

Summary
Defective apoptosis might be involved in the pathogenesis of multiple sclerosis (MS). We evaluated apoptosis‐related molecules in MS patients before and after autologous haematopoietic stem cell transplantation (AHSCT) using BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide (CY)‐based conditioning regimens. Patients were followed for clinical and immunological parameters for 2 years after AHSCT. At baseline, MS patients had decreased proapoptotic BAD, BAX and FASL and increased A1 gene expression when compared with healthy counterparts. In the BEAM group, BAK, BIK, BIMEL, FAS, FASL, A1, BCL2, BCLXL, CFLIPL and CIAP2 genes were up‐regulated after AHSCT. With the exception of BIK, BIMEL and A1, all genes reached levels similar to controls at day + 720 post‐transplantation. Furthermore, in these patients, we observed increased CD8+ Fas+ T cell frequencies after AHSCT when compared to baseline. In the CY group, we observed increased BAX, BCLW, CFLIPL and CIAP1 and decreased BIK and BID gene expressions after transplantation. At day + 720 post‐AHSCT, the expression of BAX, FAS, FASL, BCL2, BCLXL and CIAP1 was similar to that of controls. Protein analyses showed increased Bcl‐2 expression before transplantation. At 1 year post‐AHSCT, expression of Bak, Bim, Bcl‐2, Bcl‐xL and cFlip‐L was decreased when compared to baseline values. In summary, our findings suggest that normalization of apoptosis‐related molecules is associated with the early therapeutic effects of AHSCT in MS patients. These mechanisms may be involved in the re‐establishment of immune tolerance during the first 2 years post‐transplantation.
Keywords: apoptosis‐related molecules, autologous haematopoietic stem cell transplantation, autoreactive cells, immune tolerance, multiple sclerosis
Introduction
Multiple sclerosis (MS) is a chronic autoimmune disease caused by autoreactive lymphocytes that cross the blood–brain barrier, thereby promoting inflammation, degeneration and axonal damage 1, 2. More than 2·5 million people worldwide are affected with MS, which is currently the most common cause of neurological disability in young adults 3. The pathogenesis of MS has been linked to immunogenetic susceptibility 4, environmental factors 5, 6, immunoregulatory defects, including loss of tolerance 7, 8, and decreased numbers of regulatory T cells 9.
Some studies have implicated defective apoptosis during central and peripheral tolerance processes in autoimmune disease pathogenesis 7, 10. Apoptosis plays an important role during lymphocyte development and, when functionally impaired, may predispose to defective tolerance processes 11, 12. Central tolerance is determined by mitochondria‐dependent apoptosis (intrinsic apoptotic pathway), which involves components of the Bcl‐2 family (Bad, Bak, Bax, Bid, Bik, Bim, Bok, Boo, BMF, Noxa, Puma, A1, Bcl‐2, Bcl‐w, Bcl‐xL and Mcl‐1) 13, 14. In the periphery, autoreactive T cells can be silenced by anergy or deleted by apoptosis through activation‐induced cell death (AICD), with the participation of death receptor family proteins of the extrinsic apoptotic pathway (Fas, FasL, c‐Flip, Faim, Dr4, Dr5 and TRAIL) 13, 15.
Abnormal apoptosis has already been implicated in the pathogenesis of several autoimmune diseases, including systemic lupus erythematosus 16, 17, rheumatoid arthritis 18, 19, type 1 diabetes 20, 21, 22 and MS 23, 24, 25. Abnormal apoptosis processes in immune cells, with decreased pro‐apoptotic and increased anti‐apoptotic gene expression, are implicated in the development of autoimmunity 19, 22, 26.
Studies in the experimental autoimmune encephalomyelitis (EAE) mouse model have demonstrated the association of deregulated apoptosis with maintenance of autoreactive T cells and demyelination in the central nervous system 27, 28, 29, 30. Defective apoptosis has been also associated with MS pathophysiology in humans 31, 32. Furthermore, autoreactive T cells from MS patients are more resistant to apoptosis due to deregulation of cell death regulators Fas and FasL and Bcl‐2 family molecules 33, 34, 35.
Conventional therapies used for MS treatment include immunomodulators [interferon (IFN)‐β, co‐polymers], immunosuppressive drugs (cyclosporin, cyclophosphamide, azathioprine, mitoxantrone) and corticosteroids for acute relapses 36. More recently, monoclonal antibodies and other therapeutic agents targeting specific immune components and adhesion molecules of the blood–brain barrier, such as natalizumab and fingolimod, among others, have been added to the list 37, 38. However, a number of MS patients do not respond to conventional treatments, showing disease progression. Since 1997, high‐dose immunosuppression followed by autologous hematopoietic stem cell transplantation (AHSCT) has been implemented as a new investigational therapeutic strategy for refractory patients with autoimmune diseases, including MS 39, 40, 41, 42.
Immune monitoring studies have indicated that AHSCT may reset the immune system, decrease lymphocyte autoreactivity and restore immune tolerance, thereby inducing disease remission in MS patients 43, 44, 45. Nevertheless, although the therapeutic efficacy of AHSCT in MS patients has been demonstrated clearly, the immune mechanisms involved in this therapy are not yet understood completely, especially those associated with diverse clinical outcomes.
Here, we evaluated the expression of apoptosis‐related molecules in MS pathogenesis and their modulation during the 2‐year follow‐up after AHSCT in MS patients conditioned with two different regimens, BCNU, Etoposide, AraC and Melphalan (BEAM)/horse anti‐human thymocyte globulin (hATG) or cyclophosphamide (CY)/rabbit ATG.
