). In short, approximately 300 bp of homologous sequence immediately to the 5′ or 3′ of the STP2 open reading frame (ORF) was amplified by PCR and cloned, using an overlap PCR approach, between the ApaI/XhoI and SacII/SacI sites of pSFSII, respectively. The resulting SAT1-FLP cassette was used to transform C. albicans SC5314 and stp2Δ (strain SVC17) cells by electroporation with selection on YPD-Nou. Genomic DNA was isolated, and cassette integration into the selected candidates was confirmed via PCR. To eliminate the nourseothricin selection marker, the mutant strain was induced in YNB medium supplemented with 1% maltose overnight, and the nourseothricin-sensitive (Nous) colonies were selected. This process was repeated to generate two independent homozygous disruptants of the AHR1 gene in a wild-type background, SVC22 and SVC23 (ahr1Δ::FTR/ahr1Δ::FTR), and in the stp2Δ strain background, SVC25 and SVC26 (ahr1Δ::FTR/ahr1Δ::FTR/stp2Δ::FTR/stp2Δ::FTR/stp2Δ::FTR).
Complementation of the mutant strain was performed by cloning the entire AHR1 ORF into the ApaI and XhoI sites of pAG6, a SAT1-marked version of CIp10. This plasmid was linearized with StuI and used to transform ahr1Δ mutant cells to generate strain SVC27 (RPS10/rps10::CIp10-AHR1-SAT1).
Alkalinization and ammonia release assays.
Alkalinization experiments were performed as previously described (8, 12) in YNB medium, pH 4.0, with 1% Casamino Acids as the sole carbon source. C. albicans cells were grown in YPD medium overnight, washed, and diluted to an optical density at 600 nm (OD600) of 0.2 in the alkalinization medium. Cells were incubated at 37°C with aeration for up to 24 h. Growth was measured via determination of the OD600, and the culture pH was measured using a standard pH electrode.
Ammonia release by C. albicans cells was assessed via an acid trap assay as previously described (12). In brief, cells were grown in rich medium overnight, washed in distilled H2O, and resuspended to an OD600 of 1.0. Five microliters from the cell suspensions was spotted onto solid YAC medium, pH 4.0. Reservoirs containing 10% citric acid were affixed to the petri dish lid directly underneath the colonies. Cells were incubated at 37°C, and samples from the acid trap were collected at the indicated times. Ammonia was quantified using Nessler's reagent. Experiments were performed at least in triplicate, and the data were analyzed using Prism (version 5.0; GraphPad) software.
Hyphal morphogenesis of phagocytosed C. albicans cells.
To assess the interaction of single C. albicans cells with the macrophages, we seeded 5 × 105 J774A.1 macrophages to glass coverslips in a 12-well plate and incubated them overnight in RPMI medium at 37°C in 5% CO2. C. albicans cells were grown in YPD medium overnight, diluted 1:100 in fresh YNB medium, and grown for 3 h at 30°C. Cells were then washed in distilled water, resuspended in phosphate-buffered saline (PBS), and stained with 1 μM FITC-concanavalin A for 15 min, followed by extensive washing in PBS. C. albicans cells were cocultured with the macrophages at a 1:1 ratio at 37°C for 90 min. Nonphagocytosed C. albicans cells were stained with 35 μg/ml calcofluor white, followed by three washes with PBS to remove the excess dye. The cocultures were then fixed with 2.7% paraformaldehyde for 20 min at room temperature. Images of the Candida-macrophage interaction were taken using an Olympus IX81 automated inverted microscope. The cellular morphology of the C. albicans cells was scored manually (by counting the number of yeast cells versus the number of germinated cells). To calculate the hyphal length, we used SlideBook (version 6.0) software, where we drew a line from the middle of the cell to the tip of the emerging hyphae. At least 50 cells per strain were scored. Results from at least 3 experiments were analyzed using a one-way analysis of variance (ANOVA) paired with Tukey's multiple-comparison test using Prism (version 5.0) software.
Macrophage lysis and IL-1β release assays.
