

ABSTRACT
Bacterial persisters are a subpopulation of cells that can tolerate lethal concentrations of antibiotics. However, the possibility of the emergence of genetically resistant mutants from antibiotic persister cell populations, upon continued exposure to lethal concentrations of antibiotics, remained unexplored. In the present study, we found that Mycobacterium tuberculosis cells exposed continuously to lethal concentrations of rifampin (RIF) or moxifloxacin (MXF) for prolonged durations showed killing, RIF/MXF persistence, and regrowth phases. RIF-resistant or MXF-resistant mutants carrying clinically relevant mutations in the rpoB or gyrA gene, respectively, were found to emerge at high frequency from the RIF persistence phase population. A Luria-Delbruck fluctuation experiment using RIF-exposed M. tuberculosis cells showed that the rpoB mutants were not preexistent in the population but were formed de novo from the RIF persistence phase population. The RIF persistence phase M. tuberculosis cells carried elevated levels of hydroxyl radical that inflicted extensive genome-wide mutations, generating RIF-resistant mutants. Consistent with the elevated levels of hydroxyl radical-mediated genome-wide random mutagenesis, MXF-resistant M. tuberculosis gyrA de novo mutants could be selected from the RIF persistence phase cells. Thus, unlike previous studies, which showed emergence of genetically resistant mutants upon exposure of bacteria for short durations to sublethal concentrations of antibiotics, our study demonstrates that continuous prolonged exposure of M. tuberculosis cells to lethal concentrations of an antibiotic generates antibiotic persistence phase cells that form a reservoir for the generation of genetically resistant mutants to the same antibiotic or another antibiotic. These findings may have clinical significance in the emergence of drug-resistant tubercle bacilli.
KEYWORDS: Mycobacterium tuberculosis, rifampicin, moxifloxacin, hydroxyl radical, persistence phase cells, antibiotic resistance, Mycobacterium tuberculosis
INTRODUCTION
Antibiotic persister cells constitute a subpopulation of bacteria that can tolerate lethal concentrations of antibiotic and give rise to antibiotic-sensitive bacteria upon its withdrawal (1). Reintroduction of a lethal concentration of the antibiotic would again enrich the antibiotic-tolerant persister population (2). Thus, antibiotic persister cells essentially would always remain tolerant in the continued presence of lethal concentrations of the antibiotic. They are believed to arise stochastically or deterministically through phenotypic variation against lethal concentrations of antibiotics (3–6). Several mechanisms, which include expression of toxin-antitoxin systems, generation of reactive oxygen species (ROS), and stochastic changes in gene expression, have been proposed as reasons for the formation of antibiotic persister cells (4, 5, 7, 8). Due to antibiotic tolerance, bacterial persister cells are believed to be the major reason for the recalcitrance of infection, with the requirement for an extended antibiotic regimen in many chronic infections, like tuberculosis (TB).
The presence of persistent populations of tubercle bacilli had been documented about a century ago in TB patients, in the autopsy samples of lung and lymph nodes from humans who died of reasons other than TB, and in animal models during the latter half of the last century (reviewed in references 9 and 10). Sputum and blood samples from TB patients under treatment and with destructive cavities in the lungs showed the presence of persistent tubercle bacilli (11–15). Among the animal models, Mycobacterium tuberculosis persister cells have been found against anti-tuberculosis drugs in the lungs and spleen of mice (16–20), guinea pigs (21–27), macrophages (28, 29), in vitro cultures (30–32), and the environment (33). These antibiotic persister cells from human tissue samples and the animal models could be cultured to get an infectious, drug-susceptible population of tubercle bacilli (13, 18, 19, 34). Thus, the phenomenon of persistence of M. tuberculosis and other mycobacteria against antibiotics has been observed in TB patients, animal models, and in vitro systems. Although the persister cell population was believed to give rise to a drug-sensitive population, the possibility of the emergence of drug-resistant bacilli from the persister cell population has remained unexplored.
Generation of drug-resistant and multidrug-resistant (MDR) M. tuberculosis cells showing resistance to single (drug-resistant) and multiple antibiotics, such as rifampin (RIF) and isoniazid (INH) (i.e., MDR), is one of the major challenges faced in the treatment of tuberculosis. M. tuberculosis is known to attain resistance to most of the drugs used for the treatment of tuberculosis (35). The emergence of M. tuberculosis strains that are resistant to rifampin, isoniazid, and any fluoroquinolone and to at least one of the three injectable second-line drugs (i.e., amikacin, kanamycin, or capreomycin), which are called extensively drug-resistant TB (XDR-TB) mutants, has also been reported (36). According to the recent WHO report on TB, 20% of the retreatment cases harbor MDR-TB, in contrast to 3.3% of new cases (36, 37). It has been demonstrated for Escherichia coli, Staphylococcus aureus, and Pseudomonas aeruginosa that sublethal concentrations of antibiotics can cause the emergence of antibiotic-resistant mutants through the generation of reactive oxygen species (ROS) (38–41), in addition to several other modes of generation of antibiotic resistance in M. tuberculosis (42) and other bacteria (43). Although the mechanisms by which M. tuberculosis gains resistance against antibiotics is known, the causes underlying these mechanisms need further investigation, which will have significance in the clinical scenario of the emergence of antibiotic-resistant strains of tubercle bacilli in patients who do not follow a complete regimen of treatment.
Since the incidences of MDR-TB are found largely in the retreatment cases, wherein the patients might not have complied with the treatment regimen, it is possible that the antibiotic persister cells have a role in generating the antibiotic-resistant mutants. Also, since TB treatment involves a prolonged regimen, it may be relevant to find out whether antibiotic-resistant mutants can emerge from the antibiotic persister cell population in the continued presence of lethal concentrations of antibiotics. In this regard, it has been postulated that the antibiotic persister cells could behave as an evolutionary reservoir for the emergence of antibiotic-resistant mutants (2). In line with these possibilities, in the present study, we investigated whether antibiotic-resistant mutants of M. tuberculosis could emerge de novo from the antibiotic persister cell population upon prolonged exposure of the bacilli to lethal concentrations of RIF and moxifloxacin (MXF). Consistent with this hypothesis, we found de novo emergence of mutants genetically resistant to both antibiotics at high frequency from the persistence phase of M. tuberculosis cells exposed to RIF for prolonged periods. The M. tuberculosis cells in the RIF persistence phase were found to be carrying elevated levels of hydroxyl radical, which inflicted genome-wide mutations. This facilitated isolation of mutants genetically resistant to the same antibiotic (RIF) or another antibiotic (MXF). Thus, the present study reveals that M. tuberculosis bacilli that are resistant to antibiotics can emerge from the persistence phase cells formed in response to prolonged exposure of the cells to lethal concentrations of the antibiotics.
RESULTS
M. tuberculosis cells exposed to lethal concentrations of RIF showed killing, persistence, and regrowth phases.
In order to expose M. tuberculosis cells to RIF, we first determined the minimal bactericidal concentration (MBC) of RIF, which was defined as the lowest concentration of the antibiotic in the medium that decreased the bacterial population by 2 log10 or more after 6 days of incubation (28). By this definition, the 1× MBC for RIF against the M. tuberculosis cells was found to be 0.1 μg/ml (see Fig. S1 in the supplemental material). Three independent cultures (R1, R2, and R3, which are biological replicates) of M. tuberculosis cells were exposed to 1 μg/ml (10× MBC) and 2 μg/ml (20× MBC) of RIF for 18 days under shaking conditions at 37°C. From day 0 onwards, different dilutions of the cultures (after washing the cells) were plated on RIF-free plates and undiluted culture on RIF-containing (5 μg/ml, 50× MBC) plates and incubated in a CO2 incubator at 37°C until colonies appeared. The colonies, which sometimes took 2 to 3 months to appear, were counted and the CFU per milliliter was determined from the RIF-free and RIF-containing plates.
The CFU profile of the M. tuberculosis cells on the RIF-free plate from the R1, R2, and R3 cultures, upon exposure to 1 μg/ml (10× MBC) of RIF for 18 days under shaking conditions at 37°C, showed three distinct phases (Fig. 1A, red line). The first phase, from day 0 to day 10 of exposure, was characterized by a steady and steep decrease of about a 5-log10 reduction (from about 108 to 103) in CFU (Table S3). This phase was called the killing phase due to the drastic decrease in the CFU. The killing phase was followed by a phase (from day 10 to day 15) without any appreciable change in the CFU, which was maintained at about 103 to 104 CFU/ml (Fig. 1A, red line; Table S3). We called this the RIF persistence phase, since the cells were found to persist against a lethal concentration of RIF, as determined in previous studies (9–34). Although the concentration of RIF was found to decrease slowly and steadily over 18 days, about 8× to 6× MBC of RIF was still present during the RIF persistence phase (Fig. 1A, blue line). The RIF persistence phase was followed by a phase (from about day 15 to day 18) showing a notable increase in the CFU on RIF-free plates (Fig. 1A, red line). This third phase was called the regrowth phase, as there was an increase in the CFU, from about 104 to 106 CFU/ml, on RIF-free plates (Fig. 1A, red line). The increase in the CFU, when plated on RIF-free plates, was contributed by the cells that were surviving under the lethal concentration of about 4× to 2× MBC of RIF (0.4 to 0.2 μg/ml; Fig. 1A, blue line) during the regrowth phase.
FIG 1.
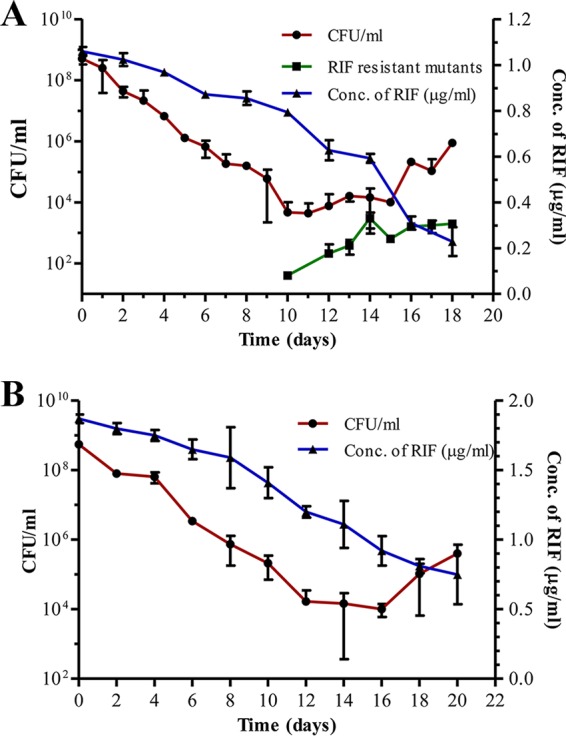
RIF susceptibility profile of M. tuberculosis cells during extended exposure to lethal concentrations of RIF. (A) RIF susceptibility profile of M. tuberculosis cells exposed to 1 μg/ml (10× MBC) RIF for 18 days when plated on RIF-free plates (•; red line). The CFU of the cells on RIF-containing (50× MBC) plates (■; green line) and the concentration of RIF during the course of the experiment (right y axis; ▲; blue line) are shown. (B) RIF susceptibility profile of M. tuberculosis cells exposed to 2 μg/ml (20× MBC) of RIF (•; red line) and concentration of RIF during the course of the experiment (right y axis; ▲; blue line).
Exposure of M. tuberculosis culture, generated from a single RIF-sensitive colony, to a higher concentration of 2 μg/ml of RIF (20× MBC) also gave a similar triphasic profile of the bacterial response to the antibiotic (Fig. 1B, red line). Here also, the killing phase showed about a 5-log10 reduction (from 108 to 104) in the CFU up to about day 12, when the RIF concentration declined from 20× MBC to 12× MBC (Fig. 1B, blue line). The killing phase was followed by the RIF persistence phase from day 12 to day 16 (Fig. 1B; Table S4). Here, the CFU did not show appreciable change, and it was maintained at about 103 to 104 CFU/ml (Fig. 1B, red line). A high concentration of about 1.2 to 0.8 μg/ml RIF (12× to 8× MBC) was still present during this phase (Fig. 1B, blue line). The regrowth phase, showing an increase in the CFU (on RIF-free plates), was found from about day 16 to day 20 (Fig. 1B, red line). The RIF concentration was still 9× to 7× MBC during this phase (Fig. 1B, blue line). Thus, it may be noted that the increase in the CFU on RIF-free plates was given by the population that was surviving under the lethal concentration of 9× to 7× MBC during this phase.