Material and methods
Patients and controls
A total of 18 patients (10 females), with median age of 42 (ranging from 21–54 years), and progressive MS forms, previously refractory to first‐line therapy (intravenous corticosteroids, β‐IFN, glatiramer acetate and immunosuppressive drugs), were recruited for treatment with BCNU, Etoposide, AraC and Melphalan (BEAM)/hATG (BEAM group; n = 7) or cyclophosphamide (CY)/rATG (CY group; n = 11) conditioning regimens, followed by infusion of autologous hematopoietic stem cells (HSCs) at the Bone Marrow Transplantation Unit of the Ribeirão Preto Medical School, University of São Paulo, Brazil (trial registration in clinicaltrials.gov identifier: NCT00273364) (Table 1). Diagnosis of MS was confirmed by physicians from the Neurology Department of the same hospital, according to Poser's criteria 46. Inclusion criteria comprised patients aged from 18 to 60 years, with Expanded Disability Status Scale (EDSS) score between 3 and 6·5, and evidence of disease progression in the last 6 months. Exclusion criteria were co‐morbidities (infectious diseases or haematological, kidney, pulmonary, hepatic and heart dysfunctions), cognitive or psychiatric disorders, pregnancy or acute relapses in the last 30 days. All MS patients must have failed first‐line MS therapy and were off treatment before AHSCT. Specifically, IFN‐β and glatiramer acetate were stopped for at least 30 days before the beginning of the transplant procedure.
Table 1.
Clinical data from multiple sclerosis patients before and after autologous haematopoietic stem cell transplantation
| Controls gender/age (years) | Patients gender/age (years) | MS subtypes | Disease duration (years) | EDSS pre‐AHSCT | EDSS day + 180/ day + 360/day+ 720 post‐AHSCT | MRI pre‐AHSCT/post‐AHSCT | Previous therapy | Clinical response at day + 720 post‐AHSCT |
|---|---|---|---|---|---|---|---|---|
| M/37 | M/37 | SP | 14 | 6·5 | 6·5/6·5/6·0 | Inactivity/inactivity | STER/IFN/GA | Non‐progression |
| M/48 | M/52 | SP | 09 | 6·5 | 6·0/6·0/7·5 | Inactivity/n.d. | STER/IFN/GA | Progression |
| M/52 | M/50 | SP | 09 | 6·5 | 8·0/7·5/7·5 | Inactivity/inactivity | STER/IFN/GA | Progression |
| F/42 | F/43 | SP | 12 | 6·5 | 8·0/7·5/7·5 | Inactivity/inactivity | STER/IFN/GA | Progression |
| F/42 | F/42 | SP | 11 | 7·0 | 7·0/9·0/9·0 | Inactivity/n.d. | STER/IFN/GA | Progression |
| M/20 | M/31 | PP | 04 | 6·5 | 6·5/6·5/6·5 | Activity/inactivity | STER/IFN/GA | Non‐progression |
| M/54 | M/52 | PP | 11 | 6·5 | 6·5/6·5/7·5 | Inactivity/n.d. | STER/IFN/GA | Progression |
| F/47 | F/44 | SP | 20 | 6·5 | 6·5/6·5/6·5 | Inactivity/inactivity | STER/IFN/GA | Non‐progression |
| F/48 | F/48 | SP | 10 | 4·5 | 2·5/3·0/3·0 | Inactivity/inactivity | STER/IFN/GA | Non‐progression |
| M/55 | M/54 | SP | 09 | 6·5 | 6·5/6·0/6·0 | Inactivity/inactivity | STER/IFN/GA | Non‐progression |
| F/33 | F/36 | SP | 14 | 6·5 | 6·5/7·0/8·0 | Inactivity/inactivity | STER/IFN/GA | Progression |
| F/32 | F/32 | SP | 06 | 6·0 | 6·0/6·0/6·0 | Inactivity/inactivity | STER/IFN/GA | Non‐progression |
| F/40 | F/42 | SP | 10 | 6·0 | 6·0/6·0/6·0 | Inactivity/inactivity | STER/IFN/GA | Non‐progression |
| M/47 | M/49 | PP | 10 | 6·5 | 6·5/6·5/6·5 | Inactivity/inactivity | STER/IFN/GA | Non‐progression |
| F/36 | F/38 | PP | 09 | 6·5 | 6·5/6·5/6·5 | Inactivity/inactivity | STER/IFN/GA | Non‐progression |
| F/36 | F/36 | SP | 11 | 5·0 | 2·5/2·5/2·5 | Inactivity/inactivity | STER/IFN/GA | Non‐progression |
| M/33 | M/33 | SP | 11 | 6·5 | 6·5/6·5/7·0 | Activity/inactivity | STER/IFN/GA | Progression |
| F/30 | F/31 | SP | 06 | 6·5 | 5·5/5·0/4·5 | Activity/inactivity | STER/IFN/GA | Non‐progression |
M = male; F = female; MS = multiple sclerosis; SP = secondary progressive; PP = primary progressive; EDSS = expanded disability status scale; MRI = magnetic resonance imaging; activity = contrast enhancement of any lesion; inactivity = no contrast enhancement; n.d. = not determined; STER = intravenous (i.v.) corticosteroids; IFN = β interferon; GA = glatiramer acetate. Progression or improvement was considered when the EDSS scores, respectively, increased or decreased by 0·5 points when the initial score was ≥ 5.5 or by 1·0 or more points if the initial score was < 5·0, remaining unchanged for at least 3 months. Patients were treated with BCNU, Etoposide, AraC and Melphalan/horse anti‐human thymocyte globulin (BEAM/hATG) (BEAM group; patients 1–7) or cyclophosphamide/rabbit ATG (CY/rATG) (CY group; patients 8–18) conditioning regimens. Based on EDSS changes after autologous haematopoietic stem cell transplantation (AHSCT), from baseline to last visit, patients were classified, retrospectively, as: progression = patients who presented an increase in the EDSS scores; non‐progression = patients whose EDSS scores either decreased or remained stable.
