

Abstract
Accumulating evidence has shown that the lung is one of the target organs for microangiopathy in patients with either type 1 or type 2 diabetes mellitus (DM). Diabetes is associated with physiological and structural abnormalities in the diabetic lung concurrent with attenuated lung function. Despite intensive investigations in recent years, the pathogenic mechanisms of diabetic lung injury remain largely elusive. In this review, we summarize currently postulated mechanisms of diabetic lung injury. We mainly focus on the pathogenesis of diabetic lung injury that implicates key pathways, including oxidative stress, non-enzymatic protein glycosylation, polyol pathway, NF-κB pathway, and protein kinase c pathway. We also highlight that while numerous studies have mainly focused on tissue or cell damage in the lung, studies focusing on mitochondrial dysfunction in the diabetic lung have remained sketchy. Hence, further understanding of mitochondrial mechanisms of diabetic lung injury should provide invaluable insights into future therapeutic approaches for diabetic lung injury.
Keywords: diabetic hyperglycemia, diabetic lung injury, diabetes mellitus, mitochondria, oxidative stress
Diabetes mellitus (DM), characterized by persistent blood hyperglycemia, is a leading cause of morbidity and mortality in the world. A recent report released by the World Health Organization reveals that there were 1.5 million (2.7%) deaths caused by diabetes in 2012, up from 1.0 million (2.0%) in 2000. The major cause of death in diabetic patients is glucotoxicity-induced complications. There are now increasing evidence showing that lung is also one of the target organs for diabetic microangiopathy in patients with either type 1 or type 2 DM [1-3]. Because the lung microvascular system has huge reserve function, diabetic lung damage is quite subclinical and often ignored by patients and physicians. With continued increase in the occurrence of diabetes in an aging population, more and more pulmonary dysfunction is likely to be attributed to diabetic pulmonary complications. Pulmonary disease associated with diabetes includes a predisposition to infections and chronic obstructive pulmonary disease such as pneumonia, asthma, pulmonary fibrosis, and pulmonary tuberculosis as well as impaired breathing during sleeps [4-11]. Furthermore, it has been reported that incidence death due to pulmonary diseases among Japanese diabetic patients has been found to be greater than 50% [12]. When compared with healthy subjects, patients with type 1 or type 2 DM are at increased risk for respiratory tract infections and the risk further increases with repeated occurrence of common infections [13, 14]. Relative risks of developing pulmonary tuberculosis of all types and bacteriologically confirmed cases were 3.47 times and 5.15 times higher, respectively, in diabetic patients than in matched healthy controls [15-18].
Therefore, elucidating the pathogenesis of the diabetic lung injury has become an important research topic. Several concepts of pathogenesis such as oxidative stress, non-enzymatic glycation of proteins, and the polyol pathway have been identified to be involved in the etiology of diabetic lung injury. In this paper, we attempt to provide an overview of the potential and major biochemical mechanisms of morphological changes and pulmonary dysfunction implicated in diabetic lung injury. A deeper understanding of the underlying mechanisms should provide invaluable insights into novel approaches for attenuating diabetic lung injury in the future. It should be noted that this review is by no means to exhaust all the possible mechanisms of diabetic lung injury documented in the literature.
Morphological changes in diabetic lung injury
Numerous studies indicate physiological and structural abnormalities in diabetic lungs. A histological investigation in rabbit lung shows that diabetic rabbits exhibit morphological abnormalities within 3 weeks of diabetes induction [19]. It has been reported that diabetic hyperglycemia damages the respiratory system due to the pulmonary interstitial injury caused by microangiopathy, which in the meantime could also contribute to autonomic neuropathy [20]. It has also been reported that there is a widely increase in the volume proportion of alveolar wall and alveoli per unit volume, in the relative amounts of collagen, elastin, and basal laminae, and in the surface-to-volume ratio of the lungs of the diabetic rats [21]. The basal membranes were thickened, along with an intense inflammatory reaction in diabetic lungs [22]. The structures of lung tissue and lamellar bodies showed collapse in DM group [23], neutrophil infiltration or aggregation and alveolar wall thickening in lung tissue were significantly higher in the DM group than in the control group [24]. In diabetic lung models, histological examination with Sirius red and eosin and hemotoxylin staining showed fibrosis along with massive inflammatory cell infiltration [25]. The number of tiny villus and the quantity of osmiophilic multilamellar body decreased markedly, while hyperplasia was found in collagen fiber [26]. The mechanisms underlying morphological changes in diabetic lung injury might be the following: (1) Activation of NADPH oxidase that mediates oxidative and nitrosative damage [25]. (2) Activation of the polyol pathway that is one of the most popular candidate mechanisms to explain the cellular toxicity of diabetic hyperglycemia. When glucose concentration in the cell becomes too high, aldose reductase is activated to reduce glucose to sorbitol [27]. Sorbitol can induce cellular osmotic pressure, leading to cell death [28]. On the other hand, as the polyol pathway also consumes NADPH and produces NADH, cellular redox imbalance can occur and trigger oxidative stress, leading to changes in cell membrane integrity and function. (3) Generation of advanced glycation end-products (AGEs) that can impair protein structure and function.
Pulmonary dysfunction in diabetic lung injury
Decreased lung function is associated with diabetes, both cross-sectionally and longitudinally [29-33]. It has been found that pulmonary function abnormalities appeared within 3 years of diabetes diagnosis in 51.2% of children with type 1 diabetes [34] and there is also a statistically significant increase in total airway resistance in children with type 1 diabetes [35]. Airflow restriction is a predictor of demise in type 2 diabetes after adjusting for other recognized risk factors [36]. One study in the Strong Heart Study involving 2,396 adults shows a significantly decreased pulmonary function in American Indians with DM; moreover, lung functional impairment was found to already appear in this ethnic group before the development of overt DM [37].