The 10× MBC of RIF ensured that a microbicidal concentration (4× to 2×) of the antibiotic always remained in the medium, even during prolonged exposure of the cells for 18 to 20 days (Fig. 1A and B). The continuous presence of such high concentrations of RIF for prolonged durations in the liquid culture was maintained to find out whether RIF-resistant mutants could emerge when exposed to lethal concentrations of the antibiotic. Otherwise, the RIF concentration would have declined to sublethal levels, where the possibility of ROS generation and consequential mutagenesis would have occurred, as reported previously (38–41). The use of 5 μg/ml of RIF (50× MBC) on the agar plate selected genuine RIF-resistant mutants, as most of the RRDR mutations impart a high level of RIF resistance to M. tuberculosis (44, 45). Also, such a high MBC of RIF was used to avoid any possible reduction in the RIF levels during the course of long incubation periods of 2 to 3 months on agar plates.
Emergence of RIF-resistant M. tuberculosis mutants from the RIF persistence phase cells.
While the CFU on RIF-free plates did not show appreciable change during day 10 to day 15, a period that spanned the RIF persistence phase, plating of the RIF-exposed cultures on 50× MBC RIF plates from day 0 to day 18 showed about a 2-log10 increase in the CFU only during the persistence phase (Fig. 1A, green line). On RIF plates, the increase in the CFU of the cells from the RIF persistence phase indicated the emergence of RIF-resistant mutants (Fig. 1A, green line). These RIF-resistant cells constituted, for example, only about 27.45%, 15.80%, and 39.25% of the day 14 RIF persistence phase cultures of R1, R2, and R3 samples, respectively (Table S5). Similarly, while 27.45% of the cells from the day 14 R1 culture could form colonies on 50× MBC RIF plates, only 10.50% and 3.39% of the cells from day 12 and day 13 of the RIF persistence phase of R1 culture could form colonies (Table S5). From some days' RIF persistence phase cultures, colonies grew on RIF-free plates but not on 50× MBC RIF plates, while from some other days' cultures colonies grew in the 50× MBC plates but not in the RIF-free plates (Table S5; see Discussion for explanation). The emergence of different proportions of RIF-resistant colonies on 50× MBC RIF plates from the three RIF persistence phase cultures of the same day or from the same culture on different days showed that different proportions of the RIF persistence phase cells might have gained RIF resistance on different days of the RIF persistence phase. These observations suggested that the emergence of RIF-resistant mutants occurs in the RIF persistence phase.
From the regrowth phase of day 15 to day 18, where the cells were surviving in the presence of a RIF concentration of about 4× to 2× MBC in liquid culture (Fig. 1A, blue line), the CFU of the cells on the 50× MBC plates were low, showing low levels of emergence of RIF-resistant mutants (Fig. 1A, green line). For instance, only 0.68% of the day 16 R2 culture, 7.67% and 12.1% of the day 17 R1 and R3 cultures, and 1.69% of the day 18 R3 culture formed colonies on 50× MBC RIF plates (Table S5). At the same time, on RIF-free plates, the cells from the cultures of day 15 to day 18 showed about a 2-log10 increase (104 to 106) in CFU (Fig. 1A, red line). This indicated that these cultures contain RIF persistence cells that had not yet gained resistance and RIF-resistant cells with various MICs, as reported previously (46), which could tolerate the concentration of RIF in the culture. On the contrary, on the 50× MBC RIF plate only those resistant cells capable of tolerating such a high concentration of RIF would have grown to form colonies. Thus, the differences in the CFU of the regrowth-phase cells on 50× MBC RIF and RIF-free plates indicated that these cultures contain different proportions of low-MIC, RIF-resistant cells and the cells that had not gained RIF resistance, with both contributing to the CFU on RIF-free plates.
M. tuberculosis cells exposed to lethal concentration of MXF also showed killing, persistence, and regrowth phases.
A triphasic response, similar to that shown by the M. tuberculosis cells exposed to 10× MBC of RIF, was shown by the M. tuberculosis cells in the three independent cultures exposed to the continued presence of lethal concentrations of MXF (1 μg/ml; 2× MBC) (Fig. S2) (47). The CFU, as determined by plating on MXF-free plates, declined drastically up to day 10 of the exposure, indicating the killing phase (Fig. S2, red line, and Table S6). From day 10 to day 16 of the exposure, the CFU in the MXF-free plate was found more or less unchanged (Fig. S2, red line, and Table S6), although the MXF concentration continued to be about 1.5× MBC (Fig. S2, blue line). This indicated the MXF persistence phase. As in the case of RIF-exposed cells, from day 16 to day 18, when the cells were surviving under about 2× MBC of MXF, the CFU on MXF-free plates was found to increase. This was the regrowth phase of the culture. A high MBC of MXF was used in the cultures and on the plate for the same reasons as those mentioned in the case of RIF.
Emergence of MXF-resistant M. tuberculosis mutants from the MXF persistence phase cells.
The MXF-exposed cultures from day 0 to day 18, when plated on 4× MBC MXF plates, showed an increase in the CFU from about day 12 until almost day 16 that spanned the MXF persistence phase (Fig. S2, green line, and Table S7). The increase in the CFU indicated the emergence of MXF-resistant mutants (Fig. S2, green line). These MXF-resistant cells constituted, for example, about 20%, 10%, and 80% (which later became 145% due to more colonies that emerged after 4 months postplating) of the day 15 cultures of M1, M2, and M3 samples, respectively (Table S7). Similarly, 4.17% of the cells from day 12 and 20% and 60% of the cells from days 15 and 16 of M1 culture from the MXF persistence phase formed colonies on 4× MBC MXF plates (Table S7). From some other days' cultures in the MXF persistence phase, colonies could be obtained only on the 4× MBC plates but not on the MXF-free plates and vice versa (Table S7; see Discussion for explanation). Thus, as in the case of RIF persistence phase cultures, different proportions of 4× MBC MXF-resistant cells were observed from different days' (day 12 to day 16) cultures and from the same day of the three MXF persistence phase cultures.
However, the cells from the regrowth phase (day 16 to day 18), which were surviving in the presence of about 2× MBC MXF in liquid culture (Fig. S2, blue line), showed only a marginal increase in the CFU on the 4× MBC MXF plates, indicating the continued but low level of emergence of MXF-resistant mutants (Fig. S2, green line, and Table S7). For example, about 14.67% and 53.33% of the cells from the M1-17 and M3-17 cultures, 14.64% of M1-18 cultures, and 39.1% of M2-18 cultures formed colonies on 4× MBC MXF plates (Table S7), whereas on MXF-free plates, the cells from the regrowth phase showed about a 2-log10 increase (102 to 104) in the CFU (Fig. S2, red line). Thus, the differences in the CFU of the regrowth-phase cells on 4× MBC MXF and MXF-free plates implied the existence of different proportions of MXF-resistant cells with different MICs and MXF-tolerant persisting cells.
M. tuberculosis RIF-resistant mutants emerging from the RIF persistence phase cells had mutations at the RRDR of the rpoB gene.
Mutations in the rifampin resistance-determining region (RRDR) of the rpoB gene are known to confer resistance to high concentrations of RIF (48–51). Sixteen RIF-resistant mutants isolated from the RIF persistence phase of the R1, R2, and R3 cultures (day 12 samples of R1 and R2 and day 13 samples of R3) and grown in 50× MBC RIF medium showed mutations in the RRDR of the rpoB gene (Fig. 2). Five different kinds of mutations could be observed among the 16 different isolates, at the nucleotide locations 968, 1333, and 1349 of the rpoB gene (numbering based on M. tuberculosis sequence). These mutations involved C→T or C→G changes, resulting in Thr323-Met, His445-Asp, His445-Tyr, Ser450-Leu, and Ser450-Trp changes in the amino acids (Fig. 2). The C→T change at the 968th nucleotide position constituted 6.25% of the mutations (not shown in the schematic diagram, as it is outside the RRDR of the rpoB gene). These specific mutations have been reported in the rpoB gene of the RIF-resistant M. tuberculosis isolates from TB patients in different countries (52–59). Thus, the RIF-resistant mutants isolated from the RIF persistence phase were genetically resistant mutants and not phenotypically resistant cells. Incidentally, the C→T or C→G changes indicated that the mutations were brought about by oxidative stress (60, 61) within the cells.
FIG 2.

Schematic diagram of the RRDR of the rpoB gene and the list of mutations found in M. tuberculosis colonies isolated from the persistent phase of RIF treatment. (A) Genetic map of the RRDR of rpoB, with the location of the mutations found in RIF-resistant mutants obtained from the persistence phase during prolonged exposure of M. tuberculosis cells to RIF. The different-colored stars represent different kinds of mutations, and the number within the parentheses shows the percentage of that kind of mutation. The nature of the nucleotide changes are indicated. (B) The list of RIF-resistant mutations selected from the RIF persistence phase from three independent cultures (R1, R2, and R3) during RIF treatment. The letter “B” indicates the batch of the colony on the plate (batchwise colony formation was observed on the agar plate during the persistent phase, reflecting their growth rate, and the colonies formed at a given time were considered a batch). Five different kinds of mutations detected (Ser450-Leu, Ser450-Trp, His445-Asp, His445-Tyr, and Thr323-Met) in the RRDR of the rpoB gene are indicated.
M. tuberculosis RIF-resistant mutants were not preexisting but were formed de novo.
Although the triphasic response of the bacilli to RIF showed the emergence of RIF-resistant mutants, it was not clear whether they emerged de novo from the killing or RIF persistence phase or from the preexisting and probably slow-growing mutants in the population. In order to resolve these possibilities, a Luria-Delbruck (LD) fluctuation test (62) was performed in a modified format. Since the kind of mutations obtained indicated that they were oxidative stress driven (60, 61), the emergence of RIF-resistant mutants was monitored in the LD experiment in the absence and presence of thiourea (TU), which is a hydroxyl radical quencher (63). RIF-resistant colonies could be observed in the 10 individual cultures (not exposed to TU), specifically from the RIF persistence phase from day 10 of the exposure onwards on almost all days (Fig. 3A). While the natural frequency of RIF resistance in M. tuberculosis mid-log-phase cells was determined to be (5.51 ± 3.34) × 10−9, the frequency of emergence of RIF-resistant mutants from the RIF persistence phase was found to be (8.92 ± 0.12) × 10−4, which is about 5-log10 higher in magnitude (Fig. 3A). This clearly indicated that the mutants have been generated de novo from the RIF persistence phase cells.
FIG 3.

Luria-Delbruck (modified) experiment showing M. tuberculosis RIF-resistant mutant generation and the response of the bacilli to RIF in the absence and presence of thiourea (TU). RIF-resistant mutant generation in M. tuberculosis cells upon exposure to RIF in the absence (A) and in the presence (B) of TU. Yellow coloration represents cultures exposed to TU. The numbers in the red boxes indicate the numbers of resistant colonies obtained from that particular culture upon plating the cells on 50× MBC RIF plates. (C) RIF susceptibility kinetics of RIF-exposed M. tuberculosis cells in the absence and presence of TU during the LD experiment. The right y axis represents the RIF concentration. Additional cultures (2 to 3) after day 17 of plating were used for RIF assay on day 18, and for this reason results for RIF assay are shown for 18 days, although the experiment was completed on day 17.
On the contrary, only a few RIF-resistant colonies were observed in the TU-exposed samples from the RIF persistence phase (Fig. 3B). Accordingly, the frequency of generation of RIF-resistant mutants in the presence of TU was (2.28 ± 3.58) × 10−5, which was about 40-fold less than the frequency of generation of RIF-resistant mutants in the absence of TU [(8.92 ± 0.12) × 10−4] (Fig. 3B). This indicated the involvement of hydroxyl radical in the generation of RIF resistance mutations. However, interestingly, the RIF susceptibility kinetics of the TU-exposed and the TU-unexposed cultures from the RIF persistence phase were comparable (Fig. 3C). These comparable kinetics of RIF susceptibility and the TU sensitivity in the emergence of RIF-resistant mutant populations from the RIF persistence phase cultures (Fig. 3A, B, and C) showed that the RIF-susceptible population might be those cells that had not yet incurred mutations in the rpoB gene. The consistent emergence of only a certain proportion of the RIF persistence phase population as RIF-resistant mutants supports this inference. From the RIF persistence phase, the TU-exposed cells formed small-size colonies with delay, unlike the cells not exposed to TU.