For mobilization of haematopoietic stem cells, patients were treated with cyclophosphamide (CY, 2 g/m2) followed by granulocyte colony‐stimulating factor (G‐CSF) injections until reaching 103 leucocytes/mm3, and at least 10 CD34+ cells/mm3. Patients transplanted from 2001 to 2004 were conditioned subsequently with BEAM/hATG at a total dose of 60 mg/kg. Further patients were treated with CY/rATG (200mg/kg cyclophosphamide plus rabbit ATG at the total dose of 4·5 mg/kg). The reason that conditioning regimens were changed was excessive toxicity observed in the former, as reported previously 47. All procedures were approved by the institutional ethics committee (process number 14105/06). Informed consent was obtained from MS patients and controls at enrolment.
Peripheral blood mononuclear cells (PBMCs) were collected from MS patients at the following time‐points: baseline (pre‐mobilization), 180 days (day + 180), 360 days (day + 360), 540 days (day + 540) and 720 days (day + 720) after AHSCT. The control group included 18 healthy subjects (10 females), with a median age of 41 years (ranging from 20 to 55). Control and transplant groups of patients were age‐ and sex‐matched.
Neurological disability was assessed through EDSS by a single certified neurologist who was not blinded to patient treatment. Disease progression and improvement were considered when the EDSS scores, respectively, increased or decreased by 0·5 points when the initial score was ≥ 5·5 or by 1·0 or more points if the initial score was < 5·0, remaining unchanged for at least 3 months. Based on EDSS alterations after AHSCT from baseline to last visit, patients were classified retrospectively as: progression – patients who presented an increase in the EDSS scores; and non‐progression – patients whose EDSS scores either decreased or remained stable. Patients with progression of disability after AHSCT were not treated with any immunomodulatory or immunosuppressive drugs, except for two patients who received intravenous methylprednisolone pulses. In these cases, blood samples were collected at least 30 days after the last steroid infusion. Table 1 summarizes the demographic features of MS patients and controls.
Isolation of PBMCs
PBMCs from MS patients and healthy subjects were isolated from heparinized blood (20 ml) by centrifugation on a Ficoll‐Hypaque density gradient (d = 1·077; Amersham‐Pharmacia, Uppsala, Sweden). A total of 1·0 × 107 cells were resuspended in a guanidine isothyocyanate phenol solution (TRIzol; GIBCO BRL Life Technologies, Grand Island, NY, USA) and frozen at −80°C until RNA extraction.
RNA extraction and cDNA synthesis
Total RNA was isolated by TRIzol methodology according to the manufacturer's instructions (GIBCO BRL Life Technologies, Birmingham, AL, USA) and quantified by spectrophotometry. RNA concentration was adjusted to 1·0 μg/μl and cDNA synthesized using 2·0 μg of RNA by employing a high‐capacity cDNA archive kit (Applied Biosystems, Foster City, CA, USA), following the manufacturer's protocol. cDNA synthesis reaction was performed at 25°C for 10 min and then at 37°C for 2 h on a 9700 GeneAmp polymerase chain reaction (PCR) System (Applied Biosystems).
Relative quantification of gene expression by real‐time PCR
Relative quantification of pro‐ and anti‐apoptotic genes from the Bcl‐2 family (intrinsic pathway), death receptor family (extrinsic pathway) and the inhibitors of apoptosis proteins (IAP) family was performed with SYBR green PCR Master Mix Kit (Applied Biosystems) on a 7500 real‐time PCR system (Applied Biosystems). The PCR mixture consisted of 7·5 μl of SYBR green PCR Master Mix, 10·0 μM of forward and reverse primers, 4·0 ηg of cDNA and 4·5 μl of deionized water to a final volume of 15 μl. The PCR conditions comprised 1 cycle at 50°C for 2 min, 95°C during 10 min and 50 cycles at 95°C during 15 s, 54–62°C for 25 s (annealing temperatures were determined for each gene) and 72°C for 34 s.
The evaluated pro‐ and anti‐apoptotic genes, sequence primers, annealing temperatures and amplicon size are described in Table 2. GAPDH (glyceraldehyde‐3‐phosphate dehydrogenase) and ACTB (actin beta) were used as housekeeping genes and the relative expression of the studied target genes were obtained after normalizing using the geometric average of the housekeeping genes mRNA levels. All reactions were performed in duplicate and the gene expression was calculated by the relative expression units (REU) method 48.
Table 2.