Lung ventilatory dysfunction
Ventilation is an important reflection of lung function. Diabetic patients exhibit a high risk of pulmonary disorders that are often associated with restrictive impairment of lung function. Many cross-sectional studies have consistently shown that when compared with healthy subjects, patients with diabetes have significant decreases in many parameters including lower vital capacity, medium expiratory flow, expiratory residual volume, the total lung capacity (TLC), forced vital capacity (FVC), and forced expiratory volume in one second (FEV1) [38-41][26, 27]. It has also been reported that the restrictive, but not the obstructive ventilatory dysfunction, is associated with development of prediabetes and precedes the development of type 2 diabetes [42]. Moreover, it has been found that the early stage of diabetes modulates the bronchial reactivity to both acetylcholine and isoproterenol by disrupting the nitric oxide (NO), KATP channels, and cyclooxygenase pathways in guinea pigs [43]. There have been several hypotheses proposed on the mechanism of ventilatory dysfunction in the diabetic lung: (1) Respiratory muscle strength decreased, which was significantly related to attenuated lung volumes and ill metabolic control in type 2 diabetes [44-46]. (2) The restrictive type of pulmonary impairment is likely due to non-enzymatic glycosylation of pulmonary proteins and subsequent accumulation of collagen in the lungs and chest wall [30]. Moreover, glycosylation of elastin fibers can also cause improper cross-linking and emphysema-like diminution in alveolar surface area, resulting in stiffening of the lungs, reduced elastic recoil, impaired vascular diffusion, and inflammatory changes in lungs [47, 48]. (3) Nearly one-third of subjects with diabetes showed abnormal ventilatory response to hypoxia, hypercapnia, or exercise, a symptom of autonomic neuropathy [20]. Moreover, neuroadrenergic bronchopulmonary denervation may also occur in diabetic patients with autonomic neuropathy [49]. Indeed, it has been reported that neuroadrenergic denervation of the lung is associated with the decline of respiratory functional indexes in diabetic patients [50], leading to lung diastolic stress disorder [49]. (4) Non-adrenergic non-cholinergic neurotransmitter release is decreased due to diabetic autonomic neuropathy [51], which could deregulate the pulmonary vascular tone and pulmonary ventilation.
Pulmonary diffusing capacities
Transfer of oxygen and carbon dioxide over the alveolocapillary membrane is the main function of the lung. Lung diffusing capacity for carbon monoxide (DLCO) is a measure of gas conductance across alveolar tissue membrane into capillary erythrocytes and subsequent chemical binding to hemoglobin. DLCO is influenced by alveolar-capillary membrane conductance and pulmonary capillary blood volume, both of which are reduced in adults with type 1 diabetes [52]. Evidence shows that children with type 1 diabetes also had impaired pulmonary functions including DLCO, airway resistance (Raw), alveolar volume (AV), and DLCO/AV ratio [53]. It has been demonstrated that there is a significant decrease in the ratio between DLCO and alveolar ventilation in pulmonary gas exchange in the diabetic group when compared with that in healthy controls [54]. DLCO is predominantly due to a low membrane diffusing capacity (DMCO) [55] while in type 2 diabetes both DMCO and capillary blood volume are reduced [38]. Interestingly, at peak exercise, type 1 diabetic subjects demonstrated a decreased DLCO when corrected for cardiac output (DLCO/Q) [56]. Additionally, a decrease in DMCO when corrected for cardiac output (DMCO/Q) was also present and the decrease in diffusing capacity was associated with a reduction in oxygen saturation. It is suggested that the limitation in gas transfer becomes functionally important as the transit time of red blood cells through the lung is shortened [56].
Etiopathogenesis of diabetic lung injury
There is increasingly a large body of supporting data that may provide mechanisms of pathogenesis of diabetic lung injury. However, a detailed elucidation of each mechanism remains challenging. Long lasting hyperglycemia triggers upregulation of a variety of pathways, including oxidative stress, non-enzymatic glycation of proteins, polyol pathway and sorbitol production, NF-κB pathway, and activation of the protein kinase C pathway (Fig. 1). Here we review these mainstream mechanisms that have been implicated in diabetic lung injury.
Figure 1.
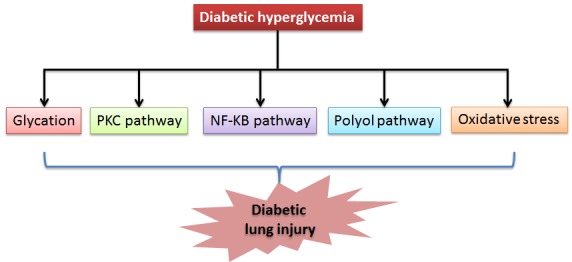
Hyperglycemia-upregulated pathways that are potentially involved in diabetic lung injury. These include protein glycation, PKC pathway, NF-KB pathway, polyol pathway, and oxidative stress. It should be noted that these pathways may be inter-related. For example, the polyol pathway can also contribute to oxidative stress.
Oxidative stress
Previous experimental and clinical studies indicate that hyperglycemia-induced overproduction of superoxide plays a key role in the pathogenesis and development of diabetic complications in all kinds of tissue injuries [57-59]. Sustained hyperglycemia produces excessive reactive oxygen species (ROS) and reactive nitrogen species (RNS) that cannot be overwhelmed by antioxidants, resulting in damage to DNA, lipids, and proteins [60-63]. Evidence indicates that in diabetic lung, the activity of superoxide dismutase (SOD) was decreased, while the contents of nitric oxide (NO) and malondialdehyde (MDA) were significantly increased [23, 26]. As NO and superoxide can react simultaneously to produce peroxynitrite that is very damaging [64], any evidence that NO concentration is elevated in the diabetic lung would suggest that NO/peroxynitrite pathway is a mechanism by which lung is injured. The pulmonary lipid peroxidation, glutathione peroxidase activities and inducible nitric oxide synthase (iNOS) were all found to be increased in streptozotocin diabetic rats [65]. Immunohistochemical staining in the pulmonary bronchial epithelium and capillary endothelium in the DM group indicates an elevated level of iNOS expression [65]. It has also been shown that 8-OHdG increases in patients with type 2 diabetes [66], demonstrating the occurrence of DNA damage in the diabetic lungs. Clinic data illustrates that tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), interleukin-10 (IL-10), and MDA levels in serum were all significantly increased in diabetic patients who show diabetic lung injury [67]. Additionally, it has been reported that in type 2 diabetes patients with pneumonia, levels of toll-like receptor 2/4 in peripheral blood monocytes and serum surfactant protein A were altered [68]. All these studies demonstrate the involvement of oxidative damage in diabetic lung injury.