Fluctuation in the number of RIF-resistant mutants emerging from the different individual cultures on different days and from different cultures on the same day during the RIF persistence phase indicated that the mutants were formed de novo independently from the persistence phase and that they were not preexistent in the culture. While the low numbers of resistant mutants during the initial period of the persistence phase suggested their de novo generation, the high numbers of resistant mutants at later periods during the persistence phase also indicated de novo generation and subsequent multiplication of the resistant mutants that arose earlier. All 21 mutants isolated from the different cultures from different days of exposure to RIF in the LD experiment showed point mutations at the RRDR of the rpoB gene (Fig. 4). Most of the mutations were present at four specific nucleotide positions in the rpoB gene (1295, 1333, 1334, and 1349), with C→T, C→G, A→T, or A→G changes, yielding six different types of amino acid changes (Gln432-Leu, His445-Arg, His445-Asp, His445-Tyr, Ser450-Leu, and Ser450-Trp). Identical mutations have been found in the RIF-resistant mutants of virulent M. tuberculosis cells isolated from TB patients from different countries (52–59), indicating their clinical relevance. Further, the reduction in the emergence of RIF-resistant mutants in the presence of the hydroxyl radical quencher TU indicated the involvement of hydroxyl radical, a potent mutagen (64), in the generation of the mutations. Since the emergence of the RIF-resistant mutants was found in the RIF persistence phase, it was likely that the mutations were inflicted by hydroxyl radical during the RIF persistence phase of the cultures. Therefore, we examined the RIF persistence phase intact cells and their lysates for the presence of hydroxyl radical.
FIG 4.

Schematic diagram of the mutations in the RRDR of rpoB and list of mutations found in colonies isolated from LD experiment. (A) Genetic map of the mutations in the RRDR of rpoB from mutants isolated from the LD experiment. The colored stars represent different kinds of mutations, and the number within the parentheses shows the percentage of each kind of mutation. (B) List of the mutations at the nucleotide and amino acid levels. Six different mutations were detected (Gln432-Leu, His445-Arg, His445-Asp, His445-Tyr, Ser450-Leu, and Ser450-Trp) in the RRDR of rpoB.
High levels of hydroxyl radical in the RIF persistence phase M. tuberculosis cells and cell lysates.
We used electron paramagnetic resonance (EPR), which is the gold standard method for the measurement of hydroxyl radical (65, 66), to determine and quantitate the levels of hydroxyl radical in the RIF persistence phase cells, in the absence and presence of TU, in comparison to the hydroxyl radical levels in the mid-log-phase cells (not exposed to RIF). Analysis of the lysates of the RIF persistence phase M. tuberculosis cells, from day 13 of exposure to RIF, showed the characteristic strong signal specific to 5,5-dimethyl-1-pyrroline N-oxide (DMPO)-OH adduct compared to that of the mid-log-phase cells (Fig. 5A and B). On the contrary, from the day 13 culture of the RIF persistence phase, the lysates of the cells exposed to the hydroxyl radical quencher TU (63) did not show the signal (Fig. 5C). The levels of the hydroxyl radical present in the lysates of the cells from day 13 of the RIF persistence phase was significantly higher than that found in the lysates of the mid-log-phase control cells (Fig. 5D). The presence of elevated levels of hydroxyl radical in the RIF persistence phase M. tuberculosis cell lysates strongly supported the possibility of hydroxyl radical-mediated generation of RIF-resistant mutations.
FIG 5.

EPR spectra of DMPO-OH adduct in the lysates of RIF-exposed M. tuberculosis cells (n = 2). (A to C) Representative EPR profile of DMPO-OH adduct in the lysates of the mid-log phase (MLP), persister (RIF Per), and TU-treated RIF persister (RIF Per TU) cells. (D) Bar graph representing the levels of DMPO-OH adduct in the cell lysates of RIF-exposed M. tuberculosis cells in the absence and presence of TU and of MLP cells (control). An asterisk indicates a P value of ≤0.05. Statistical significance was calculated using Student's t test.
Since high levels of hydroxyl radical were detected in the cell lysates, its presence in the intact cells was determined in the cells from the killing, RIF persistence, and regrowth phases in comparison to that in the mid-log-phase cells (not exposed to RIF). Flow cytometry of the cells stained with a hydroxyl radical-specific dye, 3′-(p-hydroxyphenyl) fluorescein (HPF) (67–69), which is a fluorescein derivative that is nonfluorescent until it reacts with hydroxyl radical, was used for the determination of hydroxyl radical in intact cells. Upon oxidation by hydroxyl radical, HPF gains an excitation maximum of 490 nm and emission maximum of 520 nm (67). The density plots, the histogram overlay, and quantitation of the fluorescence of equivalent numbers of HPF-stained M. tuberculosis cells from the mid-log, killing, persistence, and regrowth phases showed significantly higher levels of fluorescence in the RIF persistence phase cells than in the cells from the mid-log, killing, and regrowth phases (Fig. 6). The RIF persistence phase cells showed a clean single peak of fluorescence, indicating that all of the surviving cells in the persistence phase are carrying elevated levels of hydroxyl radical. The killing phase cells showed significantly greater fluorescence than the mid-log-phase cells (Fig. 6E and F), whereas the regrowth-phase cells, which would be proliferating like normal cells, irrespective of the presence of RIF, showed fluorescence comparable to that of the mid-log-phase cells (Fig. 6F). A gradual but significant increase in the hydroxyl radical levels was observed during the exposure of the mid-log-phase cells to 10× MBC RIF in the killing phase, through the RIF persistence phase, where the fluorescence reached its maximum level, and into the regrowth phase, where the fluorescence declined to basal levels comparable to that of the mid-log-phase cells (Fig. 6F and G). The positive correlation of high levels of HPF fluorescence with the RIF persistence phase cells showed that the generation of hydroxyl radical was maximum in the RIF persistence phase cells. This correlation was consistent with the formation of de novo RIF-resistant mutants from the RIF persistence phase cells in the Luria-Delbruck experiment (Fig. 3A).
FIG 6.

Flow cytometry analysis of intact HPF-stained M. tuberculosis cells during RIF exposure (n = 3 independent biological samples). Shown are representative density plots of HPF-stained M. tuberculosis cells from the RIF-unexposed mid-log phase (control) (A), RIF-exposed killing phase (B), day 12 of RIF persistence phase (C), and regrowth phase (D). (E) Histogram overlay of the HPF fluorescence of the respective density plots. (F) Bar graph representing the average median HPF fluorescence normalized [HPF median fluo. (Nor.)] to its respective autofluorescence control. (G) The overlay of the HPF fluorescence of the samples from different days of cell exposure to 10× MBC rifampin on the CFU graph (red line) depicted in Fig. 1A, showing high levels of fluorescence (high levels of OH radical generation) in the cells during the RIF persistence phase and reduced levels in the killing phase and regrowth phase. The y axis on the left represents CFU/ml, and the y axis on the right represents normalized median fluorescence of HPF by flow cytometry. An asterisk indicates a P value of ≤0.05. Statistical significance was calculated using the paired t test.
High oxidative status of M. tuberculosis cells in the RIF persistence phase.
The presence of elevated levels of hydroxyl radical in the M. tuberculosis RIF persistence phase cells exposed to lethal concentrations of RIF indicated high oxidative status of the intracellular environment. We measured the overall oxidative status of the intracellular environment using a reduction-oxidation status-sensitive green fluorescent protein indicator (roGFP) that is responsive to the redox status of mycothiol (MSH) (70), which plays a significant role in maintaining the redox status of mycobacterial cytoplasm (71–73). Redox-sensitive GFP (roGFP2) has two excitation maxima, one at 405 nm for the oxidized state and the other at 488 nm for the reduced state (74). The ratio of the excitation maxima values (405 nm/488 nm) is an indication of the redox status of the protein. The fusion of mycoredoxin (Mrx1) to roGFP2 reflects the real-time redox status of roGFP2 in mycobacteria, as previously demonstrated (70).
The mid-log-phase M. tuberculosis cells carrying genome-integrated Mrx1-roGFP2 showed a low ratio of fluorescence corresponding to the 405 nm/488 nm excitation maxima, indicating a highly reduced environment (Fig. 7A and G). The ratio of fluorescence corresponding to the 405 nm/488 nm excitation maxima of Mrx1-roGFP2 gradually increased during the period of exposure to RIF, reaching the highest value in the RIF persistence phase cells, followed by a reduction in the regrowth-phase cells (Fig. 7B to D and G). The fluorescence ratio of the RIF persistence phase cells was significantly higher than that of the mid-log-phase cells (Fig. 7G). The histogram overlays of the roGFP2 fluorescence of the oxidized and reduced forms of Mrx1-roGFP2 showed that the general oxidative status of the cells in the RIF persistence and killing phases were comparable and higher than those of the cells in the regrowth and mid-log phases (Fig. 7E, F, and G). Although Mrx1-roGFP2 would be indicative of the overall redox status of the cells, the relatively high oxidative status of the RIF persistence phase cells correlated well with the elevated levels of hydroxyl radical in them (Fig. 5 and 6). Thus, the continued and prolonged exposure to RIF changed the intracellular redox status of the bacilli to a relatively high oxidized status in the RIF persistence phase cells, which later returned to a less oxidized status once the cells had acquired RIF resistance and come out of the persistence phase into the regrowth phase.
FIG 7.

Detection of oxidative stress status in RIF-exposed M. tuberculosis cells using redox-sensitive roGFP2. (A to D) Representative density plots of the flow cytometry profile of the M. tuberculosis-Mrx1-roGFP2 integrant in the different samples showing redox changes in bacteria during RIF exposure. (E and F) Corresponding histogram overlay of fluorescence from Mrx1-roGFP2 in the reduced and oxidized states. Ex., excitation. (G) Bar graph showing the ratiometric changes in the median values of roGFP2 (excitation at 405 and 488 nm) for the same experiment. Double asterisks indicate a P value of ≤0.01. Statistical significance was calculated using the paired t test.
Selection of MXF-resistant mutants from RIF persistence phase cells.
Since RIF resistance mutations appeared to have been generated by hydroxyl radical, which is not a sequence-specific mutagen (75), it was quite possible that the genome incurred mutations elsewhere as well, besides at the RRDR of the rpoB gene. If this is the case, then it must be possible to select de novo resistant mutants to other antibiotics as well from the RIF persistence phase cells. In order to verify this contention, an LD fluctuation test (62) was performed essentially as mentioned before, except that the RIF-exposed cells in the presence and absence of TU were plated on MXF plates (MXF, 2 μg/ml; 4× MBC) (47), in addition to using RIF plates (5 μg/ml; 50× MBC), to score for the resistant mutants against the respective antibiotic and on antibiotic-free plates to determine CFU. The LD experiment profile revealed that the MXF-resistant mutants could be selected from the RIF persistence phase cells (Fig. 8A). The fluctuation in the number of MXF-resistant colonies from different individual cultures of different days and from different cultures of the same day revealed their independent de novo generation. The reduction in the emergence of MXF-resistant mutant colonies in the presence of TU (Fig. 8B) once again confirmed the involvement of hydroxyl radical in the generation of the resistant mutants. As in the earlier experiment (see Fig. 3), higher numbers of RIF-resistant mutants emerged from the TU-unexposed RIF persistence phase cells than from the TU-exposed RIF persistence phase cells (Fig. 8C and D). These observations were consistent with the contention that genome-wide high levels of random mutations generated by the hydroxyl radical formed in response to exposure to RIF would facilitate selection of de novo resistant mutants to MXF from the RIF persistence phase cells.
FIG 8.

Luria-Delbruck (modified) experiment showing the emergence of RIF-resistant and MXF-resistant mutants from RIF persistence phase M. tuberculosis cells in the absence and presence of TU. (A and B) MXF-resistant mutants formed from the RIF persistence phase M. tuberculosis cells in the absence and presence of TU. The numbers in green boxes indicate the numbers of MXF-resistant mutants obtained from the respective cultures by plating the cells on a 4× MBC MXF plate. (C and D) RIF-resistant mutants formed from the RIF persistence phase M. tuberculosis cells in the absence and presence of TU, respectively. The numbers in the red boxes indicate the numbers of RIF-resistant mutants obtained from the respective culture.
M. tuberculosis MXF-resistant mutants emerging from the RIF persistence phase cells had mutation at the QRDR of gyrA.
Nine MXF-resistant mutants from the LD experiment showed a single type of mutation, G→A at the 280th nucleotide position, GAC→AAC, in the gyrA gene, yielding an Asp→Asn change at the 94th amino acid position at the quinolone resistance-determining region (QRDR) of gyrA (Table S8). Earlier studies on MXF-resistant or ciprofloxacin-resistant clinical isolates of M. tuberculosis cells with high MBC for MXF or ciprofloxacin have shown an identical Asp94→Asn mutation in the gyrA gene (76–82). The frequency of formation of MXF-resistant mutants from the RIF persistence phase cells was found to be (5.45 ± 1.69) × 10−4, which was 4-log10 higher than the natural mutation frequency of 10−8 for M. tuberculosis cells against fluoroquinolones, as reported by other groups (76, 81, 82). Such a significant change in the mutation frequency implied the involvement of a potent mutagen such as hydroxyl radical (64). Although MXF-resistant mutants emerged from the RIF persistence phase of M. tuberculosis cultures exposed to lethal concentrations of RIF due to high levels of hydroxyl radical-mediated mutagenesis, the RRDR of rpoB of the MXF-resistant mutants did not carry any mutation. We did not screen for the possibility of a RIF- and MXF-resistant double mutant, as the frequency of generation of such a mutant would be 10−8 since the frequency of generation of the individual mutants was 10−4 each. Nevertheless, the emergence of RIF-resistant and MXF-resistant mutants from the RIF persistence phase cells implied that the RIF persistence phase cells formed upon exposure to lethal concentrations of RIF are a pool of hydroxyl radical-generated mutants from which mutants resistant to RIF and to other antibiotics, such as MXF, could be selected.