Primer sequences, amplicon size and annealing temperature of apoptosis‐related genes
| mRNA targets | Oligonucleotides (5′–3′) | Amplicon (bp) | AT (ºC) |
|---|---|---|---|
| BAD |
F: CCG AGT GAG CAG GAA GAC TC R: GGT AGG AGC TGT GGC GAC T |
209 bp | 60 |
| BAK |
F: TTT TCC GCA GCT ACG TTT TT R: TGG TGG CAA TCT TGG TGA AGT |
201 bp | 60 |
| BAX |
F: CCC TTT TGC TTC AGG GTT TC R: TCT TCT TCC AGA TGG TGA GTG |
500 bp | 56 |
| BID |
F: GCT TCC AGT GTA GAC GGA GC R: GTG CAG ATT CAT GTG TGG ATG |
203 bp | 62 |
| BIK |
F: TCT GCA ATT GTC ACC GGT TA R: TTG AGC ACA CCT GCT CCT C |
193 bp | 60 |
| BIMEL |
F: TCT GCA ATT GTC ACC GGT TA R: AAG ATG AAA AGC GGG GCT CT |
196 bp | 59 |
| BOK |
F: TTT TCC GCA GCT ACG TTT TT R: TGG TGG CAA TCT TGG TGA AGT |
249 bp | 59 |
| NOXA |
F: AGC TGG AAG TCG AGT GTG CT R: ACG TGC ACC TCC TGA GAA AA |
167 bp | 54 |
| A1 |
F: GGC TGG CTC AGG ACT ATC R: CCA GTT AAT GAT GCC GTC |
227 bp | 50 |
| BCL‐2 |
F: ACG AGT GGG ATG CGG GAG ATG TG R: GCG GTA GCG GCG GGA GAA GTC |
241 bp | 67 |
| BCLW |
F: AGT TCG AGA CCC GCT TCC R: CCC GTC CCC GTA TAG AGC |
308 bp | 60 |
| BCL‐XL |
F: CTG AAT CGG AGA TGG AGA CC R: TGG GAT GTC AGG TCA CTG AA |
211 bp | 60 |
| MCL1 |
F: AGA AAG CTG CAT CGA ACC AT R: CC AGC TCC TAC TCC AGC AAC |
183 bp | 56 |
| FAS |
F: CAA GGG ATT GGA ATT GAG GA R: TGG AAG AAA AAT GGG CTT TG |
203 bp | 55 |
| FASL |
F: AGG AAA GTG GCC CAT TTA AC R: CAA GAT TGA CCC CGG AAG TA |
176 bp | 55 |
| c‐FLIPL |
F: GCC GAG GCA AGA TAA GCA R: GCC CAG GGA AGT GAA GGT |
464 bp | 54 |
| c‐IAP1 |
F: AGT CTT GCT CGT GCT GGT TT R: ATG GAC AGT TGG GAA AAT GC |
550 bp | 59 |
| c‐IAP2 |
F: AGT CTT GCT CGT GCT GGT TT R: TGC TTT TGC CAG ATC TGT TG |
432 bp | 58 |
| ACTB |
F: GCC CTG AGG CAC TCT TCC A R: CCA GGG CAG TGA TCT CCT TCT |
192 bp | 58 |
| GAPDH |
F: GCC TCA AGA TCA TCA GCA ATG C R: CAT GGA CTG TGG TCA TGA GTC CT |
111 bp | 60 |
F = forward primer; R = reverse primer; bp = base pairs; AT = annealing temperature.
Immunophenotypical analysis
PBMCs from MS patients and healthy individuals (5 ml) were collected in ethylenediamine tetraacetic acid (EDTA) (8·55 mg/tube) for blood counts and immunophenotyping. Briefly, 100 μl of peripheral mononuclear suspension (1·0 × 106) were labelled with 5 μl of monoclonal antibody and incubated for 15 min at 4°C in the dark. Frequencies of T lymphocyte subsets expressing Fas or FasL apoptosis‐related molecules (CD3+CD4+Fas+, CD3+CD8+Fas+, CD3+CD4+FasL+, CD3+CD8+FasL+) were analysed.
After incubation, cells were centrifuged for 5 min at 500 g, washed twice with fluorescence activated cell sorter (FACS) buffer [phosphate‐buffered saline (PBS), 0·2% fetal bovine serum, 0·02% sodium azide], resuspended in 200 μl of FACS buffer and analysed by flow cytometry. Five thousand mononuclear cells were acquired using a FACSCanto Flow Cytometer (Becton‐Dickinson, San Diego, CA, USA) and analysed by dot‐plot using Diva version 6.0 software (Becton‐Dickinson). Results are shown as absolute cell numbers (cells/μl), which were calculated by multiplying the percentage of positive cells by the lymphocytes numbers/μl, obtained from blood cell counts of peripheral blood samples.
Protein extraction and Western blotting analysis
Proteins were isolated by the TRIzol extraction method, according to the manufacturer's instructions (Gibco BRL Life Technologies). Protein samples were diluted in sodium dodecyl sulphate 1% and quantified by using the bicinchoninic acid assay (BCA) Protein Assay Kit (Pierce, Rockford, IL, USA). A total of 30 μg of protein were separated using 10 or 15% polyacrylamide gel electrophoresis and blotted onto polyvinylidene difluoride (PVDF) membranes using standard procedures. Following overnight incubation at 4°C with a blocking cocktail (5% light milk powder, Tris‐HCl pH 7·5, 0·1% Tween 20 buffer), membranes were incubated with specific primary antibodies for 24 h at 4°C.
We used monoclonal antibodies anti‐Bad, anti‐Bax, anti‐Bid and anti‐Bcl‐2 (BD Pharmingen, San Jose, CA, USA), polyclonal rabbit antibodies anti‐Bak (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti‐Bim (BD Pharmingen), anti‐Bcl‐xL (BD Pharmingen) and anti‐c‐FlipL (Calbiochem, La Jolla, CA, USA). Primary antibodies were diluted at 1 : 1000 in blocking buffer plus 0·01% sodium azide. Following three washes in TBS‐Tween buffer, membranes were labelled for 45 min with secondary antibody conjugated with horseradish peroxidase anti‐mouse or anti‐rabbit (GE Healthcare, Little Chalfont, UK). Bands were detected by the enhanced chemiluminescence (ECL)‐plus system (GE Healthcare) and quantified densitometrically by Alphaease software (Alpha Innotech, San Leandro, CA, USA). Polyclonal rabbit antibody anti‐γ‐tubulin and monoclonal anti‐β‐actin were used as endogenous controls. Results are presented by ratio of the integrated density value (IDV) of target proteins by the endogenous control IDV.
Statistical analysis
The Mann–Whitney t‐test was used to compare gene expression of MS patients with healthy subjects and MS patients at day + 720 post‐AHSCT with controls. Longitudinal analyses of gene expression in MS patients treated with BEAM or CY/AHSCT were performed by a mixed‐effects linear regression model. For multiple comparisons, we used orthogonal contrasts and the results were obtained using sas ® version 9 through proc npar1way. P‐values lower than 0·05 were considered statistically significant.