Non-enzymatic protein glycosylation
Nonenzymatic glycosylation is accelerated by oxidative stress and elevated levels of aldoses [69]. AGEs are a heterogeneous group of modified proteins, lipids and nucleic acids arising from intracellular hyperglycemia via a non-enzymatic Maillard reaction [63]. This modification can result in changes in tissue/cellular properties by forming crosslinks [70] that impair the biological functions of the target proteins [71]. Tissues exposed to continuous state of high blood glucose can exhibit non-enzymatic glycosylation of proteins, whereby AGEs are eventually formed [72]. It has been suggested that formation and accumulation of AGEs are involved in the pathogenesis of diabetic vascular complications [73]. As reactive oxygen intermediates are involved in the formation of AGE structures such as carboxymethyllysine [74], the interaction of AGE modified proteins with macrophages might further induce macrophage ROS production, thereby contributing to the development of pulmonary fibrosis [75].
Therefore, hyperglycemia-accelerated formation of nonenzymatic advanced glycosylation end products in tissues has been suggested as the central pathologic features of diabetic complications [76]. Interaction of AGEs with the receptor for advanced glycation end-products (RAGE) has been shown to activate down-stream signaling pathways and evoke inflammatory responses in vascular wall cells [77, 78]. Glycation can upregulate transforming growth factor-β intermediate and lead to increased synthesis of collagen, laminin, and fibronectin in the extracellular matrix [79, 80].
Polyol pathway
Polyol pathway is one of the major sources of ROS production in diabetes [81]. Hyperglycemia decreases NAD+ levels by activation of the polyol pathway and by over-activation of poly (ADP-ribose)-polymerase (PARP) [82]. The pulmonary arteries from diabetic rats showed impaired relaxant response to acetylcholine and decreased vasoconstrictor response to N-omega-nitro-L-arginine methyl ester (L-NAME) that is a widely used NOS inhibitor. It has also been suggested that diabetes induces pulmonary artery endothelial dysfunction by enhancing superoxide production [83]. In experimental diabetic lung in rabbits, it was found that glutathione peroxidase (GSH-Px) activity, glutathione reductase (GSSG-R) activity, and ascorbic acid level decreased while the concentration of lipid peroxidation products increased [84]. Therefore, it is conceivable that the lowered level of NAD+ in the diabetic lung may aggravate cellular and tissue damage. The levels of free 15-F2t-isoprostane were increased in lung tissues in diabetic rats along with a significantly increased membrane translocation of the NADPH oxidase subunit p67phox and increased expression of the membrane-bound subunit p22phox in diabetic lung [85]. Additionally, damaged mitochondria can also generate an excess of superoxide that can mediate tissue injury in diabetes. Studies conducted in diabetic rats have revealed significant generation of mitochondrial superoxide at the site of NADH/ubiquinone oxidoreductase (complex I) [86, 87].
NF-κB pathway
The nuclear factor (NF)-κB signaling pathway is involved in regulating gene expression in early inflammatory responses. NF-κB pathway is a primary target for activation by hyperglycemia, oxidative stress, and inflammatory cytokines [88]. The expression of TNF-α, IL-1β, IL-6 and other pro-inflammatory cytokines are regulated downstream of NF-κB [89]. NF- κB has been shown to activate the genes encoding pro-inflammatory cytokines, cell adhesion molecules, nitric oxide synthase, and cyclooxygenase-2 [90]. In the lung tissues of diabetic rats, it has been reported that while the levels of IκB were declined when compared with that in the control group, the levels of phosphorylated IκB and nuclear NF-κB were actually elevated. Meanwhile, the mRNA levels and protein levels of iNOS and cyclooxygenase-2 were also up-regulated in the lung tissue of diabetic animals [91]. Additionally, protein carbonyl content was higher in diabetic lungs, but SOD and GSH activities were lower [22]. Hence, it seems that lungs are exposed to oxidative stress mediated by NF- κB activation in diabetes.
Protein Kinase C pathways
Protein kinase C (PKC) is a family of enzymes that are involved in regulating the function of numerous proteins. PKC has been associated with vascular alterations such as increase in permeability, contractility, extracellular matrix synthesis, cell growth and apoptosis, angiogenesis, leukocyte adhesion, and cytokine activation and inhibition. It has been demonstrated that diabetic rats exhibited a significant decrease in LPS-induced phosphorylation of extracellular signal-regulated kinase, protein kinase C α and δ, p38, and protein kinase B [92]. Additionally, expression of iNOS and levels of IL-6 and cyclooxygenase are also decreased in the lung homogenates derived from diabetic rats [92]. In particular, PKC activation by hyperglycemia can activate NADPH oxidase that generates more ROS and accentuates oxidative stress [93]. Therefore, PKC dysregulation could be involved in diabetic lung injury.
Role of mitochondrial dysfunction in diabetic lung injury
Mitochondria are important organelles for cell survival [94, 95]. Mitochondria play important roles in intracellular energy generation, in modulation of apoptosis, and in redox-dependent intracellular signaling [96]. Mitochondria can be both a source of ROS and a target of oxidative damage during oxidative stress. Hence, numerous studies attempting to link mitochondria to the complications of diabetes has focused on the role of mitochondrial ROS [97-100]. Prolonged hyperglycemia results in an over-generation of ROS by the mitochondria, which in turn may contribute significantly to the development of diabetic complications [100]. Interestingly, AMP kinase (AMPK) seems to be involved in the pathogenesis of diabetic complications. It has been reported in diabetes that C-peptide induces AMPKα activation and inhibits hyperglycemia-induced ROS production, mitochondrial membrane potential collapse, mitochondrial fission, and endothelial cell apoptosis [101]. It has also been demonstrated that increased mitochondrial ROS in diabetes is maintained by a feed-forward AMPK activation cascade [102]. Interestingly, basal ROS concentration was increased in lymphocytes from type 2 DM and triiodothyronine (T3) significantly stimulated ROS concentration in type 2 DM patients. Thus an increased mitochondrial sensitivity for T3 may be a significant factor responsible for increased ROS production in diabetic patients [103].
