

Significance
Ovulation is thought to be divergent from insects to mammals. In the latter case, ovulation is regulated by luteinizing hormone and the steroid hormone progesterone, neither of which is found in insects so far. This paper reports a role of ecdysteroids in Drosophila ovulation. Similar to the action of progesterone in mammals, 20-hydroxyecdysone produced in Drosophila follicle cells activates one of its receptors, EcR.B2, to allow proper activation of matrix metalloproteinase and follicle rupture. The conservation of steroid signaling in ovulation makes Drosophila a simple model to define the mechanism of steroid action in ovulation.
Keywords: steroid signaling, ecdysone, ovulation, Shade, EcR isoforms
Abstract
Although ecdysteroid signaling regulates multiple steps in oogenesis, it is not known whether it regulates Drosophila ovulation, a process involving a matrix metalloproteinase-dependent follicle rupture. In this study, we demonstrated that ecdysteroid signaling is operating in mature follicle cells to control ovulation. Moreover, knocking down shade (shd), encoding the monooxygenase that converts ecdysone (E) to the more active 20-hydroxyecdysone (20E), specifically in mature follicle cells, blocked follicle rupture, which was rescued by ectopic expression of shd or exogenous 20E. In addition, disruption of the Ecdysone receptor (EcR) in mature follicle cells mimicked shd-knockdown defects, which were reversed by ectopic expression of EcR.B2 but not by EcR.A or EcR.B1 isoforms. Furthermore, we showed that ecdysteroid signaling is essential for the proper activation of matrix metalloproteinase 2 (Mmp2) for follicle rupture. Our data strongly suggest that 20E produced in follicle cells before ovulation activates EcR.B2 to prime mature follicles to be responsive to neuronal ovulatory stimuli, thus providing mechanistic insights into steroid signaling in Drosophila ovulation.
Ovulation is crucial for reproduction and requires the proteolytic breakdown of the ovarian follicle for releasing a fertilizable oocyte (1, 2). This process consists of a complex series of events tightly coordinated by a network of paracrine and endocrine signals, most notably in mammals by steroid progesterone signaling, in mature follicles (3–5). Pharmacological inhibition of progesterone synthesis or genetic disruption of the progesterone receptor results in an ovulation failure due to a lack of follicle wall proteolysis or follicle rupture (6, 7). The signaling mechanism by which progesterone regulates a spatiotemporal proteolysis leading to a successful follicle rupture is largely unknown.
Recent work in Drosophila has demonstrated some striking similarities between Drosophila and mammalian ovulation. Drosophila ovaries are organized into ovarioles, which consist of a string of egg chambers developing through 14 different stages into mature follicles (stage 14) (8). Like mammals, mature oocytes in Drosophila are wrapped in a layer of somatic follicle cells. During ovulation, posterior follicle cells are partially broken down to allow oocytes to be released into the oviduct, whereas the residual follicle cells develop into a corpus luteum (9). This follicle rupture also relies on proteolytic enzymes, like matrix metalloproteinase 2 (Mmp2), which is specifically expressed in the posterior follicle cells of stage-14 egg chambers (9). Mmp2 activation is stimulated by the monoamine octopamine (OA), which is equivalent to norepinephrine in mammals and is essential for ovulation (10, 11). OA binds to its receptor Octopamine receptor in mushroom body (Oamb) in mature follicle cells to induce an intracellular calcium rise, Mmp2 activation, and follicle rupture (12). Drosophila ovulation is also regulated by multiple ovarian extrinsic factors, including secretions from the oviduct (13), female reproductive glands (14), and male accessory glands (15, 16). However, a role for steroid signaling in Drosophila ovulation has never been established.
In Drosophila, the major steroid hormones are ecdysteroids, including ecdysone (E) and the most biologically active form 20-hydroxyecdysone (20E). Many enzymes involved in the ecdysone biosynthesis pathway have been identified, including Phantom (Phm), Disembodied (Dib), Shadow (Sad), and Shade (Shd), which carry out the final four steps of 20E synthesis (17–20). In addition, enzymes encoded by spook (spo) and shroud (sro) act more upstream for chemical reactions designated as the black box (21, 22). Ecdysteroids signal through a heterodimeric nuclear receptor complex comprised of Ultraspiracle (Usp) and Ecdysone receptor (EcR) (23, 24). The latter has three alternative splicing isoforms (EcR.A, EcR.B1, and EcR.B2), which vary at their N-terminal regions (25). All three isoforms have identical DNA binding and ligand binding sequences, but differ in transcriptional activation and repression (26). Ecdysteroids have long been known to play essential roles in developmental transitions, such as larval molting and metamorphosis (27, 28). In contrast, the roles of ecdysteroids in adult physiology have just started to emerge (29–39).
In this study, we characterized the role of ecdysteroid signaling in late oogenesis, mainly as a key component in mediating ovulation. The enzyme Shd, which converts E to 20E, is dramatically up-regulated in mature follicle cells. Disruption of ecdysteroid signaling in these follicle cells blocked OA-induced follicle rupture and ovulation. Ecdysteroid signaling is essential for proper activation of Mmp2 enzymatic activity. Our work thus demonstrates the role of ecdysteroid signaling in Drosophila ovulation, reminiscent to progesterone signaling in mammalian ovulation.
Results
E-20-Monooxygenase Shade Is Required in Mature Follicle Cells for Ovulation.
Shd, the E-20-monooxygenase, converts E to 20E to elicit steroid signaling during early development (19). To investigate the role of steroid signaling in ovulation, we characterized the expression pattern of Shd in late oogenesis. Shd is enriched in follicle cells at stage 10 (Fig. 1A) and down-regulated to an undetectable level at stage 13 (Fig. 1B). At stage 14, Shd is dramatically up-regulated in all follicle cells (Fig. 1C).
Fig. 1.

Shade is required in mature follicle cells for ovulation. (A–D) Shd protein expression (green) in egg chambers of control (A–C; R44E10-Gal4 driving UAS-RFP, 44E10 > RFP) and shd-knockdown (D; 44E10 > shd-i2) flies. RFP is in red, and DNA staining with DAPI is shown in blue. The stage of the egg chambers is indicated, and the Insets show the higher magnification of green channel in the outlined area. (E–H) The quantification of egg-laying number (E and F), egg-laying time (G), and mature follicles (H) with specified genotypes. R44E10-Gal4 was used. The number of females analyzed is shown in parentheses, and the error bars are SE for egg-laying number (E and F), 95% confidence interval for egg-laying time (G), and SD for mature follicles (H). Table S1 for statistical analysis. ***P < 0.001, **P < 0.01, and *P < 0.05.