Results
Clinical aspects
Patients were evaluated thoroughly for clinical and laboratory aspects before, during and periodically after transplantation for the whole follow‐up period. Seven of the 18 patients presented worsening of the EDSS scores at 2 years after transplantation when compared to baseline, and were thus labelled as the ‘progression’ group. The 11 remaining patients were responsive to the procedure, experiencing either improvement or stabilization of the neurological disability. There were no significant differences in the response rate between patients treated with either BEAM/hATG or CY/rATG conditioning regimens.
One patient experienced severe transplant‐related toxicity, with bilateral bacterial pneumonia treated in the intensive care unit with broad‐spectrum antibiotics under mechanical ventilation. This patient recovered fully from the complication after a few weeks of treatment. Three other patients presented mild allergic reactions to ATG infusions, which were treated successfully with steroids and histamine blockers. One patient presented severe hypotension during etoposide infusion, reversed with volume expansion. Three patients developed non‐complicated, dermatome‐limited herpes zoster rash several months after AHSCT. Two patients who had previous history of urinary dysfunction and recurrent infections developed lower urinary tract infections after transplant and required antibiotic treatment followed by continuous prophylaxis. There were no deaths due to transplant toxicity or disease progression in any of the studied patients. None of patients were treated with disease‐modifying drugs during the length of this study.
Defective apoptosis‐related gene expression in MS patients
To evaluate the apoptosis profile in MS patients, we analysed the expression of pro‐ and anti‐apoptotic genes of the intrinsic and extrinsic pathways, and from IAP family members in PBMCs isolated from MS patients and healthy subjects. We observed a significant decrease in pro‐apoptotic BAD (median: 0·032, P < 0·01), BAX (0·003, P = 0·01) and FASL (6·966, P = 0·03) expression in the PBMCs of MS patients when compared with controls (BAD: 1·423; BAX: 9·625; FASL: 18·368) (Fig. 1a–c). In contrast, significant increase in BID (101·629, P = 0·03) and BOK (4·867, P < 0·01) expression was observed in MS patients compared with controls (BID: 13·499; BOK: 1·493) (Fig. 1d,e).
Figure 1.
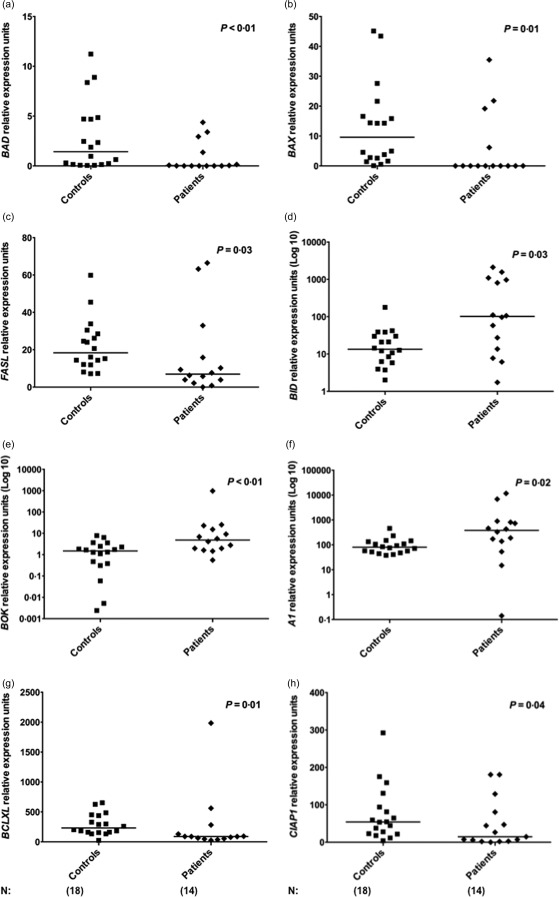
Deregulated expression of apoptosis‐related genes in multiple sclerosis (MS) patients. (a–e) Expression of BAD, BAX, FASL, BID and BOK pro‐apoptotic genes in peripheral blood mononuclear cells (PBMCs) from healthy controls (n = 18) and MS patients (n = 14). (f–h) Expression of A1, BCLXL and CIAP1 anti‐apoptotic genes in controls and patients. Apoptosis‐related gene expression was evaluated by real‐time polymerase chain reaction (PCR). Statistical analyses were performed by Mann–Whitney U‐test. Significance was set at P < 0·05. Horizontal bars represent median values. Symbols correspond to individual subjects.
Furthermore, the anti‐apoptotic gene A1 (median 381·789, P = 0·02) was up‐regulated significantly in MS patients compared with healthy individuals (A1: 79·972) (Fig. 1f), while BCL‐XL (88·376, P = 0·01) and c‐IAP1 (11·145, P = 0·04) were down‐regulated in patients compared with controls (BCL‐XL: 232·31; c‐IAP1: 54·135) (Fig. 1g‐h). Supporting information, Table S1 shows gene expression values for all 18 genes evaluated in patients at the pretransplantation period (BEAM/hATG or CY/hATG groups) and controls. Figure 2a summarizes comparative gene expression data from MS patients and controls.
Figure 2.