Moreover, hyperglycemia-induced over-production of mitochondrial ROS may lead to collapse of mitochondrial transmembrane potential and opening of the mitochondrial permeability transition pore (MPTP) [100, 104, 105]. The occurrence of apoptosis via cytochrome c release induced by MPTP opening may lead to more ROS generation [106, 107], forming a vicious cycle that eventually results in cell death [108] (Fig. 2). Conversely, therapeutic inhibition of mitochondrial ROS by antioxidants such as mito-TEMPO can decrease adverse cellular changes and mitigate cellular dysfunction in diabetic mice [109]. Thus, antioxidants targeting mitochondria could be an effective therapeutic approach for diabetic complications in the lung [110].
Figure 2.
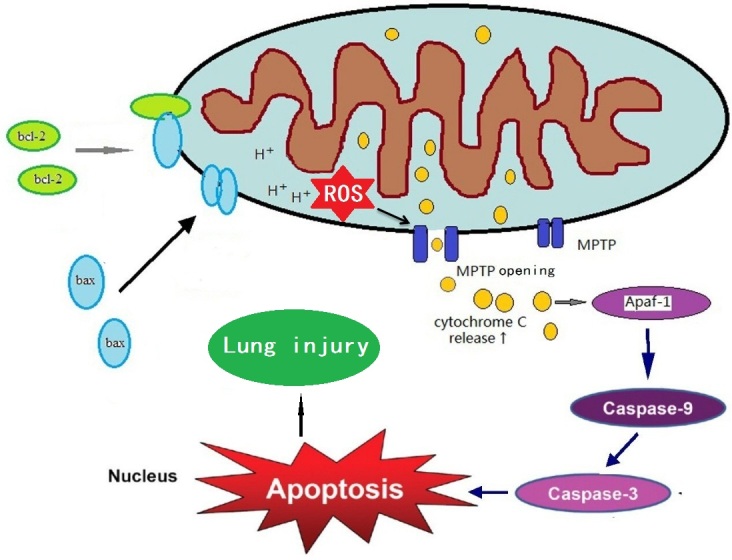
Possible role of mitochondrial dysfunction in diabetic lung injury. Show is the mitochondrial elements that are involved in cell death. Initial production of mitochondrial ROS can lead to changes in membrane potential and opening of mitochondrial permeability transition pore (MPTP) opening. MPTP opening could further enhance mitochondrial ROS production, forming a vicious cycle that eventually leads to cell death and lung dysfunction.
Based on research in other diabetic organs or tissues, we envision that defective or insufficient mitochondrial function plays potentially a pathogenic role in diabetic lung injury. Therefore, qualitative and quantitative analysis on functional perturbations of mitochondria needs to be conducted in the diabetic lung. In particular, the state of both mitochondrial transmembrane potential and the opening of MPTP in the diabetic lung injury need to be assessed.
Summary
While numerous studies have shown that the lung is one of the target organs of diabetes, the biochemical pathogenesis of diabetic lung injury remains largely unexplored. In this review, we have summarized experimental and clinical evidence demonstrating diabetic lung dysfunction manifested by morphological and pathological changes. As sustained hyperglycemia could result in oxidative stress which contributes to tissue damage, oxidative stress has been postulated to be a major contributing factor in diabetic lung injury. Nonetheless, the role of mitochondria, a major source of ROS and oxidative stress, in diabetic lung injury has yet to be fully explored. Therefore, more studies should focus on mitochondrial dysfunction in the diabetic lung in the future, which will offer invaluable insights into design of therapeutic approaches for diabetic lung injury.
Footnotes
Conflict of interest
None
References
- [1].Pitocco D, Fuso L, Conte EG, Zaccardi F, Condoluci C, Scavone G, et al. (2012). The diabetic lung--a new target organ? Rev Diabet Stud, 9(1): 23-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kuitert LM (2008). The lung in diabetes--yet another target organ? Chron Respir Dis, 5(2): 67-68. [DOI] [PubMed] [Google Scholar]
- [3].Sandler M (1990). Is the lung a ’target organ’ in diabetes mellitus? Arch Intern Med, 150(7): 1385-1388. [PubMed] [Google Scholar]
- [4].Hsiao YT, Cheng WC, Liao WC, Lin CL, Shen TC, Chen WC, et al. (2015). Type 1 Diabetes and Increased Risk of Subsequent Asthma: A Nationwide Population-Based Cohort Study. Medicine (Baltimore), 94(36): e1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Black MH, Anderson A, Bell RA, Dabelea D, Pihoker C, Saydah S, et al. (2011). Prevalence of asthma and its association with glycemic control among youth with diabetes. Pediatrics, 128(4): e839-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Carlsson AC, Wandell P, Osby U, Zarrinkoub R, Wettermark B, Ljunggren G (2013). High prevalence of diagnosis of diabetes, depression, anxiety, hypertension, asthma and COPD in the total population of Stockholm, Sweden - a challenge for public health. BMC Public Health, 13: 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].d’Annunzio G, Tosca MA, Pistorio A, Silvestri M, Romanisio G, Lorini R, et al. (2015). Type 1 diabetes mellitus and asthma: A follow-up study. Allergol Immunopathol (Madr), 43(2): 225-227. [DOI] [PubMed] [Google Scholar]
- [8].Kent BD, Grote L, Ryan S, Pepin JL, Bonsignore MR, Tkacova R, et al. (2014). Diabetes mellitus prevalence and control in sleep-disordered breathing: the European Sleep Apnea Cohort (ESADA) study. Chest, 146(4): 982-990. [DOI] [PubMed] [Google Scholar]
- [9].Wu Z, Guo J, Huang Y, Cai E, Zhang X, Pan Q, et al. (2016). Diabetes mellitus in patients with pulmonary tuberculosis in an aging population in Shanghai, China: Prevalence, clinical characteristics and outcomes. J Diabetes Complications, 30(2): 237-241. [DOI] [PubMed] [Google Scholar]
- [10].Ehrlich SF, Quesenberry CP Jr., Van Den Eeden SK, Shan J, Ferrara A (2010). Patients diagnosed with diabetes are at increased risk for asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, and pneumonia but not lung cancer. Diabetes Care, 33(1): 55-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hu Y, Ma Z, Guo Z, Zhao F, Wang Y, Cai L, et al. (2014). Type 1 diabetes mellitus is an independent risk factor for pulmonary fibrosis. Cell Biochem Biophys, 70(2): 1385-1391. [DOI] [PubMed] [Google Scholar]
- [12].Goto Y, Sato SI, Masuda M (1974). Causes of death in 3151 diabetic autopsy cases. Tohoku J Exp Med, 112(4): 339-353. [DOI] [PubMed] [Google Scholar]