This prominent up-regulation of Shd in mature follicle cells prompted us to investigate its role in ovulation. With R44E10-Gal4 transcription factor driving UAS-shdRNAi expression in mature follicle cells (12), shd expression is severely knocked down in mature follicles but normal in early egg chambers (Fig. 1D and Fig. S1). Female flies with shd knockdown laid significantly fewer eggs in 2 d (Fig. 1E). This egg-laying defect can be completely rescued by ectopic expression of shd in mature follicle cells (Fig. 1F). A similar egg-laying defect was observed when shd was knocked down by R47A04-Gal4 (Fig. S2), another Gal4 line overlapped with R44E10-Gal4 in mature follicle cells (9). These data suggest that Shd functions in mature follicle cells to regulate egg laying.
Fig. S1.
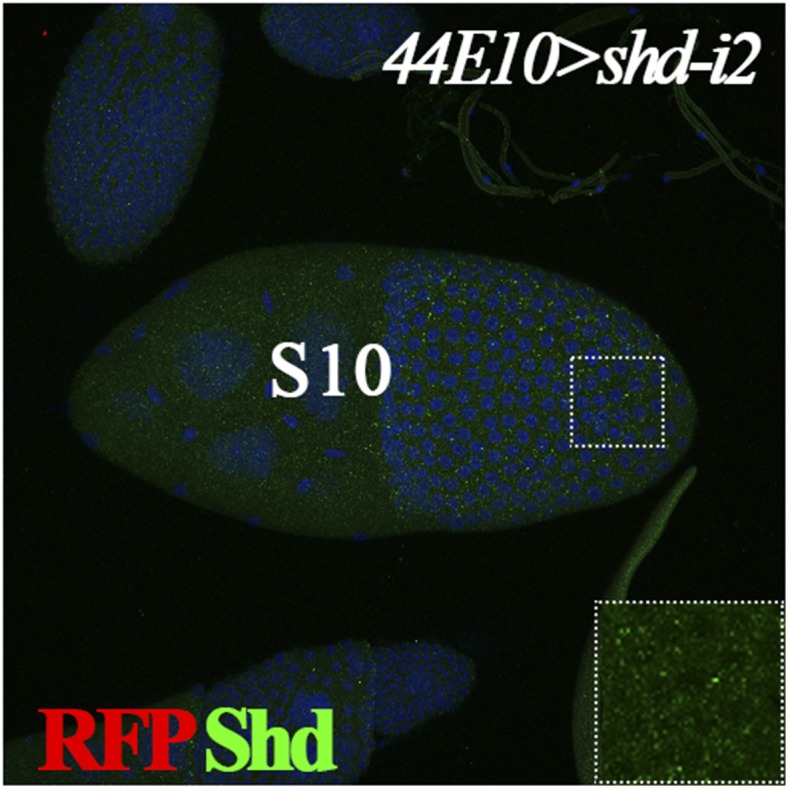
Shd expression in stage-10 egg chamber. Expression of Shd (green) is normal in stage-10 follicles from females with shd knockdown using R44E10-Gal4. R44E10-Gal4 is not expressed in stage 10 as indicated by UAS-RFP expression (in red). DNA staining with DAPI is shown in blue. Inset shows the higher magnification of green channel in the outlined area.
Fig. S2.

Fecundity of females with shd knockdown using R47A04-Gal4. Females with shd knockdown lay significantly less eggs than control. Student’s t test was used (***P < 0.001, *P < 0.05).
The egg laying consists of ovulation (releasing eggs from follicles), egg transport through the oviduct, and oviposition (releasing eggs from the uterus to the external environment). To determine which steps are altered when shd is knocked down, we investigated the distribution of eggs in the female reproductive tract (Table S1) and calculated the average time of each egg spent in the ovulation, in the oviduct, or in the uterus. When shd was knocked down, the time required for ovulation was significantly longer than controls (Fig. 1G and Table S1). The fourfold increase in ovulation time could not be solely explained by the slight reduction of mature follicles in the ovaries (Fig. 1H), but rather by an ovulation defect (see below). These data suggest that up-regulation of Shd in mature follicle cells is essential for normal ovulation.
Table S1.
Effect of ecdysteroid signaling on egg laying, egg distribution in the reproductive tract, and egg-laying time
| Genotype | Egg laying in 2 d† | Egg distribution in 6 h | Egg laying time, min | ||||||
| n | Eggs per female per day | n | Uterus with egg, % | Oviduct with egg, % | Total time | Ovulation time | Oviduct time | Uterus time | |
| UAS-dcr2/+; 44E10-Gal4/+ (Ore-R) | 100 | 74.2 ± 0.9 | 90 | 60.0 ± 10.1 | 5.6 ± 4.7 | 17.8 ± 0.2 | 6.1 ± 1.7 | 1.0 ± 0.8 | 10.7 ± 1.8 |
| UAS-dcr2/shd-i1; 44E10-Gal4/+ | 75 | 36.2 ± 1.4*** | 60 | 31.7 ± 11.8 | 5.0 ± 5.5 | 36.5 ± 1.4 | 23.1 ± 4.5*** | 1.8 ± 2.0 | 11.6 ± 4.3 |
| UAS-dcr2/+; 44E10-Gal4/shd-i2 | 75 | 37.6 ± 0.6*** | 60 | 21.7 ± 10.4 | 11.7 ± 8.1 | 35.1 ± 0.6 | 23.4 ± 4.2*** | 4.1 ± 2.9 | 7.6 ± 3.7 |
| UAS-dcr2/phm-i; 44E10-Gal4/+ | 50 | 51.9 ± 0.6*** | 25 | 56.0 ± 19.5 | 8.0 ± 10.6 | 25.4 ± 0.3 | 9.2 ± 4.8 | 2.0 ± 2.7 | 14.2 ± 4.9 |
| UAS-dcr2/dib-i; 44E10-Gal4/+ | 25 | 50.6 ± 4.1*** | 28 | 46.4 ± 18.5 | 7.1 ± 9.5 | 26.1 ± 2.1 | 12.1 ± 4.9 | 1.9 ± 2.5 | 12.1 ± 4.9 |
| EcRA483T/SM6b | 50 | 82.9 ± 2.1 | 62 | 54.8 ± 12.4 | 3.2 ± 4.4 | 15.9 ± 0.4 | 6.7 ± 2.0 | 0.5 ± 0.7 | 8.7 ± 2.0 |
| EcRA483T/EcRM554fs | 45 | 19.6 ± 2.1*** | 46 | 23.9 ± 12.3 | 6.5 ± 7.1 | 67.5 ± 7.4 | 45.5 ± 10.4*** | 4.4 ± 4.8 | 16.1 ± 8.5 |
All data are mean ± 95% confidence interval. Student's t test was used for egg laying, and Z score test was used for egg laying time assuming normal distribution. ***P < 0.001.
One day = 22 h at 29 °C.
Shade Synthesizes 20-Hydroxyecdysone to Regulate Follicle Rupture.