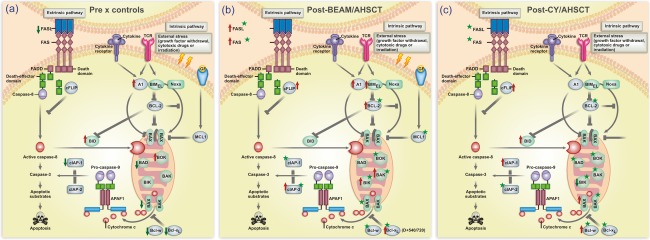
Schematic diagram of the extrinsic and intrinsic apoptotic pathways highlighting results of the study. (a) Alterations of gene expression in peripheral blood mononuclear cells (PBMCs) from multiple sclerosis (MS) patients compared with those from healthy subjects. Red arrows indicate up‐regulated genes and green arrows indicate down‐regulated genes. (b) Abnormalities of gene expression in PBMCs from MS patients after BCNU, Etoposide, AraC and Melphalan/horse anti‐human thymocyte globulin autologous haematopoietic stem cell transplantation (BEAM/hATG AHSCT) (day + 720) compared with pre‐AHSCT values. (c) Abnormalities of gene expression in PBMCs from MS patients after cyclophosphamide (CY)/rabbit ATG (rATG) AHSCT (day + 720) compared with pre‐AHSCT values. In (b) and (c), red arrows indicate up‐regulated genes and green arrows indicate down‐regulated genes in MS patients following transplantation. Stars indicate modulated genes that reached healthy control levels at day + 720. [Colour figure can be viewed at wileyonlinelibrary.com]
Apoptosis‐related gene expression is modulated in MS patients after BEAM/hATG followed by AHSCT
Once we detected abnormal expression of several apoptosis‐related genes in MS patients at baseline, we evaluated the effect of BEAM/AHSCT therapy on the expression of those genes after transplantation.
The pro‐apoptotic BAK (median pre‐AHSCT: 5·408; P < 0·01), BIK (0·747; P = 0·02), BIMEL (28·487; P < 0·01), FAS (53·659; P = 0·03) and FASL (7·676; P = 0·01) gene expressions were increased after AHSCT, mainly at day + 540 (FAS: 174·712; FASL: 29·565) and day + 720 (BAK: 603·477; BIK: 4·392; BIMEL: 172·688; FASL: 35·837) (Fig. 3a–e). Similarly, a significant increase in the anti‐apoptotic A1 (median pre‐AHSCT: 173·596; P = 0·02), BCL‐2 (0·815; P = 0·04), BCL‐XL (47·522; P < 0·01), c‐FLIPL (21·004; P = 0·03) and c‐IAP2 (14·731; P = 0·04) gene expressions was detected at day + 360 (c‐FLIPL: 95·651), day + 540 (c‐IAP2: 31·553) and day + 720 (A1: 374·090; BCL‐2: 12·353; BCL‐XL: 295·262) (Fig. 3f–j).
Figure 3.
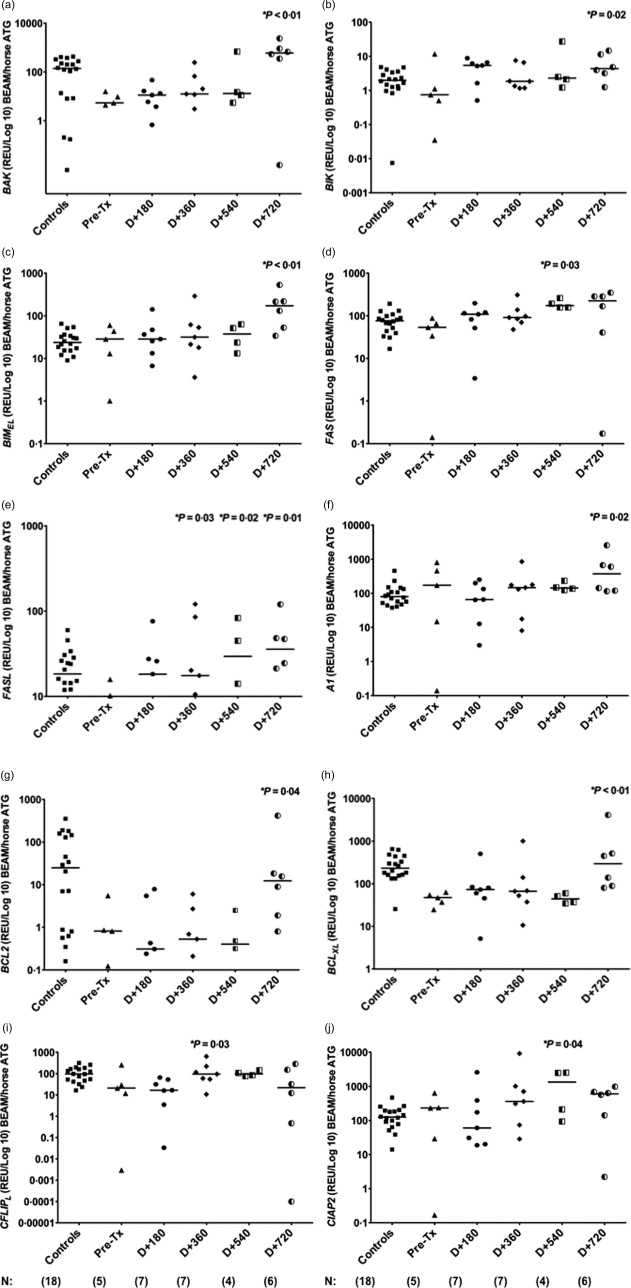
Modulation of apoptosis‐related genes in multiple sclerosis (MS) patients after AHSCT with BCNU, Etoposide, AraC and Melphalan/horse anti‐human thymocyte globulin autologous haematopoietic stem cell transplantation (BEAM/hATG AHSCT) conditioning regimen. (a–e) Expression of BAK, BIK, BIMEL, FAS and FASL pro‐apoptotic genes in peripheral blood mononuclear cells (PBMCs) from healthy controls (n = 18) and MS patients (n = 5) before and at day + 180 (n = 7), day + 360 (n = 7), day + 540 (n = 4) and day + 720 (n = 6) post‐AHSCT with BEAM/hATG conditioning regimen. (f–j) Expression of A1, BCL2, BCLXL, CFLIPL and CIAP2 anti‐apoptotic genes in PBMCs from healthy controls and MS patients before and at day + 180, day + 360, day + 540 and day + 720 post‐AHSCT with BEAM/hATG conditioning regimen. Apoptosis‐related gene expression was evaluated by real‐time polymerase chain reaction (PCR). Statistical analyses were performed by linear regression model with mixed effects. For multiple comparisons, we used orthogonal contrasts post‐test. Significance was set at P < 0·05. Horizontal bars represent medians. Symbols correspond to individual subjects.