- [13].Klekotka RB, Mizgala E, Krol W (2015). The etiology of lower respiratory tract infections in people with diabetes. Pneumonol Alergol Pol, 83(5): 401-408. [DOI] [PubMed] [Google Scholar]
- [14].Muller LM, Gorter KJ, Hak E, Goudzwaard WL, Schellevis FG, Hoepelman AI, et al. (2005). Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin Infect Dis, 41(3): 281-288. [DOI] [PubMed] [Google Scholar]
- [15].Silwer H, Oscarsson PN (1958). Incidence and coincidence of diabetes mellitus and pulmonary tuberculosis in a Swedish county. Acta Med Scand Suppl, 335: 1-48. [PubMed] [Google Scholar]
- [16].Thanh NP, Khue PM, Sy DN, Strobel M (2015). [Diabetes among new cases of pulmonary tuberculosis in Hanoi, Vietnam]. Bull Soc Pathol Exot. [DOI] [PubMed] [Google Scholar]
- [17].Balakrishnan S, Vijayan S, Nair S, Subramoniapillai J, Mrithyunjayan S, Wilson N, et al. (2012). High diabetes prevalence among tuberculosis cases in Kerala, India. PLoS One, 7(10): e46502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim SJ, Hong YP, Lew WJ, Yang SC, Lee EG (1995). Incidence of pulmonary tuberculosis among diabetics. Tuber Lung Dis, 76(6): 529-533. [DOI] [PubMed] [Google Scholar]
- [19].McCloud LL, Parkerson JB, Zou L, Rao RN, Catravas JD (2004). Reduced pulmonary endothelium-bound angiotensin converting enzyme activity in diabetic rabbits. Vascul Pharmacol, 41(4-5): 159-165. [DOI] [PubMed] [Google Scholar]
- [20].Williams JG, Morris AI, Hayter RC, Ogilvie CM (1984). Respiratory responses of diabetics to hypoxia, hypercapnia, and exercise. Thorax, 39(7): 529-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kida K, Utsuyama M, Takizawa T, Thurlbeck WM (1983). Changes in lung morphologic features and elasticity caused by streptozotocin-induced diabetes mellitus in growing rats. Am Rev Respir Dis, 128(1): 125-131. [DOI] [PubMed] [Google Scholar]
- [22].Eren G, Cukurova Z, Hergunsel O, Demir G, Kucur M, Uslu E, et al. (2010). Protective effect of the nuclear factor kappa B inhibitor pyrrolidine dithiocarbamate in lung injury in rats with streptozotocin-induced diabetes. Respiration, 79(5): 402-410. [DOI] [PubMed] [Google Scholar]
- [23].Hu JF, Zhang GJ, Wang L, Kang PF, Li J, Wang HJ, et al. (2014). Ethanol at low concentration attenuates diabetes induced lung injury in rats model. J Diabetes Res, 2014: 107152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Alkan M, Celik A, Bilge M, Kiraz HA, Kip G, Ozer A, et al. (2015). The effect of levosimendan on lung damage after myocardial ischemia reperfusion in rats in which experimental diabetes was induced. J Surg Res, 193(2): 920-925. [DOI] [PubMed] [Google Scholar]
- [25].Yang J, Tan Y, Zhao F, Ma Z, Wang Y, Zheng S, et al. (2011). Angiotensin II plays a critical role in diabetic pulmonary fibrosis most likely via activation of NADPH oxidase-mediated nitrosative damage. Am J Physiol Endocrinol Metab, 301(1): E132-144. [DOI] [PubMed] [Google Scholar]
- [26].Liang JQ, Ding CH, Ling YL, Xu HB, Lu P, Xian XH (2007). [The protective function of puerarin to the injury of the lung and its mechanisms during diabetes]. Zhongguo Ying Yong Sheng Li Xue Za Zhi, 23(3): 355-358. [PubMed] [Google Scholar]
- [27].Luo X, Wu J, Jing S, Yan LJ (2016). Hyperglycemic Stress and Carbon Stress in Diabetic Glucotoxicity. Aging Dis, 7(1): 90-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wu J, Jin Z, Zheng H, Yan LJ (2016). Sources and implications of NADH/NAD+ redox imbalance in diabetes and its complications. Diabetes Metab Syndr Obes, 9: 145-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Teeter JG, Riese RJ (2008). Cross-sectional and prospective study of lung function in adults with type 2 diabetes: the Atherosclerosis Risk in Communities (ARIC) study: response to Yeh et al. Diabetes Care, 31(10): e82. [DOI] [PubMed] [Google Scholar]
- [30].Yeh HC, Punjabi NM, Wang NY, Pankow JS, Duncan BB, Cox CE, et al. (2008). Cross-sectional and prospective study of lung function in adults with type 2 diabetes: the Atherosclerosis Risk in Communities (ARIC) study. Diabetes Care, 31(4): 741-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dennis RJ, Maldonado D, Rojas MX, Aschner P, Rondon M, Charry L, et al. (2010). Inadequate glucose control in type 2 diabetes is associated with impaired lung function and systemic inflammation: a cross-sectional study. BMC Pulm Med, 10: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lange P, Groth S, Kastrup J, Mortensen J, Appleyard M, Nyboe J, et al. (1989). Diabetes mellitus, plasma glucose and lung function in a cross-sectional population study. Eur Respir J, 2(1): 14-19. [PubMed] [Google Scholar]
- [33].Engstrom G, Hedblad B, Nilsson P, Wollmer P, Berglund G, Janzon L (2003). Lung function, insulin resistance and incidence of cardiovascular disease: a longitudinal cohort study. J Intern Med, 253(5): 574-581. [DOI] [PubMed] [Google Scholar]
- [34].Suresh V, Reddy A, Mohan A, Rajgopal G, Satish P, Harinarayan C, et al. (2011). High prevalence of spirometric abnormalities in patients with type 1 diabetes mellitus. Pediatr Endocrinol Diabetes Metab, 17(2): 71-75. [PubMed] [Google Scholar]
- [35].van Gent R, Brackel HJ, de Vroede M, van der Ent CK (2002). Lung function abnormalities in children with type I diabetes. Respir Med, 96(12): 976-978. [DOI] [PubMed] [Google Scholar]
- [36].Davis WA, Knuiman M, Kendall P, Grange V, Davis TM, Fremantle Diabetes S (2004). Glycemic exposure is associated with reduced pulmonary function in type 2 diabetes: the Fremantle Diabetes Study. Diabetes Care, 27(3): 752-757. [DOI] [PubMed] [Google Scholar]