C. albicans-induced macrophage lysis was assessed by measuring the release of lactate dehydrogenase (LDH) using a CytoTox96 kit (Promega) as previously described (8). Briefly, J774A.1 macrophages were seeded at 2.5 × 105 cells per well of a 96-well plate in RPMI medium containing 50 ng/ml LPS and incubated overnight at 37°C in 5% CO2. Then, the medium was replaced by phenol red-free RPMI medium with or without 100 μM the caspase-1 inhibitor z-YVAD-fmk (APExBIO, Houston, TX) and the macrophages were incubated for 2 h. C. albicans cells were grown to log phase in YNB medium, washed in PBS, and cocultured with macrophages at a 3:1 ratio for 5 h. The vATPase inhibitor BafA was added at a final concentration of 50 nM 30 min prior to coculture and removed at 1 h of coculture, where indicated above, followed by incubation in BafA-free medium. At the end of the 5-h coculture period, a 40-μl aliquot of the culture supernatant from each well was transferred into a 96-well plate for analysis of LDH release and another 40-μl aliquot was transferred into a separate 96-well plate to be analyzed further for IL-1β release using a ReadySetGo ELISA kit (eBioscience, San Diego, CA). Samples for cytokine analysis were frozen at −20°C until use. The IL-1β concentration in the coculture supernatants was determined according to the manufacturer's protocol. The amount of LDH released by infected macrophages relative to the maximum amount of LDH released from lysed macrophages was then calculated according to the manufacturer's protocol and corrected for the amount of LDH spontaneously released by the macrophages or C. albicans alone. The experiment was performed in triplicate, and the data were analyzed with a one-way ANOVA paired with Tukey's multiple-comparison test using Prism (version 5.0) software.
LR assays.
LysoTracker red (LR) assays were performed as previously described (12), with the following modifications. J774A.1 macrophages were seeded onto glass coverslips in 12-well tissue culture plates at 5 × 105 cells/ml and allowed to adhere overnight at 37°C in 5% CO2. Next, 50 nM LR DM99 (Molecular Probes) was added to fresh RPMI medium, and the mixture was incubated for 2 h. The vATPase inhibitor BafA was added to the macrophages at a final concentration of 50 nM 30 min prior to the coculture, where indicated above. C. albicans cells were grown overnight in YPD medium, diluted 1:100 in fresh YNB medium, and grown for 3 h at 30°C. Cells were then washed in PBS, stained with 1 μM FITC for 15 min, and washed in PBS to remove excess dye. Cells were diluted to 1 × 106 cells/ml in phenol red-free RPMI medium without serum and cocultured with macrophages for 60 min. Cultures were stained with calcofluor white (35 μg/ml for 30 s) to label nonphagocytosed cells, washed extensively in PBS, and then fixed in 2.7% paraformaldehyde. The cocultures were then imaged at ×60 magnification. To estimate the relative phagosomal pH, the signal intensities of both FITC and tetramethyl rhodamine isocyanate (TRITC) were plotted along a line drawn transversely across the short axis of the cell using SlideBook (version 6.0) software. The average LR signal intensity was calculated for a region of 10 pixels (1 μm) immediately outside the fungal cell, whose boundary was determined by the slope of the FITC signal. The results from 3 independent experiments, each consisting of least 50 cells per condition, were plotted in a box-and-whiskers plot, where the boxes were set at 25 to 75% and the whiskers were set at 5 to 95%. The plotted data were analyzed using a one-way ANOVA with Tukey's multiple-comparison test.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to M. Wellington for the helpful technical guidance and to M. Gustin, E. Garbe, R. Martin, and other members of the M. C. Lorenz lab for the insightful discussions.
This work was supported by NIH award R01AI075091 to M.C.L.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00832-16.
REFERENCES
- 1.Lorenz MC, Bender JA, Fink GR. 2004. Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryot Cell 3:1076–1087. doi: 10.1128/EC.3.5.1076-1087.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramirez MA, Lorenz MC. 2007. Mutations in alternative carbon utilization pathways in Candida albicans attenuate virulence and confer pleiotropic phenotypes. Eukaryot Cell 6:280–290. doi: 10.1128/EC.00372-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barelle CJ, Priest CL, MacCallum DM, Gow NA, Odds FC, Brown AJ. 2006. Niche-specific regulation of central metabolic pathways in a fungal pathogen. Cell Microbiol 8:961–971. doi: 10.1111/j.1462-5822.2005.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piekarska K, Mol E, van den Berg M, Hardy G, van den Burg J, van Roermund C, MacCallum D, Odds F, Distel B. 2006. Peroxisomal fatty acid beta-oxidation is not essential for virulence of Candida albicans. Eukaryot Cell 5:1847–1856. doi: 10.1128/EC.00093-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ene IV, Adya AK, Wehmeier S, Brand AC, MacCallum DM, Gow NA, Brown AJ. 2012. Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen. Cell Microbiol 14:1319–1335. doi: 10.1111/j.1462-5822.2012.01813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu W, Solis NV, Ehrlich RL, Woolford CA, Filler SG, Mitchell AP. 2015. Activation and alliance of regulatory pathways in C. albicans during mammalian infection. PLoS Biol 13:e1002076. doi: 10.1371/journal.pbio.1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zakikhany K, Naglik JR, Schmidt-Westhausen A, Holland G, Schaller M, Hube B. 2007. In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cell Microbiol 9:2938–2954. doi: 10.1111/j.1462-5822.2007.01009.x. [DOI] [PubMed] [Google Scholar]