Furthermore, we evaluated the outcomes of initially deregulated apoptosis‐related gene expressions in MS patients after BEAM/hATG and AHSCT. At 2 years post‐transplantation, expression levels of proapoptotic BAK, BIK, BOK, FAS and FASL genes in MS patients were similar to those from controls (median controls, BAK: 139·956; BIK: 1·989; BOK: 1·492; FAS: 76·422; FASL: 18·367). Expression of BAD, BAX, BID, BIMEL and NOXA was also modulated after transplantation, although not reaching control levels (median controls, BAD: 1·423; BAX: 9·625; BID: 13·498; BIMEL: 23·778; NOXA: 17·562). Additionally, at day + 720, expression of the anti‐apoptotic genes A1, BCL‐2, BCLW, BCL‐XL, MCL1, c‐IAP1 and c‐IAP2 was normalized in MS patients, similar to healthy subjects (median controls, A1: 79·971; BCL‐2: 24·897; BCLW: 2·026; BCL‐XL: 232·310; MCL1: 7537·153; c‐IAP1: 54·135; c‐IAP2: 125·443). Finally, the expression of c‐FLIPL in MS patients at day + 720 was different from that found in controls (median controls, c‐FLIPL: 96·838). Supporting information, Tables S2–S19, show gene expression values for all 18 genes evaluated in patients (BEAM/hATG or CY/hATG groups) and controls. Figure 2b summarizes the results of comparative gene expression data from MS patients before and after BEAM/hATG followed by AHSCT.
Apoptosis‐related gene expression is modulated in MS patients after CY/rATG followed by AHSCT
We observed increased expression of pro‐apoptotic genes BAX (median pre‐AHSCT: 0·011; P < 0·01) and BID (58·014; P < 0·01) at day + 180, day + 360, day + 540 and day + 720 post‐transplantation. Conversely, decreased expression of BIK (median pre‐AHSCT: 4·430; P < 0·01) was observed at day + 720 post‐AHSCT (Fig. 4a–c).
Figure 4.
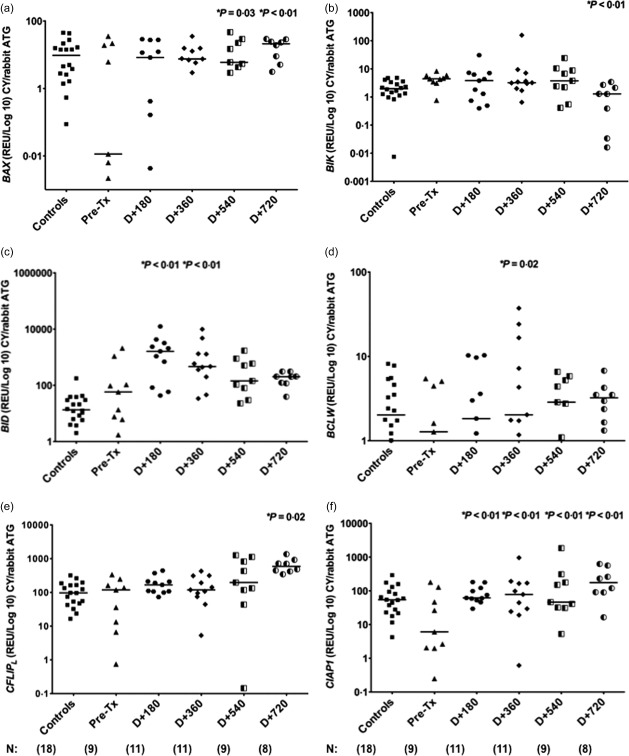
Modulation of apoptosis‐related genes in multiple sclerosis (MS) patients after autologous haematopoietic stem cell transplantation (AHSCT) with cyclophosphamide/rabbit anti‐human thymocyte globulin (CY/rATG) conditioning regimen. (a–c) Expression of BAX, BIK and BID pro‐apoptotic genes in peripheral blood mononuclear cells (PBMCs) from healthy controls (n = 18) and MS patients (n = 9) before and at day + 180 (n = 11), day + 360 (n = 11), day + 540 (n = 9) and day + 720 (n = 8) post‐AHSCT with CY/rATG conditioning regimen. (d–f) Expression of BCLW, CFLIPL and CIAP1 anti‐apoptotic genes in PBMCs from healthy controls and MS patients before and at day + 180, day + 360, day + 540 and day = 720 post‐CY/rATG conditioning regimen. Apoptosis‐related gene expression was evaluated by real‐time polymerase chain reaction (PCR). Statistical analyses were performed by linear regression model with mixed effects. For multiple comparisons we used the post‐test by orthogonal contrasts. Significance was set at P < 0·05. Horizontal bars represent medians and symbols correspond to individual subjects.
In addition, a significant increase in anti‐apoptotic BCLW (median pre‐AHSCT: 1·277; P = 0·02), c‐FLIPL (118·995; P = 0·02) and c‐IAP1 (6·086; P < 0·01) gene expressions at day + 180 (c‐IAP1: 62·026), day + 360 (BCLW: 2·034; c‐IAP1: 78·444), day + 540 (c‐IAP1: 46·341) and day + 720 (c‐FLIPL: 595·165; c‐IAP1: 175·338) was detected (Fig. 4d–f).