- [37].Yeh F, Dixon AE, Marion S, Schaefer C, Zhang Y, Best LG, et al. (2011). Obesity in adults is associated with reduced lung function in metabolic syndrome and diabetes: the Strong Heart Study. Diabetes Care, 34(10): 2306-2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fontaine-Delaruelle C, Viart-Ferber C, Luyton C, Couraud S (2016). [Lung function in patients with diabetes mellitus]. Rev Pneumol Clin, 72(1): 10-16. [DOI] [PubMed] [Google Scholar]
- [39].Aparna (2013). Pulmonary function tests in type 2 diabetics and non-diabetic people -a comparative study. J Clin Diagn Res, 7(8): 1606-1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dharwadkar AR, Dharwadkar AA, Banu G, Bagali S (2011). Reduction in lung functions in type-2 diabetes in Indian population: correlation with glycemic status. Indian J Physiol Pharmacol, 55(2): 170-175. [PubMed] [Google Scholar]
- [41].Huang H, Guo Q, Li L, Lin S, Lin Y, Gong X, et al. (2014). Effect of type 2 diabetes mellitus on pulmonary function. Exp Clin Endocrinol Diabetes, 122(6): 322-326. [DOI] [PubMed] [Google Scholar]
- [42].Kim CH, Kim HK, Kim EH, Bae SJ, Jung YJ, Choi J, et al. (2015). Association of restrictive ventilatory dysfunction with the development of prediabetes and type 2 diabetes in Koreans. Acta Diabetol, 52(2): 357-363. [DOI] [PubMed] [Google Scholar]
- [43].Saidullah B, Muralidhar K, Fahim M (2014). Onset of diabetes modulates the airway smooth muscle reactivity of guinea pigs: role of epithelial mediators. J Smooth Muscle Res, 50: 29-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fuso L, Pitocco D, Longobardi A, Zaccardi F, Contu C, Pozzuto C, et al. (2012). Reduced respiratory muscle strength and endurance in type 2 diabetes mellitus. Diabetes Metab Res Rev, 28(4): 370-375. [DOI] [PubMed] [Google Scholar]
- [45].Nakajima K, Kubouchi Y, Muneyuki T, Ebata M, Eguchi S, Munakata H (2008). A possible association between suspected restrictive pattern as assessed by ordinary pulmonary function test and the metabolic syndrome. Chest, 134(4): 712-718. [DOI] [PubMed] [Google Scholar]
- [46].Litonjua AA, Lazarus R, Sparrow D, Demolles D, Weiss ST (2005). Lung function in type 2 diabetes: the Normative Aging Study. Respir Med, 99(12): 1583-1590. [DOI] [PubMed] [Google Scholar]
- [47].Sternberg M, Cohen-Forterre L, Peyroux J (1985). Connective tissue in diabetes mellitus: biochemical alterations of the intercellular matrix with special reference to proteoglycans, collagens and basement membranes. Diabete Metab, 11(1): 27-50. [PubMed] [Google Scholar]
- [48].Hamlin CR, Kohn RR, Luschin JH (1975). Apparent accelerated aging of human collagen in diabetes mellitus. Diabetes, 24(10): 902-904. [DOI] [PubMed] [Google Scholar]
- [49].Antonelli Incalzi R, Fuso L, Giordano A, Pitocco D, Maiolo C, Calcagni ML, et al. (2002). Neuroadrenergic denervation of the lung in type I diabetes mellitus complicated by autonomic neuropathy. Chest, 121(2): 443-451. [DOI] [PubMed] [Google Scholar]
- [50].Antonelli Incalzi R, Fuso L, Pitocco D, Basso S, Trove A, Longobardi A, et al. (2007). Decline of neuroadrenergic bronchial innervation and respiratory function in type 1 diabetes mellitus: a longitudinal study. Diabetes Metab Res Rev, 23(4): 311-316. [DOI] [PubMed] [Google Scholar]
- [51].Jenkinson KM, Reid JJ (2000). Altered non-adrenergic non-cholinergic neurotransmission in gastric fundus from streptozotocin-diabetic rats. Eur J Pharmacol, 401(2): 251-258. [DOI] [PubMed] [Google Scholar]
- [52].Lee MJ, Coast JR, Hempleman SC, Baldi JC (2016). Type 1 Diabetes Duration Decreases Pulmonary Diffusing Capacity during Exercise. Respiration, 91(2): 164-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Martin-Frias M, Lamas A, Lara E, Alonso M, Ros P, Barrio R (2015). Pulmonary function in children with type 1 diabetes mellitus. J Pediatr Endocrinol Metab, 28(1-2): 163-169. [DOI] [PubMed] [Google Scholar]
- [54].Guvener N, Tutuncu NB, Akcay S, Eyuboglu F, Gokcel A (2003). Alveolar gas exchange in patients with type 2 diabetes mellitus. Endocr J, 50(6): 663-667. [DOI] [PubMed] [Google Scholar]
- [55].Shah SH, Sonawane P, Nahar P, Vaidya S, Salvi S (2013). Pulmonary function tests in type 2 diabetes mellitus and their association with glycemic control and duration of the disease. Lung India, 30(2): 108-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wheatley CM, Baldi JC, Cassuto NA, Foxx-Lupo WT, Snyder EM (2011). Glycemic control influences lung membrane diffusion and oxygen saturation in exercise-trained subjects with type 1 diabetes: alveolar-capillary membrane conductance in type 1 diabetes. Eur J Appl Physiol, 111(3): 567-578. [DOI] [PubMed] [Google Scholar]
- [57].Brownlee M (2001). Biochemistry and molecular cell biology of diabetic complications. Nature, 414(6865): 813-820. [DOI] [PubMed] [Google Scholar]
- [58].Folli F, Corradi D, Fanti P, Davalli A, Paez A, Giaccari A, et al. (2011). The role of oxidative stress in the pathogenesis of type 2 diabetes mellitus micro- and macrovascular complications: avenues for a mechanistic-based therapeutic approach. Curr Diabetes Rev, 7(5): 313-324. [DOI] [PubMed] [Google Scholar]
- [59].Lapolla A, Fedele D (1993). [Oxidative stress and diabetes: role in the development of chronic complications]. Minerva Endocrinol, 18(3): 99-108. [PubMed] [Google Scholar]