Because follicle rupture can be recapitulated by in vitro culture of mature follicles with octopamine stimulation (12), we performed the in vitro assay to determine whether shd-knockdown follicles (shd follicles) are competent for OA-induced follicle rupture. Consistent with our previous results (12), 54% of control follicles had ruptured with a 3-h OA stimulation (Fig. 2 A and C). In contrast, fewer than 15% of shd follicles had ruptured (Fig. 2 B and C). The decreased rate of follicle rupture can be fully rescued by ectopic expression of shd (Fig. 2 D–F), consistent with the egg-laying results (Fig. 1F). Altogether, these data indicate that Shd in mature follicle cells is important for follicle rupture/ovulation. It is interesting to note that knocking down upstream enzymes for E synthesis in mature follicle cells only caused a moderate reduction of egg-laying number but no statistically significant defect in follicle rupture/ovulation (Figs. 1 E and G and 2C and Table S1). Unlike Shd, Phm was highly enriched in stage-13 follicle cells (Fig. S3 A and B). These data likely suggest that these upstream enzymes function in younger follicle cells for E synthesis.
Fig. 2.

Shade is required in mature follicle cells to synthesize 20E for follicle rupture. For all follicle rupture images here and in subsequent figures, RFP (shown in red) labeling mature follicle cells was overlaid to the bright-field image of follicles (shown in blue). (A–C) shd knockdown with R44E10-Gal4 inhibits follicle rupture. Representative images (A and B) show mature follicles after 3-h culture with OA. The number of mature follicles used in each genotype is 502, 243, 204, 142, 95, 315, 195, and 276. (D–F) Shd overexpression rescues the rupture defect of shd-knockdown follicles. The number of follicles is listed in the parentheses. (G–I) 20E but not E can partially rescue rupture defect of shd-knockdown follicles after a 6-h culture. The number of follicles used in each condition in I is 480, 263, 657, 423, 87, 564, 392, 210, and 563.
Fig. S3.

Phm expression in stage-13 and stage-14 follicle cells. Endoplasmic reticulum-localized Phm (red in A and B and white in A′ and B′) is highly enriched in stage-13 (A and A′) and stage-14 (B and B′) follicle cells. Transcription factor Hindsight (Hnt; green in A and B) is marking stage-14 follicle cells, but not in stage-13 follicle cells.
To determine whether Shd regulates follicle rupture through synthesizing 20E, we set to rescue the rupture defect of shd follicles with exogenous 20E. Interestingly, supplementing with 20 nM 20E, the typical concentration found in hemolymph (40), can significantly increase the OA-induced rupture to approximately 40% in shd follicles in 3 h; however, 100 nM 20E did not elicit a better rescue, if not worse (Fig. S4 A–C). Neither did the addition of 20E affect OA-induced follicle rupture in control follicles (Fig. S4 A and B), nor was 20E alone sufficient to induce follicle rupture (Fig. S4D). Because ecdysone signaling controls transcription, we reasoned that extending the culture period may have an improved rescue effect. Thus, we performed similar experiments and examined the follicles at the end of a 6-h culture. Consistent with this idea, we observed an increased rupture rate in shd follicles to more than 50% with 20 nM 20E supplement (Fig. 2 H and I and Fig. S4E). In contrast, supplementing with 20 nM E did not elicit any rescue effect (Fig. 2 G and I). Altogether, the rescue of the rupture defect in shd follicles with 20E but not E supports the notion that Shd functions in mature follicle cells to convert E to 20E to regulate follicle rupture.
Fig. S4.

Follicle rupture in the presence of OA and 20E. (A) Quantification of follicle rupture with OA and varying concentration of 20E after a 3-h culture. (B and C) Representative images show mature follicles of control (B) and shd knockdown (C) after 3-h culture with OA and 20 nM 20E. (D) Representative image of control follicles cultured in only 20E (20 nM) showing that 20E alone does not induce follicle rupture (3.1%, n = 159). (E) Quantification of follicle rupture with OA and varying concentrations of 20E after a 6-h culture. The groups with 0 and 20 nM 20E are the same as in Fig. 2I. Student’s t test was used (**P < 0.01; *P < 0.05; ns, not significant).
EcR Is Required in Mature Follicle Cells for Follicle Rupture/Ovulation.
In Drosophila, ecdysteroids signal through the EcR/Usp complex. To examine the role of EcR in ovulation, we used a combination of temperature-sensitive (ts) and null alleles of EcR (EcRts in short) (30), which allows follicles to develop into stage 14 normally when shifted to restrictive temperature after adult eclosion. EcRts females with such treatment had a significant reduction in the egg-laying number and a slight increase in mature follicles in ovaries (Fig. 3 A and B). In addition, EcRts females displayed a sevenfold increase in ovulation time compared with control females (Fig. 3C and Table S1), supporting a role for EcR in ovulation.
Fig. 3.

EcR is required in mature follicle cells for ovulation. (A–C) The quantification of egg-laying number (A), mature follicles (B), and egg-laying time (C) in heterozygous control or EcRts females. (D–F) EcRts mature follicles are defective in OA-induced follicle rupture. Mature follicles were marked by R47A04 > RFP. (G–I) EcRDN overexpression using R44E10-Gal4 inhibits follicle rupture. The number of females or mature follicles is listed in the parentheses. ***P < 0.001.
To confirm that EcR functions to regulate follicle rupture like Shd, we isolated mature follicles from EcRts females and performed in vitro follicle rupture. Mature follicles from EcRts females showed a significant reduction in follicle rupture in comparison with controls when stimulated with OA (Fig. 3 D–F). These results demonstrate a role for EcR in follicle rupture/ovulation.
To determine whether EcR directly functions in mature follicle cells to regulate follicle rupture, we specifically disrupted EcR function in these cells by using a dominant negative form of EcR (EcR.B1DN), which binds to ecdysone-responsive genes without activating their expression (41), and tested in vitro follicle rupture. Mature follicles from two different EcR.B1DN mutants ruptured at only one-third of the rate or less of control follicles when exposed to OA (Fig. 3 G–I). This result indicates a requirement of EcR in mature follicle cells for proper follicle rupture.
EcR.B2 but Not Other Isoforms Regulates Follicle Rupture.
The EcR gene encodes three protein isoforms (EcR.A, EcR.B1, and EcR.B2), with EcR.B2 having the shortest N terminus (25). To determine which isoform functions in mature follicle cells, we examined the EcR expression by using isoform-specific antibodies (25). EcR.B1 is highly enriched in follicle cell nuclei from stage 10 to stage 12 but down-regulated to an undetectable level from stage 13–14 (Fig. 4 A and B and Fig. S5A). EcR.A has a similar expression pattern as EcR.B1 and is also not detected in stage-14 follicle cells (Fig. 4 C and D and Fig. S5B). In contrast, the antibody recognizing all three isoforms detected EcR expression in follicle cells throughout oogenesis including stage 14 (Fig. 4 E and F and Fig. S5C). These data imply that EcR.B2 is likely the isoform expressed in mature follicle cells. Unfortunately, no EcR.B2-specific antibody has been generated to allow us to detect its expression pattern directly.