- 8.Vylkova S, Lorenz MC. 2014. Modulation of phagosomal pH by Candida albicans promotes hyphal morphogenesis and requires Stp2p, a regulator of amino acid transport. PLoS Pathog 10:e1003995. doi: 10.1371/journal.ppat.1003995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danhof HA, Vylkova S, Vesely EM, Ford AE, Gonzalez-Garay M, Lorenz MC. 2016. Robust extracellular pH modulation by Candida albicans during growth in carboxylic acids. mBio 7:e01646-16. doi: 10.1128/mBio.01646-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naseem S, Araya E, Konopka JB. 2015. Hyphal growth in Candida albicans does not require induction of hyphal-specific gene expression. Mol Biol Cell 26:1174–1187. doi: 10.1091/mbc.E14-08-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vylkova S, Carman AJ, Danhof HA, Collette JR, Zhou H, Lorenz MC. 2011. The fungal pathogen Candida albicans autoinduces hyphal morphogenesis by raising extracellular pH. mBio 2:e00055-11. doi: 10.1128/mBio.00055-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danhof HA, Lorenz MC. 2015. The Candida albicans ATO gene family promotes neutralization of the macrophage phagolysosome. Infect Immun 83:4416–4426. doi: 10.1128/IAI.00984-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miramon P, Lorenz MC. 2016. The SPS amino acid sensor mediates nutrient acquisition and immune evasion in Candida albicans. Cell Microbiol 18:1611–1624. doi: 10.1111/cmi.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wellington M, Koselny K, Sutterwala FS, Krysan DJ. 2014. Candida albicans triggers NLRP3-mediated pyroptosis in macrophages. Eukaryot Cell 13:329–340. doi: 10.1128/EC.00336-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uwamahoro N, Verma-Gaur J, Shen HH, Qu Y, Lewis R, Lu J, Bambery K, Masters SL, Vince JE, Naderer T, Traven A. 2014. The pathogen Candida albicans hijacks pyroptosis for escape from macrophages. mBio 5:e00003-14. doi: 10.1128/mBio.00003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O'Meara TR, Veri AO, Ketela T, Jiang B, Roemer T, Cowen LE. 2015. Global analysis of fungal morphology exposes mechanisms of host cell escape. Nat Commun 6:6741. doi: 10.1038/ncomms7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, Fitzgerald KA. 2009. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5:487–497. doi: 10.1016/j.chom.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V, Mocsai A, Tschopp J, Ruland J. 2009. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459:433–436. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 19.Askew C, Sellam A, Epp E, Mallick J, Hogues H, Mullick A, Nantel A, Whiteway M. 2011. The zinc cluster transcription factor Ahr1p directs Mcm1p regulation of Candida albicans adhesion. Mol Microbiol 79:940–953. doi: 10.1111/j.1365-2958.2010.07504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hernday AD, Lohse MB, Fordyce PM, Nobile CJ, DeRisi JL, Johnson AD. 2013. Structure of the transcriptional network controlling white-opaque switching in Candida albicans. Mol Microbiol 90:22–35. doi: 10.1111/mmi.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Homann OR, Dea J, Noble SM, Johnson AD. 2009. A phenotypic profile of the Candida albicans regulatory network. PLoS Genet 5:e1000783. doi: 10.1371/journal.pgen.1000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reuss O, Vik A, Kolter R, Morschhauser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127. doi: 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 23.Wellington M, Koselny K, Krysan DJ. 2012. Candida albicans morphogenesis is not required for macrophage interleukin 1beta production. mBio 4:e00433-12. doi: 10.1128/mBio.00433-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis LE, Bain JM, Lowes C, Gillespie C, Rudkin FM, Gow NA, Erwig LP. 2012. Stage specific assessment of Candida albicans phagocytosis by macrophages identifies cell wall composition and morphogenesis as key determinants. PLoS Pathog 8:e1002578. doi: 10.1371/journal.ppat.1002578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fonzi WA. 2009. The protein secretory pathway of Candida albicans. Mycoses 52:291–303. doi: 10.1111/j.1439-0507.2008.01673.x. [DOI] [PubMed] [Google Scholar]
- 26.Brown AJ, Brown GD, Netea MG, Gow NA. 2014. Metabolism impacts upon Candida immunogenicity and pathogenicity at multiple levels. Trends Microbiol 22:614–622. doi: 10.1016/j.tim.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watson RT, Noble IR. 2005. The global imperative and policy for carbon sequestration. SEB Exp Biol Ser 2005:1–17. [PubMed] [Google Scholar]