- [60].Redmann M, Darley-Usmar V, Zhang J (2016). The Role of Autophagy, Mitophagy and Lysosomal Functions in Modulating Bioenergetics and Survival in the Context of Redox and Proteotoxic Damage: Implications for Neurodegenerative Diseases. Aging Dis, 7(2): 150-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yan LJ (2014). Positive oxidative stress in aging and aging-related disease tolerance. Redox Biol, 2C: 165-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yan LJ (2014). Protein redox modification as a cellular defense mechanism against tissue ischemic injury. Oxid Med Cell Longev, 2014: 343154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zheng H, Wu J, Jin Z, Yan LJ (2016). Protein Modifications as Manifestations of Hyperglycemic Glucotoxicity in Diabetes and Its Complications. Biochem Insights, 9: 1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Radi R (1998). Peroxynitrite reactions and diffusion in biology. Chem Res Toxicol, 11(7): 720-721. [DOI] [PubMed] [Google Scholar]
- [65].Di Naso FC, de Mello RN, Bona S, Dias AS, Porawski M, Ferraz Ade B, et al. (2010). Effect of Agaricus blazei Murill on the pulmonary tissue of animals with streptozotocin-induced diabetes. Exp Diabetes Res, 2010: 543926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Nishikawa T, Sasahara T, Kiritoshi S, Sonoda K, Senokuchi T, Matsuo T, et al. (2003). Evaluation of urinary 8-hydroxydeoxy-guanosine as a novel biomarker of macrovascular complications in type 2 diabetes. Diabetes Care, 26(5): 1507-1512. [DOI] [PubMed] [Google Scholar]
- [67].Xiong XQ, Wang WT, Wang LR, Jin LD, Lin LN (2012). Diabetes increases inflammation and lung injury associated with protective ventilation strategy in mice. Int Immunopharmacol, 13(3): 280-283. [DOI] [PubMed] [Google Scholar]
- [68].Que Y, Shen X (2016). Changes in blood monocyte Toll-like receptor and serum surfactant protein A reveal a pathophysiological mechanism for community-acquired pneumonia in patients with type 2 diabetes. Intern Med J, 46(2): 213-219. [DOI] [PubMed] [Google Scholar]
- [69].Baynes JW, Thorpe SR (1999). Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes, 48(1): 1-9. [DOI] [PubMed] [Google Scholar]
- [70].Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A (1986). Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science, 232(4758): 1629-1632. [DOI] [PubMed] [Google Scholar]
- [71].Hammes HP, Weiss A, Hess S, Araki N, Horiuchi S, Brownlee M, et al. (1996). Modification of vitronectin by advanced glycation alters functional properties in vitro and in the diabetic retina. Lab Invest, 75(3): 325-338. [PubMed] [Google Scholar]
- [72].Vlassara H, Bucala R, Striker L (1994). Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest, 70(2): 138-151. [PubMed] [Google Scholar]
- [73].Chilelli NC, Burlina S, Lapolla A (2013). AGEs, rather than hyperglycemia, are responsible for microvascular complications in diabetes: a “glycoxidation-centric” point of view. Nutr Metab Cardiovasc Dis, 23(10): 913-919. [DOI] [PubMed] [Google Scholar]
- [74].Nagai R, Ikeda K, Higashi T, Sano H, Jinnouchi Y, Araki T, et al. (1997). Hydroxyl radical mediates N epsilon-(carboxymethyl)lysine formation from Amadori product. Biochem Biophys Res Commun, 234(1): 167-172. [DOI] [PubMed] [Google Scholar]
- [75].Matsuse T, Ohga E, Teramoto S, Fukayama M, Nagai R, Horiuchi S, et al. (1998). Immunohistochemical localisation of advanced glycation end products in pulmonary fibrosis. J Clin Pathol, 51(7): 515-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Brownlee M, Cerami A, Vlassara H (1988). Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med, 318(20): 1315-1321. [DOI] [PubMed] [Google Scholar]
- [77].Yamagishi S, Nakamura K, Matsui T (2006). Advanced glycation end products (AGEs) and their receptor (RAGE) system in diabetic retinopathy. Curr Drug Discov Technol, 3(1): 83-88. [DOI] [PubMed] [Google Scholar]
- [78].Zong H, Ward M, Stitt AW (2011). AGEs, RAGE, and diabetic retinopathy. Curr Diab Rep, 11(4): 244-252. [DOI] [PubMed] [Google Scholar]
- [79].Goldin A, Beckman JA, Schmidt AM, Creager MA (2006). Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation, 114(6): 597-605. [DOI] [PubMed] [Google Scholar]
- [80].Throckmorton DC, Brogden AP, Min B, Rasmussen H, Kashgarian M (1995). PDGF and TGF-beta mediate collagen production by mesangial cells exposed to advanced glycosylation end products. Kidney Int, 48(1): 111-117. [DOI] [PubMed] [Google Scholar]
- [81].Yan LJ (2014). Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res, 2014: 137919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Boesten DM, von Ungern-Sternberg SN, den Hartog GJ, Bast A (2015). Protective Pleiotropic Effect of Flavonoids on NAD(+) Levels in Endothelial Cells Exposed to High Glucose. Oxid Med Cell Longev, 2015: 894597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lopez-Lopez JG, Moral-Sanz J, Frazziano G, Gomez-Villalobos MJ, Flores-Hernandez J, Monjaraz E, et al. (2008). Diabetes induces pulmonary artery endothelial dysfunction by NADPH oxidase induction. Am J Physiol Lung Cell Mol Physiol, 295(5): L727-732. [DOI] [PubMed] [Google Scholar]
- [84].Gumieniczek A, Hopkala H, Wojtowicz Z, Wysocka M (2002). Changes in antioxidant status of lung tissue in experimental diabetes in rabbits. Clinical Biochemistry, 35(2): 147-149. [DOI] [PubMed] [Google Scholar]