Fig. 4.

EcR.B2 functions in mature follicle cells for follicle rupture. (A and B) EcR.B1 protein expression (green) in late oogenesis. RFP expression driven by R44E10-Gal4 (44E10 > RFP) is shown in red, marking stage-14 egg chambers. DAPI is shown in blue. (C and D) EcR.A protein expression in late oogenesis. (E and F) EcR protein expression (including all three isoforms in green) in late oogenesis. (G) Quantification of OA-induced follicle rupture when EcR isoforms were knocked down with R44E10-Gal4. (H–J) Quantification of OA-induced rupture in follicles with misexpressing EcR isoforms and/or EcRDN. Representative images show mature follicles with R44E10-Gal4 driving EcR.B1DN2/EcR.B1 (I) and EcR.B1DN2/EcR.B2 (J) after the 3-h culture with OA. The number of follicles used in each condition is 172, 99, 265, 214, 230, 118, 187, and 183. ***P < 0.001; **P < 0.01; *P < 0.05; and ns, not significant.
Fig. S5.
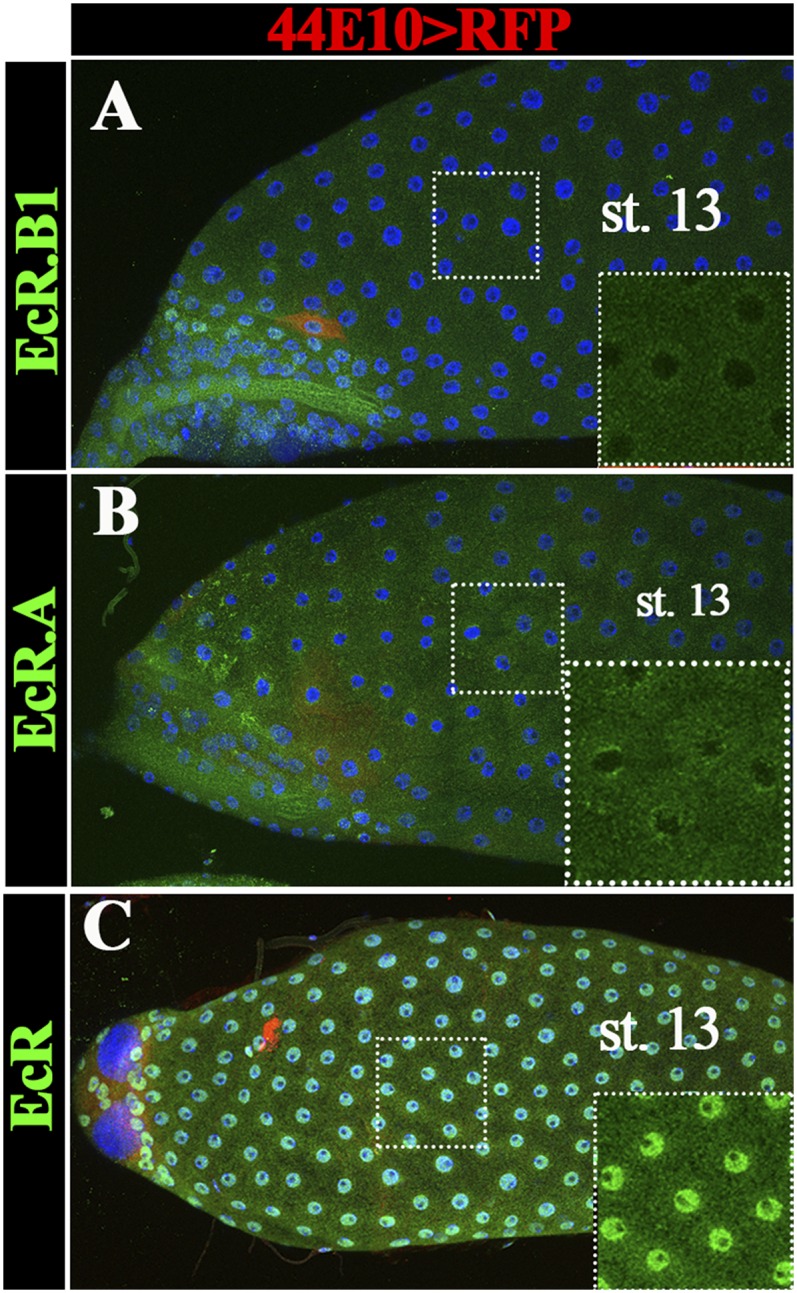
EcR isoform expression in stage-13 egg chambers. (A and B) EcR.B1 (A) and EcR.A (B) protein (green) are not expressed in stage-13 egg chambers. (C) EcR protein expression (including all three isoforms in green) is expressed in stage-13 egg chambers. R44E10-Gal4 is not expressed in stage 13 as indicated by UAS-RFP expression (in red). DNA staining with DAPI is shown in blue. Insets show the higher magnification of green channel in the outlined area.
To determine whether EcR.B2 functions in follicle rupture, we first used isoform-specific RNAi lines to knockdown EcR in mature follicle cells. EcR.A or EcR.B1 knockdown did not block OA-induced follicle rupture (Fig. 4G and Fig. S6A). No RNAi line targeting EcR.B2 is available. In contrast, RNAi targeting an EcR common region (knocking down all three isoforms) showed a decrease in follicle rupture rate (Fig. 4G and Fig. S6B). This result suggests that EcR.B2 or a combination of isoforms is required to regulate follicle rupture.
Fig. S6.

Knockdown of EcR-i causes follicle rupture defect. The representative image shows follicles after the 3-h OA stimulation. Mature follicles were expressing RNAi against either EcR.B1 (A) or EcR common region (B) in their follicle cells by using R44E10-Gal4.
To explicitly test whether 20E signals through the EcR.B2 isoform or a combination of isoforms to regulate follicle rupture, we aimed to rescue the rupture defect in EcR.B1DN mutants by overexpressing the individual EcR isoforms in mature follicle cells. Similar experiments have been performed and found that three EcR isoforms are interchangeable to support multiple organ development, whereas only EcR.A supports development of wing margins and EcR.B2 functions in larval epidermis and border cell migration (41). Interestingly, we found that overexpression of EcR.B2, but not EcR.B1 or EcR.A, completely rescued the OA-induced rupture defect seen in EcR.B1DN follicles (Fig. 4 H–J). In addition, overexpression of EcR.B2 alone in mature follicle cells did not interfere with OA-induced follicle rupture, whereas overexpression of EcR.B1 or EcR.A significantly decreased OA-induced rupture to less than 20% (Fig. 4H). Thus, the normal down-regulation of EcR.B1 and EcR.A in mature follicle cells is essential to allow proper ecdysteroid signaling for follicle development and rupture. In summary, our results demonstrate the requirement for EcR in mature follicle cells to regulate follicle rupture, and specifically, only the EcR.B2 isoform is sufficient to function in this process.