Whole-genome sequencing of M. tuberculosis RIF-resistant mutants reveals genome-wide mutations.
Selection of MXF-resistant mutants from RIF persistence phase cells indicated the possibility of mutations at multiple sites in the genome of persistence phase cells. Therefore, in order to verify whether the RIF persistence phase cells carry genome-wide mutations, we carried out whole-genome sequencing of four RRDR-specific mutation-containing RIF-resistant mutants and of the parental wild-type strain. These mutants were R1-B2-1, R1-B2-4, R2-B1-1, and R3-B1-3, which arose from RIF persistence phase cells from three independent biological replicate cultures (R1, R2, and R3) (Fig. 2). Consistent with our premise, whole-genome sequencing showed extensive mutations throughout the genome (Fig. 9A and Data Set S1) of all of the RIF-resistant mutants compared to the parental strain. The mutations in the four RIF-resistant mutants were spread throughout the 4.4-Mbp genome without any preference for a specific region in the genome (Fig. 9A and Fig. S3 to S6).
FIG 9.

Whole-genome sequencing analysis of four RIF-resistant mutants. (A) Overlaid Circos plot of four RIF-resistant mutants showing the genome-wide mutations with respect to the parental strain. (B) Bar graphs showing the kinds of base substitutions and their percentages in the RIF-resistant mutants, indicating A:T→C:G as the most prevalent mutation (69%), followed by the A:T→T:A (25%) changes. (C) Dot plot representing the position of common mutations in the four RIF-resistant mutants with respect to the parental strain, showing their independent origin. The y axis represents the position of the mutations, and the x axis indicates the number of common mutations. (D) Bar graph showing the number of genes mutated in each of the RIF-resistant mutants. (E and F) Growth curves of the RIF-resistant mutants with respect to that of the parental strain.
Out of the 12 possible base changes, an average of 69% of mutations were A→C and T→G, followed by 25% A→T and T→A combined, in all four mutants (Fig. 9B). These mutations, which together constituted 94% of all of the single-nucleotide polymorphisms (SNPs), were characteristic of oxidative stress-induced changes, as reported earlier (75, 83). The incorporation of 8-Oxo-dGTP against dA during DNA replication can lead to a specific type of A:T→C:G base substitution mutation (84). The dGTP in the nucleotide pool is more highly prone to oxidation by hydroxyl radical than dG in the DNA. This is the probable reason for the higher level of A:T→C:G than G:C→T:A mutation, which is mediated through 8-Oxo-dGTP incorporation into the DNA. In addition, the second most prevalent transversion mutation, A:T→T:A, is also characteristic of ROS-mediated base substitution mutations, as reported earlier (75).
Forty-nine genes have suffered mutations in all four resistant mutants (Fig. 9C and Data Set S2). On the other hand, the resistant mutant R3-B1-3 incurred 113 additional mutations that were not present in the other three mutants (Data Set S3). The different extents of mutations in different sets of genes of specific pathways or cellular processes in the four RIF-resistant mutants (R1-B2-1, R1-B2-4, R2-B1-1, and R3-B1-3) carrying genome-wide mutations are shown in comparison to the sequence of the parental strain (Fig. S7). Although these four different RIF-resistant mutants suffered different extents of mutation (Fig. 9D and Data Set S1), none of these incurred mutations in either gyrA or gyrB genes for MXF resistance, as they were selected for RIF resistance but not for MXF resistance. Nevertheless, genome-wide mutations in each of the RIF-resistant mutants confirmed the occurrence of genome-wide random mutagenesis due to elevated levels of hydroxyl radical generated upon exposure to lethal concentrations of RIF.
The RIF-resistant mutants incurred fitness cost.
RIF-resistant mutants of M. tuberculosis have been found to suffer from growth defects which have been attributed to the fitness cost due to mutations incurred in the rpoB gene (50, 85–89). However, compensatory mutations in the rpoA and rpoC genes have been found to restore the normal growth rate of RIF-resistant mutants (87, 88). In order to assess the fitness cost of the mutations, the growth of the four RIF-resistant mutants (R1-B2-1, R1-B2-4, R2-B1-1, and R3-B1-3), which incurred four different mutations (H445→D, H445→Y, S450→L, and S450→W, respectively; Fig. 2) in the rpoB gene, was compared with that of the parental strain. The four RIF-resistant mutants showed a compromised growth rate (Fig. 9E and F) which could be due to the fitness cost imposed by the rpoB mutations, as reported previously (50, 85–89), and probably also is due to additional genome-wide mutations in the respective mutants. Among the four RIF-resistant mutants, three mutants (R1-B2-1, R1-B2-4, and R2-B1-1), which shared mutations in 49 genes, grew much slower than the parental strain (Fig. 9E and F). R3-B1-3, which incurred 113 additional mutations that were absent from the other three mutants, was the slowest-growing mutant among them. The better-growing mutants had H445→D and S450→L mutations, while the much slower-growing mutants possessed H445→Y and S450→W mutations. Thus, while H445→D and S450→L mutations did not severely affect growth, H445→Y and S450→W mutations severely affected growth, indicating that the type of mutations in the rpoB gene also has a role in the fitness cost. Consistent with the slow growth of these mutants, none of the four mutants incurred compensatory mutations on other genes, like rpoA or rpoC (data not shown), that otherwise could have restored normal growth rates to them (87, 88).
DISCUSSION
The triphasic response of M. tuberculosis cells to prolonged exposure to lethal concentrations of RIF and MXF.
Although there have been several studies showing the presence of antibiotic persister cells in the antibiotic-exposed population of tubercle bacilli in TB patients (9–15), animal models (16–27), infected macrophages (28, 29), and in vitro cultures (30–32), the possibility of the generation of antibiotic-resistant mutants from the persistence phase cells remained mostly unexplored or unnoticed. Prolonged treatment with streptomycin for 6 months or more showed the emergence of streptomycin-resistant strains in TB patients from whom the M. tuberculosis strains isolated before treatment were sensitive to streptomycin (90, 91). In a study where macrophages infected with virulent M. tuberculosis cells were exposed to rifapentine for 28 days, with the antibiotic added on a daily basis to maintain its pharmacokinetics in the blood, the CFU showed a dramatic decrease in the first 14 days due to killing, followed by a plateau for about 7 days, probably due to persistence, and a subsequent increase in spite of high concentrations of the antibiotic (28). The emergence of antibiotic-resistant mutants in the presence of high concentrations of the antibiotic might have been one of the contributing factors for the rise in the CFU in these studies involving exposure of the bacilli to antibiotics for prolonged periods, although this possibility was not commented upon (28, 90, 91). Thus, the extracellular and intracellular virulent bacilli in TB patients and macrophages also seem to show a triphasic response to the antibiotics (28, 91). This in turn supports and validates the present study performed in vitro using the avirulent M. tuberculosis strain. Our studies performed using avirulent M. tuberculosis cells cultured in vitro are also in concurrence with the recent demonstration of the presence of persistent tubercle bacilli in the Cornell mouse model exposed to RIF, isoniazid (INH), and pyrazinamide (PZA) for 14 weeks, wherein the persistent bacilli were suggested to be the cause of the relapse of disease observed (20). It may be interesting to find out whether the persistent bacilli were the source for the drug-resistant mutants that might have contributed to disease relapse.
Recently, in chemostats, streptomycin-resistant E. coli cells, carrying mutations in the rpsL gene, were found to emerge at high frequency after a killing phase and a lull period from the cultures exposed to increasing concentrations of the antibiotic (92). It was suggested that these streptomycin-resistant E. coli mutants were preexistent in the population in the chemostat cultures but came up with higher frequency once the antibiotic-sensitive population became extinct or nearly extinct (92). In this study, the lull period was observed after the killing phase and was followed by the emergence of streptomycin-resistant mutants at high frequency. Nevertheless, it might be interesting to find out whether the lull period prior to the emergence of the mutants at high frequency was the streptomycin persistence phase and whether the emergence of the mutants was de novo and due to hydroxyl radical-mediated mutagenesis during the lull period.
High levels of oxidative stress in RIF persistence phase cells.
Gram-negative and Gram-positive bacteria that were exposed to lethal concentrations of antibiotics have been found to have a high degree of oxidative stress due to elevated levels of ROS (68, 93–97). M. tuberculosis, which was exposed to 8 μg/ml of RIF but only up to 3 days (pertaining only to the early part of killing phase) in vitro, showed generation of hydroxyl radical, the levels of which increased with the increase in RIF concentration (98). However, our study involving prolonged and continued exposure to RIF showed that the levels of hydroxyl radical generation were steadily increasing during the killing phase, reaching significantly high levels in the RIF persistence phase. The single peak of fluorescence shown by the RIF persistence phase cells (Fig. 6E) suggested that the generation of hydroxyl radical was occurring in all surviving cells, but only a proportion of them undergo hydroxyl radical-mediated mutagenesis (see Table S5). This also ruled out the possibility of the existence of another subpopulation of preexisting RIF-resistant mutants of low fluorescence in the persistence phase population. The reduction in the median value of HPF fluorescence in the regrowth-phase population indicated that the generation of hydroxyl radical by the RIF-exposed cells occurred only as long as the antibiotic was able to inflict its action on the cells.
In our study, using multiple approaches, such as EPR, HPF fluorescence, and M. tuberculosis-Mrx1-roGFP2 integrant cells, we quantitatively validated the presence of elevated levels of hydroxyl radical and the resultant highly oxidizing environment in the RIF persistence phase cells, the mutagenic ability of which remains undisputed (99–101). Earlier studies in other bacteria have also shown that while exposure to hydrogen peroxide alone brought about a 7-fold increase in the mutation rate due to hydroxyl radical, the combination of hydrogen peroxide and thiourea reduced the mutation rate from 7-fold to about 1.5-fold (38). Although the RIF susceptibility kinetics of the TU-treated and the TU-untreated cultures were comparable (Fig. 3C), there was a significant delay in colony formation in the case of TU-treated persistence phase cells on RIF-free and RIF-containing agar plates. In addition, the colonies of the TU-exposed cells were very small compared to those of the TU-untreated cells, showing their effect on growth. Further, the nature of the mutations C→T (60) and C→G (61), which were indicative of oxidation-induced changes, and the generation of elevated levels of mutants from the RIF persistence phase population correlated well with the high levels of hydroxyl radical production and high oxidation status of the RIF persistence phase cells.
Recently, it was reported that hydroxyl radicals generated against dual combinations of RIF, INH, clofazimine (CFZ), and ciprofloxacin (CIP) could be used to eradicate M. tuberculosis persisters (102). The study found that high levels of ROS, which were formed due to high levels of dissolved oxygen (DO) in the growth medium, led to killing of the bacilli exposed to dual combinations of those antibiotics over time. When the DO levels were reduced, the levels of ROS generated were also low, with the concomitant stabilization of the CFU. Higher levels of DO caused greater reduction in the CFU in all dual combinations, except in the case of CIP-INH combination against M. smegmatis. However, none of the other combinations of the antibiotics could completely sterilize either the M. smegmatis or M. tuberculosis bacilli in the given period of 6 to 8 days, as the CFU graph showed a plateau (102), probably indicating persistence. Thus, these observations do not preclude the possibility of the emergence of antibiotic-resistant bacilli from the persistence phase had the exposure been continued for more days. Nevertheless, our studies are consistent with these observations, and both studies put together suggest that even though DO content in the bacillary environment does have a role in facilitating the decrease in the CFU, after a drastic reduction in the CFU during the killing phase, the bacilli do get into persistence phase, where a portion of the persistence phase cells emerge as antibiotic-resistant mutants.
De novo generation of RIF/MXF-resistant mutants.