- 28.Hube B, Monod M, Schofield DA, Brown AJ, Gow NA. 1994. Expression of seven members of the gene family encoding secretory aspartyl proteinases in Candida albicans. Mol Microbiol 14:87–99. doi: 10.1111/j.1365-2958.1994.tb01269.x. [DOI] [PubMed] [Google Scholar]
- 29.Naglik JR, Challacombe SJ, Hube B. 2003. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev 67:400–428. doi: 10.1128/MMBR.67.3.400-428.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pascon RC, Ganous TM, Kingsbury JM, Cox GM, McCusker JH. 2004. Cryptococcus neoformans methionine synthase: expression analysis and requirement for virulence. Microbiology 150:3013–3023. doi: 10.1099/mic.0.27235-0. [DOI] [PubMed] [Google Scholar]
- 31.Kingsbury JM, Goldstein AL, McCusker JH. 2006. Role of nitrogen and carbon transport, regulation, and metabolism genes for Saccharomyces cerevisiae survival in vivo. Eukaryot Cell 5:816–824. doi: 10.1128/EC.5.5.816-824.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez FJ, Safrin S, Weycker D, Starko KM, Bradford WZ, King TE Jr, Flaherty KR, Schwartz DA, Noble PW, Raghu G, Brown KK, IPF Study Group . 2005. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med 142:963–967. doi: 10.7326/0003-4819-142-12_Part_1-200506210-00005. [DOI] [PubMed] [Google Scholar]
- 33.Lorenz MC, Fink GR. 2001. The glyoxylate cycle is required for fungal virulence. Nature 412:83–86. doi: 10.1038/35083594. [DOI] [PubMed] [Google Scholar]
- 34.Fradin C, De Groot P, MacCallum D, Schaller M, Klis F, Odds FC, Hube B. 2005. Granulocytes govern the transcriptional response, morphology and proliferation of Candida albicans in human blood. Mol Microbiol 56:397–415. doi: 10.1111/j.1365-2958.2005.04557.x. [DOI] [PubMed] [Google Scholar]
- 35.Rubin-Bejerano I, Fraser I, Grisafi P, Fink GR. 2003. Phagocytosis by neutrophils induces an amino acid deprivation response in Saccharomyces cerevisiae and Candida albicans. Proc Natl Acad Sci U S A 100:11007–11012. doi: 10.1073/pnas.1834481100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tillmann AT, Strijbis K, Cameron G, Radmaneshfar E, Thiel M, Munro CA, MacCallum DM, Distel B, Gow NA, Brown AJ. 2015. Contribution of Fdh3 and Glr1 to glutathione redox state, stress adaptation and virulence in Candida albicans. PLoS One 10:e0126940. doi: 10.1371/journal.pone.0126940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jimenez-Lopez C, Collette JR, Brothers KM, Shepardson KM, Cramer RA, Wheeler RT, Lorenz MC. 2013. Candida albicans induces arginine biosynthetic genes in response to host-derived reactive oxygen species. Eukaryot Cell 12:91–100. doi: 10.1128/EC.00290-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seider K, Heyken A, Luttich A, Miramon P, Hube B. 2010. Interaction of pathogenic yeasts with phagocytes: survival, persistence and escape. Curr Opin Microbiol 13:392–400. doi: 10.1016/j.mib.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Gow NA, Hube B. 2012. Importance of the Candida albicans cell wall during commensalism and infection. Curr Opin Microbiol 15:406–412. doi: 10.1016/j.mib.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 40.McKenzie CG, Koser U, Lewis LE, Bain JM, Mora-Montes HM, Barker RN, Gow NA, Erwig LP. 2010. Contribution of Candida albicans cell wall components to recognition by and escape from murine macrophages. Infect Immun 78:1650–1658. doi: 10.1128/IAI.00001-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rajamaki K, Nordstrom T, Nurmi K, Akerman KE, Kovanen PT, Oorni K, Eklund KK. 2013. Extracellular acidosis is a novel danger signal alerting innate immunity via the NLRP3 inflammasome. J Biol Chem 288:13410–13419. doi: 10.1074/jbc.M112.426254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mazzolla R, Barluzzi R, Brozzetti A, Boelaert JR, Luna T, Saleppico S, Bistoni F, Blasi E. 1997. Enhanced resistance to Cryptococcus neoformans infection induced by chloroquine in a murine model of meningoencephalitis. Antimicrob Agents Chemother 41:802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hair PS, Foley CK, Krishna NK, Nyalwidhe JO, Geoghegan JA, Foster TJ, Cunnion KM. 2013. Complement regulator C4BP binds to Staphylococcus aureus surface proteins SdrE and Bbp inhibiting bacterial opsonization and killing. Results Immunol 3:114–121. doi: 10.1016/j.rinim.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jorgensen I, Miao EA. 2015. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev 265:130–142. doi: 10.1111/imr.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, Timmins GS, Sander P, Deretic V. 2008. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 3:224–232. doi: 10.1016/j.chom.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES. 2010. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O'Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM. 2010. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A 107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.