Ecdysteroid Signaling Regulates Follicle Rupture by Promoting Mmp2 Activation.
OA activates the Oamb receptor in mature follicle cells, which sequentially leads to an increase of intracellular calcium, activation of Mmp activity, and follicle rupture (Fig. 5A) (12). To determine whether ecdysteroid signaling interferes with this pathway to regulate follicle rupture, we measured the activation of Mmp2 in control and steroid-defective follicles with in situ zymography. Consistent with our previous report (12), approximately 80% of control follicles showed gelatinase activity at their posterior end after OA stimulation for 3 or 6 h; however, only 40% of shd follicles showed gelatinase activity (Fig. 5 B and D and Fig. S7 A–C). This reduction of gelatinase activity can be partially rescued by supplementing 20E in the culture medium (Fig. 5 C and D). In addition, EcRts follicles also showed reduced gelatinase activity in comparison with controls after OA stimulation (Fig. S7 D–F). These data suggest that follicles lacking ecdysteroid signaling are unable to efficiently activate Mmp2 in response to OA stimulation, which may explain their poor response to OA-induced follicle rupture and ovulation.
Fig. 5.

Ecdysteroid signaling regulates OA-induced Mmp2 activation. (A) A schematic showing the model of ecdysteroid signaling in the mature follicle cell to induce follicle rupture. (B–D) In situ zymography shows the reduction of gelatinase activity (green in B and C) in shd-knockdown follicles (B), which can be rescued by 20 nM 20E (C). Follicles with posterior gelatinase activity after a 6-h stimulation were separated toward the upper region in B and C and quantified in D. (E) Quantification of Oamb and mmp2 mRNA in mature follicles of control and EcRts females with qRT-PCR. ***P < 0.001.
Fig. S7.

Ecdysteroid signaling affects Mmp2 activation but not protein expression. (A–C) In situ zymography shows the reduction of gelatinase activity (green in A and B) in shd-knockdown follicles after a 3-h OA stimulation. Follicles with posterior gelatinase activity (green in A and B) are separated toward the upper part of A and B, and the quantification is shown in C. The number of follicles analyzed is in the parentheses. (D–F) In situ zymography shows the reduction of gelatinase activity in EcRts mutant follicles after a 3-h OA stimulation. (G–I) Mmp2::GFP expression (green in G–I and G”–I′′) is detected in posterior follicle cells of control (G–G′′), shd-knockdown (H–H′′), and EcRDN2 (I–I′′) stage-14 egg chambers.
To find out where ecdysteroids interfere with the OA/Oamb-Mmp2 pathway, we examined the Mmp2 expression level with an Mmp2::GFP fusion reporter and quantitative RT-PCR (qRT-PCR). Mmp2 expression is not affected by ecdysteroid signaling at either the mRNA or protein level (Fig. 5E and Fig. S7 G–I). In addition, Oamb mRNA expression is also normal when ecdysteroid signaling is defective (Fig. 5E). Thus, ecdysteroid signaling must regulate some other components in this pathway. Overall, we conclude that ecdysteroid signaling in mature follicles regulates follicle rupture by promoting Mmp2 activation (Fig. 5A).
Discussion
Ecdysteroids Regulate Ovulation in Adult Drosophila.
Our study defined the dynamic expression pattern of Shd and EcR isoforms in late oogenesis and demonstrated the requirement of ecdysteroid signaling in ovulation. Ecdysteroids have also been shown to regulate early germ cell differentiation (35, 36), egg chamber progression at stage 8–9 (29–31), and endocycle-to-gene-amplification switch at stage10B (33). This temporal action of ecdysteroids is reminiscent to their roles in regulating multiple larval and pupal transitions (27, 28). It is, however, unclear whether egg chambers receive pulse-like ecdysteroid titers at these transitions, similar to those received during molting and metamorphosis. Unlike early development, where ecdysone is produced in the ring gland, adult female ecdysteroids are thought to be produced mainly in the ovary, because mRNAs encoding many enzymes in the ecdysone synthesis pathway are detected in follicle cells, nurse cells, or both. Because ecdysteroid secretion is mediated through a regulated vesicular trafficking mechanism (42), it is conceivable that follicle cells, nurse cells, or both could produce local pulses of ecdysteroids to promote egg chamber development at each transition during oogenesis. This hypothesis is consistent with the finding that the ligand sensor for ecdysteroids shows a stage-specific activity including stage 10 and stage 14 (32, 33). This notion is further supported by this study that up-regulation of Shd in mature follicle cells produces 20E to regulate ovulation and a previous study that local ecdysteroids produced in follicle cells regulate border cell migration (37).
Interestingly, we only observed the requirement of Shd in mature follicle cells for normal ovulation, whereas the involvement of the early enzymes such as Sad, Dib, Phm, and Spo in mature follicle cells are not decisive. It is unlikely that Shd monooxygenates another molecule, instead of E, which regulates ovulation, because 20E can rescue the defect of shd-knockdown follicles. We favor the hypothesis that early enzymes for E synthesis function in younger follicle cells (such as stage 13 where Phm protein is highly expressed; Fig. S3 A and B), which carry E into stage 14, where Shd converts them to 20E. Alternatively, E may be transported into mature follicle cells from outside, possibly from the corpus luteum where Phm is also expressed (9). It is also possible that the differential effect of shd and earlier enzymes in follicle rupture/ovulation reflects differences in the RNAi knockdown efficiency.
Isoform-Specific Roles of Ecdysone Receptor.
EcR is a typical nuclear receptor belonging to the NR1 family (43). The single EcR gene produces three alternatively spliced isoforms with variable N-terminal regions but the same C terminus, giving them the capacity to bind to the same ligands and DNA sequences. Here, we showed that the regulation of follicle rupture is not only through up-regulating 20E production in stage-14 follicles, but also likely by regulating EcR isoform expression. We discovered that both EcR.A and EcR.B1 are down-regulated before entering stage 14, unlike a previous report that both isoforms are expressed throughout oogenesis (30). This down-regulation is likely important for follicle rupture because ectopic expression of EcR.A or EcR.B1 interferes with the OA-induced follicle rupture. Our data are consistent with the idea that EcR.B2 is the only EcR isoform functioning in mature follicle cells to mediate the ecdysteroid signaling necessary for ovulation. Thus, upon 20E binding, EcR.B2 may regulate specific arrays of genes for ovulation that cannot be activated or will be interfered by other EcR isoforms. Isoform specific roles of EcR have also been reported in wing margin development and border cell migration (41); however, follicle rupture is a biological process showing interference between EcR isoforms. The specific genes regulated by EcR.B2 and how EcR.B2 fulfills such specific functions are unknown.