The possibility of the presence of preexisting mutants was ruled out by the data from the Luria-Delbruck experiment showing that the RIF/MXF-resistant mutants were formed de novo. Keeping the Luria-Delbruck plates for 6 months also did not show the emergence of RIF/MXF-resistant mutants from the early days' samples, although more colonies came up from the RIF persistence phase population. Further, the fluctuation in the number of the RIF-resistant mutant colonies formed, especially from the RIF persistence phase, ruled out the possibility of the cells in the regrowth phase being the result of catching up of slow-growing mutants present at very low abundance in the parental population. The RIF resistance mutation frequency was determined from the CFU obtained from the persistence phase in the LD experiment, since de novo formation of RIF-resistant mutants occurred during early days of the persistence phase (day 10 to day 15). This would reduce the possibility of counting enriched resistant mutants (due to proliferation of the mutants) and thereby avoid any overestimation of RIF resistance frequency. In our attempts to carry out similar experiments with INH, we found that the natural mutation frequency of M. tuberculosis cells against INH was 10−6 (data not shown), as reported previously (103). Thus, the level of preexisting INH-resistant mutants would be high in a mid-log-phase culture and would interfere with the detection of the de novo generation of INH-resistant mutants. In addition, multiple mechanisms have been cited for the emergence of INH resistance (104). These features of the response of M. tuberculosis cells against INH would have complicated the interpretations in terms of the proportion of the cells that emerged de novo as INH-resistant mutants vis-a-vis the proportion of the preexisting INH-resistant mutants. Hence, INH was not used in the present study.
We observed that the bacilli in the RIF persistence phase showed a high frequency of the emergence of RIF-resistant mutants with respect to the RIF-unexposed actively growing M. tuberculosis cells. The elevated levels of genome-wide mutations might account for the high RIF resistance frequency of 10−4 compared to the natural mutation frequency of 10−9. The high frequency of the emergence of RIF-resistant mutants in turn correlated well with the elevated levels of hydroxyl radical generation in these cells. It is possible that in some cases (e.g., 13-R3, 13-R10, and 17-R3, which were sequenced; Fig. 3 and 4) the culture contained a higher number of mutants, probably due to enrichment of an already existing mutant. On the contrary, in some cases we found multiple kinds of RIF-resistant mutants (e.g., 16-R1 and 16-R4, which were sequenced; Fig. 3 and 4), indicating that they were formed de novo and were not from an enriched population.
We observed that colony formation after exposure to RIF was delayed (5 to 6 weeks minimum), especially from the RIF persistence phase compared to the RIF-unexposed cells (mid-log phase), which usually take 4 weeks to form a visible colony on the plate. From some samples, colonies have emerged only upon prolonged incubation for 2 to 3 months. There are samples from which we could get colonies on RIF-free plates but not on RIF-containing plates and vice versa. In addition, there are time points where we did not find any colonies on RIF-free as well as RIF-containing plates (R1-15 and R2-10 in Table S5). It is interesting that some other day's samples do not show such problems. Our study showed that exposure of M. tuberculosis cells to a high MBC of RIF inflicts severe growth compromise on the cells due to hydroxyl radical-mediated genome-wide mutagenesis on genes other than rpoB as well. Thus, their growth gets affected when they were plated even on RIF-free plates and more so on RIF-containing plates. Thus, the variations in the formation of colonies indicated that the extent of growth compromise suffered by the mutants also varied due to different extents of mutagenesis inflicted by ROS. The same was also the case with the MXF-exposed samples, with the colony formation delayed much more than that in the case of RIF-exposed samples (Table S7).
The generation of very low numbers of RIF-resistant mutants even in the presence of the hydroxyl radical quencher TU might be due to mechanisms of mutagenesis involving reactive oxygen species other than hydroxyl radical (99–101). Evidence for the presence of other types of reactive oxygen species, such as superoxide anion and hydrogen peroxide (105), in bacterial cells exposed to antibiotics (96) and their ability to cause mutations (99–101) support this possibility. The high frequency of generation of the RIF/MXF-resistant M. tuberculosis mutants also argues against the possibility of their emergence due to aging of the cells during the prolonged exposure and consequential mutagenesis found in aging E. coli cells at a low frequency of 10−8 under many stress conditions (106).
Dynamic changes of the redox status in the cells exposed to RIF.
Since only about 0.001 to 0.004% of the mid-log-phase cells (Tables S3 to S5) which were exposed to lethal concentrations of RIF were present in the RIF persistence phase, one may wonder whether the hydroxyl radical detected from the RIF persistence phase cells was from live persister cells or dead/dying cells in the population. The consistently and repeatedly reproducible dynamism in the transition from the reduced status of mid-log-phase cells to the relatively high oxidized status of RIF persistence phase cells and subsequent reversal of the oxidized status to relatively less oxidized status in the regrowing cells (in the regrowth phase), which emerged from the RIF persistence phase population, strongly suggested that the changes in the oxidation status of the cells occurred in the live RIF persistence cells only. Further, although chemical reagents such as HPF may not be specific to hydroxyl radical from live cells only, the redox biosensor Mrx1-roGFP was designed to show redox changes in live cells in real time (70). Therefore, the dynamic changes in the oxidative status of Mrx1-roGFP, from reduced status in the mid-log-phase cells to highly oxidized status in the RIF persistence phase cells, with subsequent reversal to reduced status in the regrowth-phase cells, indicated that the changes occurred in live persistence phase cells only. The dynamism proposed in the renewal of INH persistence phase M. smegmatis cells in vitro (7) also supports the possibility that the changes take place in live RIF persistence phase cells only.
Implications of the emergence of RIF/MXF-resistant mutants from RIF persistence phase cells.
Whole-genome sequencing of four RIF-resistant mutants showed that 94% of all of the SNPs, constituted by A-C or T-G (69%) and A-T and T-A (25%) mutations, are characteristic of oxidative stress-induced changes, as reported earlier (75, 83). The hydroxyl radical-mediated genome-wide mutations in the four RIF-resistant mutants and the selection of MXF-resistant mutants from the RIF persistence phase cells clearly demonstrated that the RIF persistence phase cells are a source pool for the emergence of antibiotic-resistant mutants. We did not attempt to select a combined RIF-MXF double resistant mutant from the RIF persistence phase cells due to the very low combined frequency (10−4 × 10−4 = 10−8; mutation frequency of 10−4 each for the RIF- and MXF-resistant mutants; see Results) of emergence of the mutant. It was speculated that once a persistence phase cell survives the killing action of ROS, the continued exposure to the same ROS could increase the opportunity to acquire mutations and gain resistance (42).
Antibiotic persistence phase cells form a reservoir for the generation of genetically resistant mutants.
Our findings on the emergence of RIF/MXF-resistant mutants against the respective antibiotic from RIF persistence phase cells have substantiated and provided experimental proof that the antibiotic persistence phase cells are a reservoir from which mutants that are resistant to the same antibiotic or another antibiotic could emerge and get selected. Although the emergence of RIF/MXF-resistant mutants from the RIF persistence phase cells in the present work was demonstrated using M. tuberculosis cells exposed to RIF in vitro, the identity of the mutations to those that were clinically relevant in TB patients the world over (52–59, 76–82) indicated that the phenomenon of the emergence of RIF/MXF-resistant mutants from the RIF persistence phase might have been happening in vivo as well. Distinct from the previous studies, which showed emergence of genetically resistant Escherichia coli, Staphylococcus aureus, and Pseudomonas aeruginosa mutants upon short-duration exposure to sublethal concentrations of antibiotics (38–42), our study demonstrates that continuous prolonged exposure of M. tuberculosis cells to lethal concentrations of RIF causes de novo emergence of RIF- or MXF-resistant mutants from the RIF persistence phase cells at high frequency.
MATERIALS AND METHODS
Bacterial cultures.
Mycobacterium tuberculosis H37Ra cells (107) (obtained from the National JALMA Institute of Leprosy and Other Mycobacterial Diseases, Agra, India; see Table S1 in the supplemental material) were cultured in Middlebrook 7H9 broth (Difco) containing 0.2% glycerol and 0.05% Tween 80 and supplemented with 10% ADS (albumin, dextrose, and saline). The M. tuberculosis cells were plated on Middlebrook 7H10 agar and were incubated in a CO2 incubator (5% CO2, 95% humidity) at 37°C for 20 to 25 days or more, if the colonies took more time to grow, to determine CFU per milliliter. Escherichia coli JM109 (108) (Table S1) cells were grown in LB medium for the propagation and preparation of plasmids for cloning purposes.
Exposure of M. tuberculosis cells to antibiotics.
Rifampin (RIF; Sigma) and moxifloxacin (MXF; Cayman Chemical Company, USA) powders were dissolved in dimethyl sulfoxide (DMSO) to get a concentration of 2 mg/ml each. Antibiotic solutions were always prepared fresh and filter sterilized using a 0.22-μm syringe filter. Measured volumes of antibiotics were added to actively growing M. tuberculosis cultures so as to get concentrations of 1 μg/ml of both antibiotics or 2 μg/ml of RIF. RIF at 1 μg/ml amounted to 10× MBC (determined in the laboratory; Fig. S1), and 1 μg/ml for MXF was about 2× MBC for MXF, as reported previously (47). After mild sonication (3 pulses of 1 s each with a 1-s interval at 16% amplitude using a micro probe in a Vibra-Cell; Sonics & Materials, Inc., USA), serial dilution and plating were carried out in sterility-checked Middlebrook 7H10 agar supplemented with 10% ADS. The plates containing M. tuberculosis cells were sealed with Parafilm and incubated at 37°C under 5% CO2 in a CO2 incubator. Resistant bacteria were selected on Middlebrook 7H10 agar plates and supplemented with 10% ADS containing 5 μg/ml (50× MBC, per the MBC determined in the laboratory) of RIF or 2 μg/ml of MXF (4× MBC) (47).
RIF and MXF bioassays.
RIF- and MXF-sensitive Staphylococcus aureus (ATCC 25923) was used for the bioassay for RIF (109) and MXF (110). Staphylococcus aureus cells were embedded in LB agar medium, and wells were made in the agar (0.5-cm diameter). Fifty microliters of RIF or MXF solution or culture supernatant was added to the wells and incubated overnight at 37°C. The diameter of the zone of growth inhibition was measured using Vernier Calipers. A standard graph was prepared with known concentrations of RIF or MXF for calculating unknown concentrations from the diameter of the zone of growth inhibition.
Determination of MBC for RIF.
MBC was determined by incubating 1:100 dilutions of mid-log-phase cells with decreasing concentrations of RIF, from 1 μg/ml to 0.1 μg/ml, in 2-ml Cryovials for 6 days at 37°C. Cells were plated after mild sonication (3 pulses of 1 s each with a 1-s interval at 16% amplitude using a micro probe in a Vibra-Cell; Sonics & Materials, Inc., USA), followed by serial dilution on Middlebrook 7H10 agar plates before and after the treatment. MBC was defined as the lowest concentration of the antibiotic in the medium that decreased the bacterial population by 2 or more log10 units within 6 days of incubation (28).
Luria-Delbruck experiments.
In order to verify the emergence of RIF-resistant M. tuberculosis, we performed Luria-Delbruck experiments (62) in a modified form. Freshly prepared RIF (1 μg/ml) was added to two 100-ml actively growing mid-log-phase (optical density at 600 nm [OD600] of 0.6) M. tuberculosis cultures, and immediately 400-μl aliquots were made in 2-ml screwcap Cryovial tubes and incubated at 37°C and 200 rpm in a bacteriological shaker for 19 days (19 days × 13 tubes = 247 tubes). Once every 24 h from day 0 onwards, 13 tubes were randomly picked, and the entire 400-μl culture from 10 of those tubes was plated independently on individual Middlebrook 7H10 agar plates containing 5 μg/ml RIF. Cultures from the remaining three tubes were used for serial dilution (after mild sonication of 3 pulses of 1 s each with a 1-s interval at 16% amplitude using a micro probe in Vibra-Cell; Sonics & Materials, Inc., USA) and plated on RIF-free Middlebrook 7H10 agar plates.
On day 8 of the exposure, freshly prepared, filter-sterilized thiourea (TU; 150 mM final concentration) (102) was added to the rest of the tubes in one set of the samples. The TU-treated cells were washed once with Middlebrook 7H9 medium before plating on RIF-containing plates, which were sealed with Parafilm and incubated at 37°C in a CO2 incubator. RIF resistance frequency was calculated by dividing the total number of RIF-resistant mutants from the RIF-containing plates by the total number of RIF-exposed cells from RIF-free plates. For the estimation of natural RIF resistance frequency, cells from the mid-log-phase culture (OD600 of 0.6) were plated (undiluted) on Middlebrook 7H10 agar plates containing 5 μg/ml RIF. The total number of colonies obtained from RIF plates was divided by the total number of cells plated, which was estimated from the CFU per milliliter on RIF-free Middlebrook 7H10 agar plates. For the determination of the frequency of emergence of RIF-resistant mutants from the RIF persistence phase population, all of the RIF-resistant colonies from the cultures (in screwcap Cryovial tubes) of the RIF persistence phase (day 10 to day 15) were considered.