- [85].Lei SQ, Li YA, Liu HM, Yu H, Wang H, Xia ZY (2012). Effects of N-Acetylcysteine on Nicotinamide Dinucleotide Phosphate Oxidase Activation and Antioxidant Status in Heart, Lung, Liver and Kidney in Streptozotocin-Induced Diabetic Rats. Yonsei Medical Journal, 53(2): 294-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, et al. (2009). RAGE-induced Cytosolic ROS Promote Mitochondrial Superoxide Generation in Diabetes. Journal of the American Society of Nephrology, 20(4): 742-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Schmidt AM, Hori O, Cao R, Yan SD, Brett J, Wautier JL, et al. (1996). RAGE - A novel cellular receptor for advanced glycation end products. Diabetes, 45: S77-S80. [DOI] [PubMed] [Google Scholar]
- [88].Zhong P, Wu L, Qian Y, Fang Q, Liang D, Wang J, et al. (2015). Blockage of ROS and NF-kappaB-mediated inflammation by a new chalcone L6H9 protects cardiomyocytes from hyperglycemia-induced injuries. Biochim Biophys Acta, 1852(7): 1230-1241. [DOI] [PubMed] [Google Scholar]
- [89].Sun CK, Lee FY, Sheu JJ, Yuen CM, Chua S, Chung SY, et al. (2009). Early combined treatment with cilostazol and bone marrow-derived endothelial progenitor cells markedly attenuates pulmonary arterial hypertension in rats. J Pharmacol Exp Ther, 330(3): 718-726. [DOI] [PubMed] [Google Scholar]
- [90].Yagi O, Aoshiba K, Nagai A (2006). Activation of nuclear factor-kappaB in airway epithelial cells in patients with chronic obstructive pulmonary disease. Respiration, 73(5): 610-616. [DOI] [PubMed] [Google Scholar]
- [91].Zhang F, Yang F, Zhao H, An Y (2015). Curcumin alleviates lung injury in diabetic rats by inhibiting NF-kappaB pathway. Clin Exp Pharmacol Physiol. [DOI] [PubMed] [Google Scholar]
- [92].Martins JO, Ferracini M, Anger DB, Martins DO, Ribeiro LF, Jr., Sannomiya P, et al. (2010). Signaling pathways and mediators in LPS-induced lung inflammation in diabetic rats: role of insulin. Shock, 33(1): 76-82. [DOI] [PubMed] [Google Scholar]
- [93].Bey EA, Xu B, Bhattacharjee A, Oldfield CM, Zhao X, Li Q, et al. (2004). Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol, 173(9): 5730-5738. [DOI] [PubMed] [Google Scholar]
- [94].Wu J, Yan LJ (2015). Streptozotocin-induced type 1 diabetes in rodents as a model for studying mitochondrial mechanisms of diabetic beta cell glucotoxicity. Diabetes Metab Syndr Obes, 8: 181-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Jin Z, Wu J, Yan LJ (2016). Chemical Conditioning as an Approach to Ischemic Stroke Tolerance: Mitochondria as the Target. Int J Mol Sci, 17 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Onyango IG, Dennis J, Khan SM (2016). Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis, 7(2): 201-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Bonomini F, Rodella LF, Rezzani R (2015). Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis, 6(2): 109-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Victor VM, Rocha M, Herance R, Hernandez-Mijares A (2011). Oxidative stress and mitochondrial dysfunction in type 2 diabetes. Curr Pharm Des, 17(36): 3947-3958. [DOI] [PubMed] [Google Scholar]
- [99].Fernyhough P, Jonathan M (2014). Mechanisms of disease: Mitochondrial dysfunction in sensory neuropathy and other complications in diabetes. Handb Clin Neurol, 126: 353-377. [DOI] [PubMed] [Google Scholar]
- [100].Blake R, Trounce IA (2014). Mitochondrial dysfunction and complications associated with diabetes. Biochim Biophys Acta, 1840(4): 1404-1412. [DOI] [PubMed] [Google Scholar]
- [101].Bhatt MP, Lim YC, Kim YM, Ha KS (2013). C-peptide activates AMPKalpha and prevents ROS-mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes, 62(11): 3851-3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Towler DA (2013). Mitochondrial ROS deficiency and diabetic complications: AMP[K]-lifying the adaptation to hyperglycemia. J Clin Invest, 123(11): 4573-4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Anthonsen S, Larsen J, Pedersen PL, Dalgaard LT, Kvetny J (2013). Basal and T(3)-induced ROS production in lymphocyte mitochondria is increased in type 2 diabetic patients. Horm Metab Res, 45(4): 261-266. [DOI] [PubMed] [Google Scholar]
- [104].Lindblom R, Higgins G, Coughlan M, de Haan JB (2015). Targeting Mitochondria and Reactive Oxygen Species-Driven Pathogenesis in Diabetic Nephropathy. Rev Diabet Stud, 12(1-2): 134-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Yu T, Sheu SS, Robotham JL, Yoon Y (2008). Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res, 79(2): 341-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Yan LJ, Rajasekaran NS, Sathyanarayanan S, Benjamin IJ (2005). Mouse HSF1 disruption perturbs redox state and increases mitochondrial oxidative stress in kidney. Antioxid Redox Signal, 7(3-4): 465-471. [DOI] [PubMed] [Google Scholar]
- [107].Skulachev VP (1998). Cytochrome c in the apoptotic and antioxidant cascades. FEBS Lett, 423(3): 275-280. [DOI] [PubMed] [Google Scholar]
- [108].Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ (2002). Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J, 21(19): 5164-5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ni R, Cao T, Xiong S, Ma J, Fan GC, Lacefield JC, et al. (2016). Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic Biol Med, 90: 12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Sivitz WI, Yorek MA (2010). Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal, 12(4): 537-577. [DOI] [PMC free article] [PubMed] [Google Scholar]