Conserved Role of Steroids in Ovulation.
The mechanisms underlying ovulation in insects, such as Drosophila, were thought to be highly divergent from mammals, in which ovulation is regulated by luteinizing hormone and progesterone, which has not been identified in insects so far. However, our recent work demonstrated the involvement of follicle rupture and matrix metalloproteinase in Drosophila ovulation, reminiscent to mammalian ovulation (1, 9, 12). In addition, it seems that adrenergic regulation of calcium signaling is also conserved in regulating ovulation in both Drosophila and mammals (12, 44, 45). The steroid hormone progesterone and progesterone receptor signaling in preovulatory follicles is essential for mammalian ovulation (6). It remained a mystery whether steroid signaling also plays a conserved role in Drosophila ovulation. Our study solved this mystery by demonstrating an important role of ecdysteroids, the principal steroid hormones of Drosophila, in mature follicle cells for follicle rupture/ovulation. Like preovulatory follicles that only transiently up-regulate progesterone receptor in granulosa cells before ovulation (46), mature follicles of Drosophila also adjust their expression of EcR receptors to allow EcR.B2 to remain functioning and promote ovulation. It is interesting that neither progesterone nor ecdysteroid signaling regulates mmp expression (Fig. 5E) (47). The downstream targets of ecdysteroid signaling in ovulation are unclear; however, it is possible that ecdysteroids may regulate other proteases enriched in late oogenesis as progesterone signaling does (47). Future work on ecdysteroid signaling in Drosophila ovulation will thus provide insights into fundamental mechanisms of steroid signaling in ovulation.
Materials and Methods
Drosophila Genetics.
Flies were reared on standard cornmeal-molasses food at 25 °C unless otherwise indicated. EcRts is a transheterozygous combination of EcRA483T/EcRM554fs with heterozygous flies as the control (30). These animals were raised at 22 °C and shifted to 29 °C upon adult eclosion. R47A04-Gal4 and R44E10-Gal4 from the Janelia Gal4 collection (48) were used for misexpressing genes, or RNAi in mature follicle cells and their specific expression pattern in female reproductive system were characterized (9, 12). All RNAi-knockdown experiments were performed at 29 °C with UAS-dcr2. The RNAi lines used were as follows: UAS-shd-i1 (V108911), UAS-shd-i2 (V17203), UAS-sad-i (V41269), UAS-phm-i1 (V108359), UAS-phm-i2 (V6169), UAS-dib-i (V101117), and UAS-spo-i (V51170) from the Vienna Drosophila Resource Center (VDRC); and UAS-EcR-i (B9326), UAS-EcR.A-i (B9328), and UAS-EcR.B1-i (B9329) from the Bloomington Drosophila Stock Center (BDSC). The misexpressing lines used were as follows: UAS-EcR.B1DN1 (UAS-EcR.B1F645A; B6869), UAS-EcR.B1DN2 (UAS-EcR.B1W650A; B6872), UAS-EcR.A (B6470), UAS-EcR.B1 (B6469), and UAS-EcR.B2 (B6468) from the BDSC; UAS-shd (19), UAS-RFP (9). Control flies were derived from specific Gal4 drivers crossed to Oregon-R. The Mmp2::GFP fusion allele in the Mmp2 endogenous locus was used for detecting Mmp2 protein expression (9).
Egg Laying and Ovulation Time.
Analysis of egg laying and ovulation time was performed as described with minor modifications (12). In short, 5- to 6-d-old virgin females were fed with wet yeast 1 d before egg laying, and five females and 10 Oregon-R males were kept in a bottle to lay eggs on grape juice-agar plates for 2 d at 29 °C. After egg laying, ovaries were dissected and mature follicles in these ovaries were quantified. The number of eggs laid on the plates was counted and used to calculate the average time for laying an egg (egg-laying time). The egg-laying time was partitioned into the ovulation time, oviduct time, and the uterus time. The partition ratio was determined based on the percentage of females having eggs in the oviduct or uterus at 6 h after mating.
Ex Vivo Follicle Rupture, in Situ Zymography, and qRT-PCR.
Ex vivo follicle rupture and in situ zymography were performed as described (12). Five- to 6-d-old virgin females were used to isolate mature follicles according to fluorescent signals driven by R47A04-Gal4 or R44E10-Gal4. For OA-induced follicle rupture, 20 μM OA (Sigma) were supplemented in the culture medium and cultured for 3 h unless otherwise indicated. For E and 20E cultures, mature follicles were preincubated in the medium with E or 20E (Cayman Chemical) for half an hour before addition of OA. For each culture, approximately 30 mature follicles were used and the percentage of ruptured follicles (losing more than 80% follicle-cell covering the oocyte) was calculated. Data were represented as mean ± SD; and Student’s t test was used for statistical analysis.
In situ zymography for detecting gelatinase activity was performed as reported (9). DQ-gelatin conjugated with fluorescein (25 μg/mL; Invitrogen) was added into the culture media with or without OA for 3 h (or 6 h in the case of 20E rescue experiment). After a quick rinse, mature follicles with posterior fluorescent signal were directly counted.
For qRT-PCR, total RNA was extracted from isolated mature follicles with TRIzol (Invitrogen) according to the standard protocol and was used to synthesize cDNA via the SuperScript III First-Strand Synthesis System (Invitrogen) by using Oligo(dT) primers. qPCR amplication was performed with iTaq Universal SYBR Green Supermix (Bio-Rad). The primers used were as follows: OambF 5′ TGACCAACGATCGGGGTTAT 3′ and OambR 5′ ATGCGCAATATGAGCTGGGA 3′ for detecting Oamb.K3 isoform expressed in mature follicles (12), and Mmp2F 5′ TACTTGTGGCGCATTGGAAC 3′ and Mmp2R 5′ ATCGATGTGGGTCAAAGTGG 3′ for Mmp2 gene. The Rps17 is used as an internal control.
Immunostaining and Microscopy.
Immunostaining was performed by following a standard procedure (49), including fixation in 4% EM-grade paraformaldehyde (diluted from 32% stock; Electron Microscopy Sciences) for 15 min, blocking in PBTG [PBS + 0.2% Triton+ 0.5% BSA + 2% (vol/vol) normal goat serum], and primary and secondary antibody staining. Rabbit anti-Shd and anti-Phm (1:250; gifts from Michael O’Connor, University of Minnesota Twin Cities, Minneapolis), mouse anti-EcR, anti-EcR.B1, anti-EcR.A, and anti-Hnt (1:15; Developmental Study Hybridoma Bank), rabbit anti-RFP (1:4,000; MBL International) and rabbit anti-GFP (Invitrogen) were used as primary antibodies, and Alexa-488 and Alexa-546 goat secondary antibody (1:1,000, Invitrogen) were used. Images were acquired by using a Leica TCS SP8 confocal microscope or Leica MZ10F fluorescent stereoscope with a sCOMS camera (PCO.Edge), and assembled by using Photoshop software (Adobe).