Similarly, an experiment was performed to determine the MXF-resistant mutant generation from RIF persistence phase cells. The exception in this experiment was that out of the 13 tubes containing RIF-exposed cells used for plating on each day, entire cultures from five tubes were used for plating on the 2 μg/ml MXF (4× MBC)-containing plate after washing the cells with Middlebrook 7H9 medium to remove traces of RIF. Similarly, entire cultures from another five tubes were used for plating on 5 μg/ml RIF-containing plates to determine the emergence of RIF-resistant mutants. The cultures from the remaining three tubes were used for plating on antibiotic-free plates, after serial dilution, for the determination of CFU per milliliter. On the 8th day of the exposure, freshly prepared, filter-sterilized thiourea (150 mM final concentration) (102) was added into the rest of the tubes in one set of the samples. The entire culture from 5 tubes each were plated on MXF and RIF plates as described above. For the determination of the frequency of emergence of MXF-resistant mutants from the RIF persistence phase population, all of the MXF-resistant colonies from the cultures (in screwcap Cryovial tubes) of RIF persistence phase cells (day 10 to day 15) were considered. The number of cells plated was estimated from the CFU per milliliter on antibiotic-free Middlebrook 7H10 agar plates.
EPR analysis of RIF-exposed M. tuberculosis cells.
M. tuberculosis cells from mid-log phase, antibiotic-exposed RIF persistence phase, and TU-treated RIF persistence phase (150 mM TU was added on the 8th day of treatment) were harvested from 20-ml cultures, and the cell pellet was snap-frozen in liquid nitrogen and lysed using a Teflon pestle. The powdered cell lysate was resuspended in 200 μl of 100 mM sodium acetate (pH 5.2) and centrifuged at 12,000 × g for 5 min at room temperature. The supernatant (180 μl) was then mixed with 20 μl of 1 M DMPO to achieve a 100 mM final concentration (98). Samples were immediately loaded into an aqueous flat cell (ES-LC12), and a reading was taken in a JEOL JES-X3 ESR spectrometer exactly 2 min after the addition of DMPO using the following parameters: X-band frequency, 9428.401 (MHz); power, 4.00 (mW), field center, 337.275 (mT); sweep time, 2.0 (min). Electron paramagnetic resonance (EPR) signals were obtained at a g factor of ≈2. Data were processed using Wizard of Baseline and Peaks in OriginPro 8 software. Statistical significance between the samples was calculated using Student's t test.
HPF staining of M. tuberculosis cells.
Five hundred microliters of M. tuberculosis cells from each of the biological triplicates at different stages of exposure to RIF (1 μg/ml) were collected, and 1 μl of 5 mM 3′-(p-hydroxyphenyl) fluorescein (HPF; Invitrogen) (67) solution was added (to reach a 10 μM final concentration) at 25°C in the dark and incubated at 37°C for 1 h in the dark under shaking conditions (170 rpm in a bacteriological incubator). Cells were collected by centrifugation at 12,000 × g for 5 min at room temperature, resuspended in 500 μl of Middlebrook 7H9 broth, and immediately used for flow cytometry analysis. Data were collected using a Becton Dickinson FACSVerse flow cytometer with a 488-nm solid state laser and a 527/32 nm emission filter (GFP) at low or medium flow rate. About 50,000 cells were collected for each sample. The photomultiplier tube (PMT) voltage settings were 208 (forward scatter [FSC]) and 333 (side scatter [SSC]), and the median fluorescence was kept at 2-log10 fluorescence units for the autofluorescence control. The shift of fluorescence of HPF-stained cells from the autofluorescence control was determined for every time point. A FACSuite cytometer setup and tracking beads (CS&T; Becton Dickinson) were used for instrument calibration. Flow cytometry data were processed and analyzed with FACSuite software. Statistical significance between the time points was calculated using the paired t test of GraphPad Prism, version 5.0.
Cloning and expression of Mrx1-roGFP2.
Mrx1-roGFP2, along with the hsp60 promoter, was released from pMV762-Mrx1-roGFP2 (70) (kind gift from Amit Singh, Department of Microbiology and Cell Biology, Indian Institute of Science) using ClaI and XbaI and cloned into pAKMN2 (111) after removing gfpm2+ using the same restriction enzymes (Table S1). The ligated product was used to transform E. coli JM109 cells to get transformants carrying the hygromycin-resistant recombinant pAKMN2-Mrx1-roGFP2. After verifying the presence of Mrx1-roGFP2 in the recombinant vector, M. tuberculosis electrocompetent cells were transformed with the construct using the following electroporation conditions: voltage, 2.5 kV; capacitance, 25 μF; resistance, 1,000 Ω; cuvette width, 2 mm. Transformants were selected on a hygromycin-containing Middlebrook 7H10 agar plate and subcultured in the presence of hygromycin (50 μg/ml). The roGFP2 fluorescence of the M. tuberculosis integrants carrying Mrx1-roGFP2 at the mycobacteriophage L5 phage att site was verified using H2O2-treated cells as the positive control for oxidative stress and dithiothreitol (DTT)-treated cells as the negative control.
Measurement of redox status using M. tuberculosis-Mrx1-roGFP2 integrant cells.
In order to measure the redox status of roGFP2, mid-log-phase cells of M. tuberculosis integrant cultured in the absence of hygromycin were treated with 1 μg/ml of RIF and monitored for 18 days. Data were acquired from the samples at different time points using a Becton Dickinson FACSVerse flow cytometer with a 405-nm (V-500) and 488-nm (fluorescein isothiocyanate [FITC]) solid state laser and a 528/45-nm and 527/32-nm emission filter, respectively, at low or medium flow rate. At least 50,000 gated cells were considered for analysis from each sample. The PMT voltage settings were 208 (FSC) and 333 (SSC). FACSuite cytometer setup and tracking beads (CS&T; Becton Dickinson) were used for instrument calibration. Flow data were processed and analyzed with FACSuite software. The median fluorescence for V-500 and FITC was set to 2 log10 fluorescence units for RIF-treated wild-type M. tuberculosis at each time point. The biosensor response ratio from the drug-treated M. tuberculosis/pAKMN2-Mrx1-roGFP2 samples was calculated by dividing the median fluorescence obtained for 405 nm (V-500) by the median fluorescence obtained for 488 nm (FITC) and was used for plotting the graph. Statistical significance between the time points was calculated using the paired t test of GraphPad Prism, version 5.0.
Determination of mutations in the RRDR and QRDR loci.
RIF-resistant colonies and MXF-resistant colonies were isolated and inoculated into 5 μg/ml RIF-containing or 2 μg/ml MXF-containing Middlebrook 7H9 broth with ADS supplement, respectively, and grown to mid-log phase for genomic DNA isolation. The RRDR locus of rpoB and the QRDR locus of gyrA and gyrB were amplified using RRDR-specific and QRDR-specific primers (Table S2) and Phusion polymerase (Thermo Scientific, USA). The PCR products were purified using agarose gel electrophoresis and extracted from the gel with an extraction kit (GeneJET gel extraction kit; Thermo Scientific, USA), and the sequence was determined on both strands. The sequencing reactions were performed by Chromous Biotech, Bangalore, India.
Genomic DNA isolation.
Genomic DNA was isolated from wild-type M. tuberculosis cells and RIF-resistant and MXF-resistant mutants of M. tuberculosis cells using the phenol-chloroform extraction method. In brief, mid-log-phase cells from 20-ml cultures were treated with 0.2 M glycine (final concentration) overnight, harvested, resuspended in 1 ml of Tris-HCl-EDTA buffer (10 mM Tris-HCl containing 1 mM EDTA, pH 8), and digested with 5 mg/ml (final concentration) each of lysozyme and lipase simultaneously for 6 h at 37°C in a water bath. Cell lysis was carried out by adding 2% SDS (final concentration) and incubating at 55°C for 15 min. The lysate was centrifuged at 10,000 × g for 10 min at 4°C. The supernatant was used for phenol-chloroform extraction. The aqueous phase was collected, and the DNA was precipitated using 0.3 M sodium acetate (final concentration) and 2.5 volumes of 95% ethanol. DNA was washed once with 70% ethanol and dissolved in 1× Tris-HCl-EDTA buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8). RNase A (1 μl of 10-mg/ml stock solution) treatment was performed at 50°C for 30 min, and the DNA was reextracted using the phenol-chloroform method and reprecipitated as mentioned earlier. The genomic DNA thus prepared was dissolved in 1× Tris-HCl-EDTA buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8) and stored at 4°C.
Whole-genome sequencing of M. tuberculosis RIF-resistant mutants.
Whole-genome sequencing was performed by Genotypic Technologies, Bangalore, India. In brief, 2.5 μg of genomic DNA was sheared to generate fragments of approximately 200 to 600 bp in a Covaris microTUBE with the E2 system (Covaris, Inc., Woburn, MA, USA). These fragments were subjected to end-repairing, A-tailing, and ligation of the Illumina multiplexing PE adaptors using the NEXTFlex DNA sequencing kit per the manufacturer's protocol. The fragment size distribution was determined using a Bioanalyzer instrument (Agilent Technologies, Santa Clara, CA) with an Agilent high-sensitivity DNA kit (Agilent Technologies) according to the manufacturer's instructions. The resulting fragmented DNA was cleaned up using Agencourt AMPure XP SPRI beads (no. A63882; Beckman Coulter), size selected (∼300 to 600 bp) by running on a 2% low-melting-point agarose gel, and cleaned using a MinElute column (Qiagen). These adapter-ligated fragments were subjected to 10 rounds of PCR using primers provided in the NEXTFlex DNA sequencing kit. The PCR products were purified using Agencourt AMPure XP beads. Quantification and size distribution of the prepared libraries was determined using the Agilent Bioanalyzer 2100 DNA 7500 chip and Qubit fluorometer. Whole-genome sequencing was performed using Illumina NextSeq at 20× read depth. Bowtie 2-2.0.5 was used for sequence alignment. SAMtools 0.1.18 and SnpEFF 3.4 were used for variant detection.
Growth characteristics of the RIF-resistant mutants.
The wild-type and the four RIF-resistant M. tuberculosis strains were inoculated (0.1%) in drug-free 7H9 broth containing 10% ADS supplement. Optical density readings at 600 nm were taken once every 24 h and plotted against the time intervals to obtain the growth curve. In parallel, growth of the cells in a wild-type culture was also monitored in the same manner, and the values were plotted on the same graph for the RIF-resistant mutants. Care was taken to ensure that clump-free aliquots of the cultures were taken for reading during the time intervals monitored.
Supplementary Material
ACKNOWLEDGMENTS
We thank Uttara Chakraborty, Vasishta Adiga, Urvashi Chauhan, and Leepika Baid of the DBT-supported FACS facility, Biological Sciences Division, Indian Institute of Science, for technical advice and help in flow cytometry measurements. Technical help from N. Sivaramakrishnan and N. Chandrasekaran of IIT Chennai SAIF in EPR measurements is gratefully acknowledged. We also acknowledge Vikas Patil for technical help in whole-genome sequence analysis.
Funding was provided by DST-FIST, UGC Centre for Advanced Study, DBT-IISc Partnership Programme, ICMR Centre for Advance Study in Molecular Medical Microbiology, and the IISc. J.S. (DBT), S.S., R.R.N., A.P. (CSIR), and K.J. (UGC) received junior/senior research fellowships.
The funding agencies had no role in data collection and interpretation. We have no conflicts of interest to report.
On the 75th year of the genesis of the Department of Microbiology and Cell Biology (MCB), with highest respects and regards, P.A. dedicates this work as a tribute to Prof. T. Ramakrishnan (late), who led the pioneering and foundation-laying work on the biochemistry and molecular biology of Mycobacterium tuberculosis at the Indian Institute of Science, Bangalore.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01343-16.