Acknowledgments
We thank Drs. Michael O’Connor, Allan Spradling, and Gerald Rubin for sharing fly lines and antibodies; the BDSC and the VDRC for fly stocks; the Developmental Study Hybridoma Bank for antibodies; anonymous reviewers for constructive comments; Drs. Joseph LoTurco, Karen Menuz, John Peluso, and Robert Gallo for valuable discussion on the manuscript; and Dr. Wei Li, Lylah Deady, and Wei Shen for technical support and discussion. J.S. is supported by the University of Connecticut Start-up fund and NIH/National Institute of Child Health and Human Development Grant R01-HD086175.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1614383114/-/DCSupplemental.
References
- 1.Curry TE, Jr, Osteen KG. The matrix metalloproteinase system: Changes, regulation, and impact throughout the ovarian and uterine reproductive cycle. Endocr Rev. 2003;24(4):428–465. doi: 10.1210/er.2002-0005. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi T, Fujimori C, Hagiwara A, Ogiwara K. Recent advances in the understanding of teleost medaka ovulation: The roles of proteases and prostaglandins. Zoolog Sci. 2013;30(4):239–247. doi: 10.2108/zsj.30.239. [DOI] [PubMed] [Google Scholar]
- 3.Espey LL, Richards JS. Ovulation. In: Neill JD, editor. Physiology of Reproduction. 3rd Ed. Academic Press; Amsterdam: 2006. pp. 425–474. [Google Scholar]
- 4.Kim J, Bagchi IC, Bagchi MK. Control of ovulation in mice by progesterone receptor-regulated gene networks. Mol Hum Reprod. 2009;15(12):821–828. doi: 10.1093/molehr/gap082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan H-Y, Liu Z, Mullany LK, Richards JS. Consequences of RAS and MAPK activation in the ovary: The good, the bad and the ugly. Mol Cell Endocrinol. 2012;356(1-2):74–79. doi: 10.1016/j.mce.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lydon JP, et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9(18):2266–2278. doi: 10.1101/gad.9.18.2266. [DOI] [PubMed] [Google Scholar]
- 7.Hibbert ML, Stouffer RL, Wolf DP, Zelinski-Wooten MB. Midcycle administration of a progesterone synthesis inhibitor prevents ovulation in primates. Proc Natl Acad Sci USA. 1996;93(5):1897–1901. doi: 10.1073/pnas.93.5.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spradling AC. Developmental genetics of oogenesis. In: Bate M, Martinez-Arias A, editors. The Development of Drosophila Melanogaster. Cold Spring Harbor Lab Press; Cold Spring Harbor, NY: 1993. pp. 1–70. [Google Scholar]
- 9.Deady LD, Shen W, Mosure SA, Spradling AC, Sun J. Matrix metalloproteinase 2 is required for ovulation and corpus luteum formation in Drosophila. PLoS Genet. 2015;11(2):e1004989. doi: 10.1371/journal.pgen.1004989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monastirioti M. Distinct octopamine cell population residing in the CNS abdominal ganglion controls ovulation in Drosophila melanogaster. Dev Biol. 2003;264(1):38–49. doi: 10.1016/j.ydbio.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 11.Monastirioti M, Linn CE, Jr, White K. Characterization of Drosophila tyramine β-hydroxylase gene and isolation of mutant flies lacking octopamine. J Neurosci. 1996;16(12):3900–3911. doi: 10.1523/JNEUROSCI.16-12-03900.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deady LD, Sun J. A follicle rupture assay reveals an essential role for follicular adrenergic signaling in Drosophila ovulation. PLoS Genet. 2015;11(10):e1005604. doi: 10.1371/journal.pgen.1005604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim J, et al. The octopamine receptor Octβ2R regulates ovulation in Drosophila melanogaster. PLoS One. 2014;9(8):e104441. doi: 10.1371/journal.pone.0104441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun J, Spradling AC. Ovulation in Drosophila is controlled by secretory cells of the female reproductive tract. eLife. 2013;2:e00415. doi: 10.7554/eLife.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubinstein CD, Wolfner MF. Drosophila seminal protein ovulin mediates ovulation through female octopamine neuronal signaling. Proc Natl Acad Sci USA. 2013;110(43):17420–17425. doi: 10.1073/pnas.1220018110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yapici N, Kim YJ, Ribeiro C, Dickson BJ. A receptor that mediates the post-mating switch in Drosophila reproductive behaviour. Nature. 2008;451(7174):33–37. doi: 10.1038/nature06483. [DOI] [PubMed] [Google Scholar]
- 17.Chávez VM, et al. The Drosophila disembodied gene controls late embryonic morphogenesis and codes for a cytochrome P450 enzyme that regulates embryonic ecdysone levels. Development. 2000;127(19):4115–4126. doi: 10.1242/dev.127.19.4115. [DOI] [PubMed] [Google Scholar]
- 18.Warren JT, et al. Molecular and biochemical characterization of two P450 enzymes in the ecdysteroidogenic pathway of Drosophila melanogaster. Proc Natl Acad Sci USA. 2002;99(17):11043–11048. doi: 10.1073/pnas.162375799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petryk A, et al. Shade is the Drosophila P450 enzyme that mediates the hydroxylation of ecdysone to the steroid insect molting hormone 20-hydroxyecdysone. Proc Natl Acad Sci USA. 2003;100(24):13773–13778. doi: 10.1073/pnas.2336088100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warren JT, et al. Phantom encodes the 25-hydroxylase of Drosophila melanogaster and Bombyx mori: A P450 enzyme critical in ecdysone biosynthesis. Insect Biochem Mol Biol. 2004;34(9):991–1010. doi: 10.1016/j.ibmb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Ono H, et al. Spook and Spookier code for stage-specific components of the ecdysone biosynthetic pathway in Diptera. Dev Biol. 2006;298(2):555–570. doi: 10.1016/j.ydbio.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 22.Niwa R, et al. Non-molting glossy/shroud encodes a short-chain dehydrogenase/reductase that functions in the ‘Black Box’ of the ecdysteroid biosynthesis pathway. Development. 2010;137(12):1991–1999. doi: 10.1242/dev.045641. [DOI] [PubMed] [Google Scholar]
- 23.Koelle MR, et al. The Drosophila EcR gene encodes an ecdysone receptor, a new member of the steroid receptor superfamily. Cell. 1991;67(1):59–77. doi: 10.1016/0092-8674(91)90572-g. [DOI] [PubMed] [Google Scholar]