REFERENCES
- 1.Bigger JW. 1944. Treatment of staphylococcal infection with penicillin by intermittent sterilisation. Lancet 244:497–500. doi: 10.1016/S0140-6736(00)74210-3. [DOI] [Google Scholar]
- 2.Cohen NR, Lobritz MA, Collins JJ. 2013. Microbial persistence and the road to drug resistance. Cell Host Microbe 13:632–642. doi: 10.1016/j.chom.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 4.Dorr T, Vulić M, Lewis K. 2010. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol 8:e1000317. doi: 10.1371/journal.pbio.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Y, Vulić M, Keren I, Lewis K. 2012. Role of oxidative stress in persister tolerance. Antimicrob Agents Chemother 56:4922–4926. doi: 10.1128/AAC.00921-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis K. 2010. Persister cells. Annu Rev Microbiol 64:357–372. doi: 10.1146/annurev.micro.112408.134306. [DOI] [PubMed] [Google Scholar]
- 7.Wakamoto Y, Dhar N, Chait R, Schneider K, Signorino-Gelo F, Leibler S, McKinney JD. 2013. Dynamic persistence of antibiotic stressed mycobacteria. Science 339:91–95. doi: 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- 8.Maisonneuve E, Castro-Camargo M, Gerdes K. 2013. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154:1140–1150. doi: 10.1016/j.cell.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 9.Stewart GR, Robertson BD, Young DB. 2003. Tuberculosis: a problem with persistence. Nat Rev Microbiol 1:97–105. doi: 10.1038/nrmicro749. [DOI] [PubMed] [Google Scholar]
- 10.Gomez JE, McKinney JD. 2004. M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis (Edinb) 84:29–44. doi: 10.1016/j.tube.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Opie EL, Aronson JD. 1927. Tubercle bacilli in latent tuberculous lesions and in lung tissue without tuberculous lesions. Arch Pathol 4:1–21. [Google Scholar]
- 12.Robertson HE. 1933. The persistence of tuberculous infections. Am J Pathol 9:S711–S719. [PMC free article] [PubMed] [Google Scholar]
- 13.Vandiviere HM, Loring WE, Melvin I, Willis S. 1956. The treated pulmonary lesion and its tubercle bacillus II. The death and resurrection. Am J Med Sci 232:30–37. doi: 10.1097/00000441-195607000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Jindani A, Aber VR, Edwards EA, Mitchison DA. 1980. The early bactericidal activity of drugs in patients with pulmonary tuberculosis. Am Rev Respir Dis 121:939–949. doi: 10.1164/arrd.1980.121.6.939. [DOI] [PubMed] [Google Scholar]
- 15.Khomenko AG. 1987. The variability of Mycobacterium tuberculosis in patients with cavitary pulmonary tuberculosis in the course of chemotherapy. Tubercle 68:243–253. doi: 10.1016/0041-3879(87)90064-X. [DOI] [PubMed] [Google Scholar]
- 16.McCune RM, Tompsett R. 1956. Fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. I. The persistence of drug susceptible bacilli in the tissues despite prolonged antimicrobial therapy J Exp Med 104:737–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCune RM, Tompsett R, McDermott W. 1956. The fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. II. The conversion of tuberculosis infection to the latent state by the administration of pyrazinamide and a companion drug. J Exp Med 104:763–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCune RM, Feldmann FM, Lambert HP, McDermott W. 1966. Microbial persistence. I. The capacity of tubercle bacilli to survive sterilisation in mouse tissues. J Exp Med 123:445–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCune RM, Feldmann FM, Mc Dermott W. 1966. Microbial persistence. II. Characteristics of sterile state of tubercle bacilli. J Exp Med 123:469–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y, Pertinez H, Ortega-Muro F, Alameda-Martin L, Liu Y, Schipani A, Davies G, Coates A. 2016. Investigation of elimination rate, persistent subpopulation removal, and relapse rates of Mycobacterium tuberculosis by using combinations of first-line drugs in a modified Cornell mouse model. Antimicrob Agents Chemother 60:4778–4785. doi: 10.1128/AAC.02548-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith DW, Balasubramanian V, Wiegeshaus E. 1991. A guinea pig model of experimental airborne tuberculosis for evaluation of the response to chemotherapy: the effect on bacilli in the initial phase of treatment. Tubercle 72:223–231. doi: 10.1016/0041-3879(91)90013-I. [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Kelley C, Collins F, Rouse D, Morris S. 1998. Expression of katG in Mycobacterium tuberculosis is associated with its growth and persistence in mice and guinea pigs. J Infect Dis 177:1030–1035. doi: 10.1086/515254. [DOI] [PubMed] [Google Scholar]
- 23.Lenaerts AJ, Hoff D, Aly S, Ehlers S, Andries K, Cantarero L, Orme IM, Basaraba RJ. 2007. Location of persisting mycobacteria in Guinea pig model of tuberculosis revealed by r207910. Antimicrob Agents Chemother 51:3338–3345. doi: 10.1128/AAC.00276-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kashino SS, Napolitano DR, Skobe Z, Campos-Neto A. 2008. Guinea pig model of Mycobacterium tuberculosis latent/dormant infection. Microbes Infect 10:1469–1476. doi: 10.1016/j.micinf.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmad Z, Pinn ML, Nuermberger EL, Peloquin CA, Grosset JH, Karakousis PC. 2010. The potent bactericidal activity of streptomycin in the guinea pig model of tuberculosis ceases due to the presence of persisters. J Antimicrob Chemother 65:2172–2175. doi: 10.1093/jac/dkq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmad Z, Fraig MM, Pinn ML, Tyagi S, Nuermberger EL, Grosset JH, Karakousis PC. 2011. Effectiveness of tuberculosis chemotherapy correlates with resistance to Mycobacterium tuberculosis infection in animal models. J Antimicrob Chemother 66:1560–1566. doi: 10.1093/jac/dkr188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoff DR, Ryan GJ, Driver ER, Ssemakulu CC, De Groote MA, Lenaerts AJ. 2011. Location of intra- and extracellular M. tuberculosis populations in lungs of mice and guinea pigs during disease progression and after drug treatment. PLoS One 6:e17550. doi: 10.1371/journal.pone.0017550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mor N, Simon B, Mezo N, Heifets L. 1995. Comparison of activities of rifapentine and rifampin against Mycobacterium tuberculosis residing in human macrophages. Antimicrob Agents Chemother 39:2073–2077. doi: 10.1128/AAC.39.9.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L. 2011. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145:39–53. doi: 10.1016/j.cell.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Y, Mangan JA, Dhillon J, Sole KM, Mitchison DA, Butcher PD, Coates AR. 2000. Detection of mRNA transcripts and active transcription in persistent Mycobacterium tuberculosis induced by exposure to rifampin or pyrazinamide. J Bacteriol 182:6358–6365. doi: 10.1128/JB.182.22.6358-6365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh R, Barry CE III, Boshoff HI. 2010. The three RelE homologs of Mycobacterium tuberculosis have individual drug-specific effects on bacterial antibiotic tolerance. J Bacteriol 192:1279–1291. doi: 10.1128/JB.01285-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keren I, Minami S, Rubin E, Lewis K. 2011. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2:e00100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Velayati AA, Farnia P, Mirsaeidi M. 2015. Persistence of Mycobacterium tuberculosis in environmental samples. Int J Mycobacteriol 4:1. doi: 10.1016/j.ijmyco.2014.11.005.26655191 [DOI] [Google Scholar]
- 34.Hobby GL, Auerbach O, Lenert TF, Small MJ, Comer JV. 1954. The late emergence of M. tuberculosis in liquid cultures in pulmonary lesions resected from humans. Am Rev Tuberc 70:191–218. [DOI] [PubMed] [Google Scholar]
- 35.Johnson R, Streicher EM, Louw GE, Warren RM, van Helden PD, Victor TC. 2006. Drug resistance in Mycobacterium tuberculosis. Curr Issues Mol Biol 8:97–111. [PubMed] [Google Scholar]
- 36.World Health Organization. 2015. Drug-resistant TB, p 54–66. In Global tuberculosis report 2015 , 20th ed World Health Organization, Geneva, Switzerland. [Google Scholar]
- 37.Matteelli A, Centis R, D'Ambrosio L, Migliori GB. 2012. Multidrug-resistant tuberculosis today. Bull World Health Organ 90:78. doi: 10.2471/BLT.11.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohanski MA, De Pristo MA, Collins JJ. 2010. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell 37:311–320. doi: 10.1016/j.molcel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jørgensen KM, Wassermann T, Jensen PØ, Henqzuang W, Molin S, Høiby N, Ciofu O. 2013. Sublethal ciprofloxacin treatment leads to rapid development of high-level ciprofloxacin resistance during long-term experimental evolution of Pseudomonas aeruginosa. Antimicrob Agents Chemother 57:4215–4221. doi: 10.1128/AAC.00493-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li GQ, Quan F, Qu T, Lu J, Chen SL, Cui LY, Guo DW, Wang YC. 2015. Sublethal vancomycin-induced ROS mediating antibiotic resistance in Staphylococcus aureus. Biosci Rep 35:e00279. doi: 10.1042/BSR20140167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laureti L, Matic I, Gutierrez A. 2013. Bacterial responses and genome instability induced by subinhibitory concentrations of antibiotics. Antibiotics (Basel) 2:100–114. doi: 10.3390/antibiotics2010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith T, Wolff KA, Nguyen L. 2013. Molecular biology of drug resistance in Mycobacterium tuberculosis. Curr Top Microbiol Immunol 374:53–80. doi: 10.1007/82_2012_279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV. 2015. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol 13:42–51. doi: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 44.Cavusoglu C, Karaca-Derici Y, Bilgic A. 2004. In-vitro activity of rifabutin against rifampicin-resistant Mycobacterium tuberculosis isolates with known rpoB mutations. Clin Microbiol Infect 10:657–678. doi: 10.1111/j.1469-0691.2004.00894.x. [DOI] [PubMed] [Google Scholar]
- 45.Taniguchi H, Aramaki H, Nikaido Y, Mizuguchi Y, Nakamura M, Koga T, Yoshida S. 1996. Rifampicin resistance and mutation of the rpoB gene in Mycobacterium tuberculosis. FEMS Microbiol Lett 144:103–108. doi: 10.1111/j.1574-6968.1996.tb08515.x. [DOI] [PubMed] [Google Scholar]
- 46.Huitric E, Werngren J, Jureen P, Hoffner S. 2006. Resistance levels and rpoB gene mutations among in vitro selected rifampin-resistant Mycobacterium tuberculosis mutants. Antimicrob Agents Chemother 50:2860–2862. doi: 10.1128/AAC.00303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez JC, Ruiz M, Loṕez M, Royo G. 2002. In vitro activity of moxifloxacin, levofloxacin, gatifloxacin and linezolid against Mycobacterium tuberculosis. Int J Antimicrob Agents 20:464–467. doi: 10.1016/S0924-8579(02)00239-X. [DOI] [PubMed] [Google Scholar]
- 48.Jin DJ, Gross CA. 1988. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol 202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- 49.Telenti A, Imboden P, Marchesi F, Lowrie D, Cole S, Colston MJ, Matter L, Schopfer K, Bodmer T. 1993. Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet 341:647–650. doi: 10.1016/0140-6736(93)90417-F. [DOI] [PubMed] [Google Scholar]
- 50.Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJM. 2006. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312:1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- 51.Ramaswamy S, Musser JM. 1998. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis 79:03–29. doi: 10.1054/tuld.1998.0002. [DOI] [PubMed] [Google Scholar]
- 52.Ahmad S, Araj GF, Akbar K, Fares E, Chugh TD, Mustafa AS. 2000. Characterization of rpoB mutations in rifampicin resistant Mycobacterium tuberculosis isolates from the Middle East. Diagn Microbiol Infect Dis 38:227–232. doi: 10.1016/S0732-8893(00)00200-5. [DOI] [PubMed] [Google Scholar]
- 53.Siddiqi N, Shamim M, Hussain S, Choudhary RK, Ahmed N, Prachee Banerjee S, Savithri GR, Alam M, Pathak N, Amin A, Hanief M, Katoch VM, Sharma SK, Hasnain SE. 2002. Molecular characterization of multidrug-resistant isolates of Mycobacterium tuberculosis from patients in North India. Antimicrob Agents Chemother 46:443–450. doi: 10.1128/AAC.46.2.443-450.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cavusoglu C, Hilmioglu S, Guneri S, Bilgic A. 2002. Characterization of rpoB mutation in rifampicin-resistant clinical isolates of Mycobacterium tuberculosis from Turkey by DNA sequencing and line probe assay. J Clin Microbiol 40:4435–4438. doi: 10.1128/JCM.40.12.4435-4438.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yue J, Shi W, Xie JP, Li Y, Zeng E, Wang HH. 2003. Mutations in the rpoB gene in multidrug-resistant Mycobacterium tuberculosis isolates from China. J Clin Microbiol 41:2209–2212. doi: 10.1128/JCM.41.5.2209-2212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maschmann RA, Verza M, Silva MS, Sperhacke RD, Ribeiro MO, Suffys PN, Gomes HM, Tortoli E, Marcelli F, Zaha A, Rossetti ML. 2011. Detection of rifampicin-resistant genotypes in Mycobacterium tuberculosis by reverse hybridization assay. Mem Inst Oswaldo Cruz 106:139–145. doi: 10.1590/S0074-02762011000200004. [DOI] [PubMed] [Google Scholar]
- 57.Tan Y, Hu Z, Zhoa Y, Cai X, Luo C, Zou C, Liu X. 2012. The beginning of the rpoB gene in addition to the rifampicin resistant determination region might be needed for identifying rifampin/rifabutin cross resistance in multidrug resistant Mycobacterium tuberculosis isolates in southern China. J Clin Microbiol 50:81–85. doi: 10.1128/JCM.05092-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brandis G, Hughes D. 2013. Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found clinical isolates. J Antimicrob Chemother 68:2493–2497. doi: 10.1093/jac/dkt224. [DOI] [PubMed] [Google Scholar]
- 59.Koch A, Mizrahi V, Warner DF. 2014. The impact of drug resistance on Mycobacterium tuberculosis physiology: what can we learn from rifampicin? Emerg Microbes Infect 3:e17. doi: 10.1038/emi.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kreutzer DA, Essigmann JM. 1998. Oxidized, deaminated cytosines are a source of C→T transitions in vivo. Proc Natl Acad Sci U S A 95:3578–3582. doi: 10.1073/pnas.95.7.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kino K, Sugiyama H. 2001. Possible cause of G-C→C-G transversion mutation by guanine oxidation product, imidazolone. Chem Biol 8:369–378. doi: 10.1016/S1074-5521(01)00019-9. [DOI] [PubMed] [Google Scholar]
- 62.Luria SE, Delbrück M. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28:491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tadolini B, Cabrini L. 1988. On the mechanism of OH scavenger action. Biochem J 253:931–933. doi: 10.1042/bj2530931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nunoshiba T, Obata F, Boss AC, Oikawa S, Mori T, Kawanishi S, Yamamoto K. 1999. Role of iron and superoxide for generation of hydroxyl radical, oxidative DNA lesions and mutagenesis in Escherichia coli. J Biol Chem 274:34832–34837. doi: 10.1074/jbc.274.49.34832. [DOI] [PubMed] [Google Scholar]
- 65.Srinivasan C, Liba A, Imlay JA, Valentine JS, Gralla EB. 2000. Yeast lacking superoxide dismutase(s) show elevated levels of “free iron” as measured by whole cell electron paramagnetic resonance. J Biol Chem 275:29187–29192. doi: 10.1074/jbc.M004239200. [DOI] [PubMed] [Google Scholar]
- 66.Pate KT, Rangel NA, Fraser B, Clement MHS, Srinivasan C. 2006. Measuring “free” iron levels in Caenorhabditis elegans using low-temperature Fe(III) electron paramagnetic resonance spectroscopy. Anal Biochem 358:199–207. doi: 10.1016/j.ab.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Setsukinai K, Urano Y, Kakinuma K, Majima HJ, Nagano T. 2003. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem 278:3170–3175. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- 68.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cell death induced by bactericidal antibiotics. Cell 130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 69.Miller CJ, Rose AL, Waite TD. 2012. Hydroxyl radical production by H2O2 mediated oxidation of Fe(II) complexed by Suwannee River fulvic acid under circumneutral freshwater conditions. Environ Sci Technol 47:829–835. doi: 10.1021/es303876h. [DOI] [PubMed] [Google Scholar]
- 70.Bhaskar A, Chawala M, Mehta M, Parikh P, Chandra P, Bhave D, Kumar D, Carroll KS, Singh A. 2014. Reengineering redox sensitive GFP to measure mycothiol redox potential of Mycobacterium tuberculosis during infection. PLoS Pathog 10:e1003902. doi: 10.1371/journal.ppat.1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buchmeier NA, Newton GL, Koledin T, Fahey RC. 2003. Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol Microbiol 47:1723–1732. doi: 10.1046/j.1365-2958.2003.03416.x. [DOI] [PubMed] [Google Scholar]
- 72.Ung KS, Av-Gay Y. 2006. Mycothiol-dependent mycobacterial response to oxidative stress. FEBS Lett 580:2712–2716. doi: 10.1016/j.febslet.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 73.Van Laer K, Buts L, Foloppe N, Vertommen D, Van Belle K, Wahni K, Roos G, Nilsson L, Mateos LM, Rawat M, van Nuland NA, Messens J. 2012. Mycoredoxin-1 is one of the missing links in oxidative stress defense mechanism in mycobacteria. Mol Microbiol 86:787–804. doi: 10.1111/mmi.12030. [DOI] [PubMed] [Google Scholar]
- 74.Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. 2004. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem 279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 75.Sakai A, Nakanishi M, Yoshiyama K, Maki H. 2006. Impact of reactive oxygen species on spontaneous mutagenesis in Escherichia coli. Genes Cells 11:767–778. doi: 10.1111/j.1365-2443.2006.00982.x. [DOI] [PubMed] [Google Scholar]
- 76.Takiff HE, Salazar L, Guerrero C, Philipp W, Huang WM, Kreiswirth B, Cole ST, Jacobs WR Jr, Telenti A. 1994. Cloning and nucleotide sequence of Mycobacterium tuberculosis gyrA and gyrB genes and detection of quinolone resistance mutation. Antimicrob Agents Chemother 38:773–780. doi: 10.1128/AAC.38.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu C, Kreiswirth BN, Sreevatsan S, Musser JM, Drilica K. 1996. Fluoroquinolone resistance associated with specific gyrase mutations in clinical isolates of multidrug-resistant Mycobacterium tuberculosis. J Infect Dis 174:1127–1130. doi: 10.1093/infdis/174.5.1127. [DOI] [PubMed] [Google Scholar]
- 78.Von Groll A, Martin A, Jureen P, Hoffner S, Vandamme P, Portaels F, Palamino JC, da Silva PA. 2009. Fluoroquinolone resistance in Mycobacterium tuberculosis and mutations in gyrA and gyrB. Antimicrob Agents Chemother 53:4498–4500. doi: 10.1128/AAC.00287-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Devasia R, Blackman A, Eden S, Li H, Maruri F, Shintani A, Alexander C, Kaiga A, Stratton CW, Warkentin J, Tang YW, Sterling TR. 2012. High proportion of fluoroquinolone-resistant Mycobacterium tuberculosis isolates with novel gyrase polymorphisms and a gyrA region associated with fluoroquinolone susceptibility. J Clin Microbiol 50:1390–1396. doi: 10.1128/JCM.05286-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li J, Gao X, Luo T, Wu J, Sun G, Liu Q, Jiang Y, Zhang Y, Mei J, Gao Q. 2014. Association of gyrA/B mutations and resistance levels to fluoroquinolones in clinical isolates of Mycobacterium tuberculosis. Emerg Microbes Infect 3:e19. doi: 10.1038/emi.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alangaden GJ, Manavathu EK, Vakulenko SB, Zvonok NM, Lerner SA. 1995. Characterization of fluoroquinolone-resistant mutant strains of Mycobacterium tuberculosis selected in the laboratory and isolated from patients. Antimicrob Agents Chemother 39:1700–1703. doi: 10.1128/AAC.39.8.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mayer C, Takiff H. 2014. The molecular genetics of fluoroquinolone resistance in Mycobacterium tuberculosis. Microbiol Spectrum 2(4):MGM2–0009–2013. [DOI] [PubMed] [Google Scholar]
- 83.Sekiguchi M, Tsuzuki T. 2002. Oxidative nucleotide damage: consequences and prevention. Oncogene 21:8895–8904. doi: 10.1038/sj.onc.1206023. [DOI] [PubMed] [Google Scholar]
- 84.Maki H, Sekiguchi M. 1992. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 355:273–275. doi: 10.1038/355273a0. [DOI] [PubMed] [Google Scholar]
- 85.Billington OJ, McHugh TD, Gillespie SH. 1999. Physiological cost of rifampin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob Agents Chemother 43:1866–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mariam DH, Mengistu Y, Hoffner SE, Andersson DI. 2004. Effect of rpoB mutations conferring rifampicin resistance on fitness of Mycobacterium tuberculosis. Antimicrob Agents Chemother 48:1289–1294. doi: 10.1128/AAC.48.4.1289-1294.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S. 2012. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase. Nat Genet 44:106–110. doi: 10.1038/ng.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Song T, Park Y, Shamputa IC, Seo S, Lee SY, Jeon HS, Choi H, Lee M, Glynne RJ, Bernes SW, Walker JR, Batalov S, Yusim K, Feng S, Tung CS, Theiler J, Via LE, Boshoff HI, Murakami KS, Korber B, Barry CE III, Cho SN. 2014. Fitness cost of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the β′ subunit of RNA polymerase. Mol Microbiol 9:1106–1119. doi: 10.1111/mmi.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Andersson DI, Levin BR. 1999. The biological cost of antibiotic resistance. Curr Opin Microbiol 2:489–493. doi: 10.1016/S1369-5274(99)00005-3. [DOI] [PubMed] [Google Scholar]
- 90.Marshall G, Blacklock JWS, Cameron C, Capon NB, Cruickshank R, Gaddum JH, Heaf FRG, Hill AB, Houghton LE, Hoyle JC, Raistrick H, Scadding JG, Tytler WH, Wilson GS, Hart PD. 1948. Streptomycin treatment of pulmonary tuberculosis: a medical research council investigation. Br Med J 2:769–782. doi: 10.1136/bmj.2.4582.769.18890300 [DOI] [Google Scholar]
- 91.Crofton J, Mitchison DA. 1948. Streptomycin resistance in pulmonary tuberculosis. Br Med J 2:1009–1015. doi: 10.1136/bmj.2.4588.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Spagnolo F, Rinaldi C, Sajorda DR, Dykhuizen DE. 2016. Evolution of resistance to continuously increasing streptomycin concentrations in populations of Escherichia coli. Antimicrob Agents Chemother 60:1336–1342. doi: 10.1128/AAC.01359-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goswami M, Mangoli SH, Jawali N. 2006. Involvement of reactive oxygen species in the action of ciprofloxacin against Escherichia coli. Antimicrob Agents Chemother 50:949–954. doi: 10.1128/AAC.50.3.949-954.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang X, Zhao X, Malik M, Drlica K. 2010. Contribution of reactive oxygen species to pathways of quinolone-mediated bacterial cell death. J Antimicrob Chemother 65:520–524. doi: 10.1093/jac/dkp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. 2012. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dwyer DJ, Belensky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CT, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ. 2014. Antibiotics induce redox-related physiological alterations as a part of their lethality. Proc Natl Acad Sci U S A 111:E2100–E2109. doi: 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vlu K, Mironov AS, Zavigelskii GB. 2014. Role of reactive oxygen species in the bactericidal action of quinolones–inhibitors of gyrase. Mol Biol 48:990–998. doi: 10.1134/S0026893314060107. [DOI] [PubMed] [Google Scholar]
- 98.Piccaro G, Pletraforte D, Giannoni F, Mustazzolu A, Fattorirni L. 2014. Rifampicin induces hydroxyl radical formation in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:7527–7533. doi: 10.1128/AAC.03169-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. 2002. Free radical-induced damage to DNA: mechanism and measurement. Free Radic Biol Med 32:1102–1115. doi: 10.1016/S0891-5849(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 100.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. 2003. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 101.Kamiya H. 2003. Mutagenic potentials of damaged Nucleic acids produced by reactive oxygen/nitrogen species: approaches using synthetic oligonucleotides and nucleotides: survey and summary. Nucleic Acids Res 31:517–531. doi: 10.1093/nar/gkg137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grant SS, Kaufmann BB, Chand NS, Haseley N, Hung DT. 2012. Eradication of bacterial persisters with antibiotic generated hydroxyl radical. Proc Natl Acad Sci U S A 109:12147–12152. doi: 10.1073/pnas.1203735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.O'Sullivan DM, McHugh TD, Gillespie SH. 2008. The effect of oxidative stress on the mutation rate of Mycobacterium tuberculosis with impaired catalase/peroxidase function. J Antimicrob Chemother 62:709–712. doi: 10.1093/jac/dkn259. [DOI] [PubMed] [Google Scholar]
- 104.Da Silva PEA, Palomino JC. 2011. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: classical and new drugs. J Antimicrob Chemother 66:1417–1430. doi: 10.1093/jac/dkr173. [DOI] [PubMed] [Google Scholar]
- 105.Imlay JA. 2011. Cellular defences against superoxide and hydrogen peroxide. Annu Rev Biochem 77:755–776. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bjedov I, Tenaillon O, Gérard B, Souza V, Denamur E, Radman M, Taddei F, Matic I. 2003. Stress-induced mutagenesis in bacteria. Science 300:1404–1409. doi: 10.1126/science.1082240. [DOI] [PubMed] [Google Scholar]
- 107.Steenken W., Jr 1935. Lysis of tubercle bacilli in vitro. Proc Soc Exptl Biol Med 33:253–255. doi: 10.3181/00379727-33-8330P. [DOI] [Google Scholar]
- 108.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequencing of the M13mp18 and pUC9 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 109.Dickinson JM, Aber VR, Allen BW, Ellard GA, Mitchison DA. 1974. Assay for rifampicin in serum. J Clin Pathol 27:457–462. doi: 10.1136/jcp.27.6.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ince D, Zhang X, Hooper DC. 2003. Activity and resistance to moxifloxacin in Staphylococcus aureus. Antimicrob Agents Chemother 47:1410–1415. doi: 10.1128/AAC.47.4.1410-1415.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Roy S, Narayana Y, Balaji KN, Ajitkumar P. 2012. Highly fluorescent GFPm2+-based genome integration-proficient promoter probe vector to study Mycobacterium tuberculosis promoters in infected macrophages. Microb Biotechnol 5:98–105. doi: 10.1111/j.1751-7915.2011.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.