- 24.Yao T-P, Segraves WA, Oro AE, McKeown M, Evans RM. Drosophila ultraspiracle modulates ecdysone receptor function via heterodimer formation. Cell. 1992;71(1):63–72. doi: 10.1016/0092-8674(92)90266-f. [DOI] [PubMed] [Google Scholar]
- 25.Talbot WS, Swyryd EA, Hogness DS. Drosophila tissues with different metamorphic responses to ecdysone express different ecdysone receptor isoforms. Cell. 1993;73(7):1323–1337. doi: 10.1016/0092-8674(93)90359-x. [DOI] [PubMed] [Google Scholar]
- 26.Hu X, Cherbas L, Cherbas P. Transcription activation by the ecdysone receptor (EcR/USP): Identification of activation functions. Mol Endocrinol. 2003;17(4):716–731. doi: 10.1210/me.2002-0287. [DOI] [PubMed] [Google Scholar]
- 27.Thummel CS. Flies on steroids--Drosophila metamorphosis and the mechanisms of steroid hormone action. Trends Genet. 1996;12(8):306–310. doi: 10.1016/0168-9525(96)10032-9. [DOI] [PubMed] [Google Scholar]
- 28.Riddiford LM, Cherbas P, Truman JW. Ecdysone receptors and their biological actions. Vitam Horm. 2000;60:1–73. doi: 10.1016/s0083-6729(00)60016-x. [DOI] [PubMed] [Google Scholar]
- 29.Buszczak M, et al. Ecdysone response genes govern egg chamber development during mid-oogenesis in Drosophila. Development. 1999;126(20):4581–4589. doi: 10.1242/dev.126.20.4581. [DOI] [PubMed] [Google Scholar]
- 30.Carney GE, Bender M. The Drosophila ecdysone receptor (EcR) gene is required maternally for normal oogenesis. Genetics. 2000;154(3):1203–1211. doi: 10.1093/genetics/154.3.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terashima J, Takaki K, Sakurai S, Bownes M. Nutritional status affects 20-hydroxyecdysone concentration and progression of oogenesis in Drosophila melanogaster. J Endocrinol. 2005;187(1):69–79. doi: 10.1677/joe.1.06220. [DOI] [PubMed] [Google Scholar]
- 32.Hackney JF, Pucci C, Naes E, Dobens L. Ras signaling modulates activity of the ecdysone receptor EcR during cell migration in the Drosophila ovary. Dev Dyn. 2007;236(5):1213–1226. doi: 10.1002/dvdy.21140. [DOI] [PubMed] [Google Scholar]
- 33.Sun J, Smith L, Armento A, Deng WM. Regulation of the endocycle/gene amplification switch by Notch and ecdysone signaling. J Cell Biol. 2008;182(5):885–896. doi: 10.1083/jcb.200802084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernardi F, Romani P, Tzertzinis G, Gargiulo G, Cavaliere V. EcR-B1 and Usp nuclear hormone receptors regulate expression of the VM32E eggshell gene during Drosophila oogenesis. Dev Biol. 2009;328(2):541–551. doi: 10.1016/j.ydbio.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Ables ET, Drummond-Barbosa D. The steroid hormone ecdysone functions with intrinsic chromatin remodeling factors to control female germline stem cells in Drosophila. Cell Stem Cell. 2010;7(5):581–592. doi: 10.1016/j.stem.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.König A, Yatsenko AS, Weiss M, Shcherbata HR. Ecdysteroids affect Drosophila ovarian stem cell niche formation and early germline differentiation. EMBO J. 2011;30(8):1549–1562. doi: 10.1038/emboj.2011.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Domanitskaya E, Anllo L, Schüpbach T. Phantom, a cytochrome P450 enzyme essential for ecdysone biosynthesis, plays a critical role in the control of border cell migration in Drosophila. Dev Biol. 2014;386(2):408–418. doi: 10.1016/j.ydbio.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sieber MH, Spradling AC. Steroid signaling establishes a female metabolic state and regulates SREBP to control oocyte lipid accumulation. Curr Biol. 2015;25(8):993–1004. doi: 10.1016/j.cub.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ameku T, Niwa R. Mating-induced increase in germline stem cells via the neuroendocrine system in female Drosophila. PLoS Genet. 2016;12(6):e1006123. doi: 10.1371/journal.pgen.1006123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Handler AM. Ecdysteroid titers during pupal and adult development in Drosophila melanogaster. Dev Biol. 1982;93(1):73–82. doi: 10.1016/0012-1606(82)90240-8. [DOI] [PubMed] [Google Scholar]
- 41.Cherbas L, Hu X, Zhimulev I, Belyaeva E, Cherbas P. EcR isoforms in Drosophila: Testing tissue-specific requirements by targeted blockade and rescue. Development. 2003;130(2):271–284. doi: 10.1242/dev.00205. [DOI] [PubMed] [Google Scholar]
- 42.Yamanaka N, Marqués G, O’Connor MB. Vesicle-mediated steroid hormone secretion in Drosophila melanogaster. Cell. 2015;163(4):907–919. doi: 10.1016/j.cell.2015.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King-Jones K, Thummel CS. Nuclear receptors--a perspective from Drosophila. Nat Rev Genet. 2005;6(4):311–323. doi: 10.1038/nrg1581. [DOI] [PubMed] [Google Scholar]
- 44.Föhr KJ, et al. Concerted action of human chorionic gonadotropin and norepinephrine on intracellular-free calcium in human granulosa-lutein cells: Evidence for the presence of a functional alpha-adrenergic receptor. J Clin Endocrinol Metab. 1993;76(2):367–373. doi: 10.1210/jcem.76.2.8381798. [DOI] [PubMed] [Google Scholar]
- 45.Breen SM, et al. Ovulation involves the luteinizing hormone-dependent activation of G(q/11) in granulosa cells. Mol Endocrinol. 2013;27(9):1483–1491. doi: 10.1210/me.2013-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park O-K, Mayo KE. Transient expression of progesterone receptor messenger RNA in ovarian granuiosa cells after the preovulatory luteinizing hormone surge. Mol Endocrinol. 1991;5(7):967–978. doi: 10.1210/mend-5-7-967. [DOI] [PubMed] [Google Scholar]
- 47.Robker RL, et al. Progesterone-regulated genes in the ovulation process: ADAMTS-1 and cathepsin L proteases. Proc Natl Acad Sci USA. 2000;97(9):4689–4694. doi: 10.1073/pnas.080073497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pfeiffer BD, et al. Tools for neuroanatomy and neurogenetics in Drosophila. Proc Natl Acad Sci USA. 2008;105(28):9715–9720. doi: 10.1073/pnas.0803697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun J, Spradling AC. NR5A nuclear receptor Hr39 controls three-cell secretory unit formation in Drosophila female reproductive glands. Curr Biol. 2012;22(10):862–871. doi: 10.1016/j.cub.2012.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]