Furthermore, expression of BAD, BAK, BAX, BIK, BOK, NOXA, FAS and FASL pro‐apoptotic genes in MS patients were similar to control levels (P > 0·05) at day + 720 after transplantation (median controls, BAD: 1·423; BAK: 139·956; BAX: 9·625; BIK: 1·989; BOK: 1·492, NOXA: 17·562; FAS: 76·422; FASL: 18·367). Expression of BID and BIMEL genes was modulated during follow‐up, although not reaching control levels at day + 720 (median controls, BID: 13·498; BIMEL: 23·778). In addition, we demonstrated re‐establishment of normal expression of BCL‐2, BCLW, BCL‐XL, MCL1 and c‐IAP2 anti‐apoptotic genes in MS patients at day + 720 when compared to controls (median controls, BCL‐2: 24·897; BCLW: 2·026; BCL‐XL: 232·310; MCL1: 7537·153; c‐IAP2: 125·443). In the CY group, expression of A1, c‐FLIPL and c‐IAP1 genes did not reach control levels (median controls, A1: 79·971; c‐FLIPL: 96,838; c‐IAP1: 54·135) after AHSCT (Fig. 4e,f). Supporting information, Tables S2–S19, show gene expression values for all 18 genes evaluated in patients (BEAM/hATG or CY/hATG groups) and controls. Figure 2c summarizes the results of comparative gene expression data from MS patients before and after CY/rATG followed by AHSCT.
Apoptosis‐related proteins expression in MS patients before and after AHSCT
To demonstrate further deregulated expression of the pro‐apoptotic FAS and FASL genes in MS patients submitted to BEAM/AHSCT therapy, we analysed the expression of these molecules on CD4+ and CD8+ T cells by flow cytometry. We observed a significant decrease in the number of CD4+Fas+ T cells at day + 360 post‐AHSCT (median: 182·5 cells/μl, P = 0·04) compared with pretransplantation (660·5) (Fig. 5a). Conversely, we found increased numbers of CD8+Fas+ T cells at day + 180 (1110, P = 0·04) and day + 360 (795·7, P = 0·02) post‐transplantation compared with baseline (333·4) (Fig. 5b). We did not find differences in the frequencies of CD4+FasL and CD8+FasL+ subsets in MS patients after transplantation (Fig. 5c,d).
Figure 5.
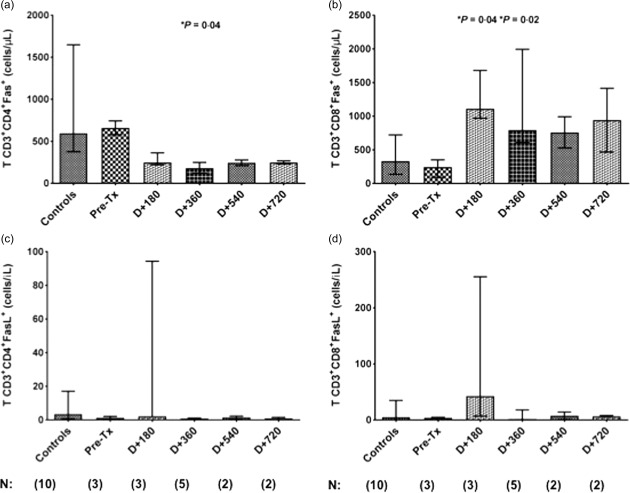
Reconstitution of CD4+Fas+ and CD8+Fas+ T cells in multiple sclerosis (MS) patients after AHSCT. (a) CD4+Fas+ T cell counts (cells/μl) in healthy controls (n = 10) and in MS patients before (n = 3) and at day (D) + 180 (n = 3), day + 360 (n = 5), day + 540 (n = 2) and day + 720 (n = 2) post‐BCNU, Etoposide, AraC and Melphalan/horse anti‐human thymocyte globulin autologous haematopoietic stem cell transplantation (BEAM/hATG AHSCT) conditioning regimen. (b) CD8+Fas+ T cell counts (cells/μl) in healthy controls and in MS patients before and at day + 180, day + 360, day + 540 and day + 720. Statistical analyses were performed by linear regression model with mixed effects. For multiple comparisons, we used the post‐test by orthogonal contrasts. Significance was set at P < 0·05. Bars represent the medians with the range.
To evaluate further whether pro‐ and anti‐apoptotic gene expression results correlated with their respective protein expressions in patients who underwent AHSCT with the CY/rATG regimen, we performed Western blotting analysis for the Bad, Bak, Bax, Bid, Bim, Bcl‐2, Bcl‐xL and c‐FlipL proteins. PBMCs from MS patients, at baseline and day + 360 post‐AHSCT, and controls were analysed.
At day + 360 post‐AHSCT, Bak (IDV median: 0·046), Bim (0·231), Bcl‐xL (0·251) and c‐FlipL (0·158) protein expressions were decreased significantly (P = 0·04 for all comparisons) compared to baseline (median Bak: 0·152, Bim: 1·564, Bcl‐xL: 0·624, cFlip‐L: 0·415). At baseline, there were no differences in Bak, Bim, Bcl‐xL and c‐FlipL protein expressions when patients were compared to controls (Fig. 6a–e). At 1 year post‐transplantation, the expression of Bak, Bim, Bcl‐xL and c‐FlipL protein was modulated during follow‐up, although not reaching control levels.
Figure 6.
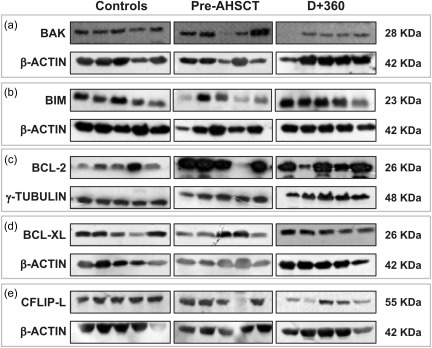
Expression of apoptosis‐related proteins in multiple sclerosis (MS) patients after cyclophosphamide/autologous haematopoietic stem cell transplantation (CY/AHSCT). (a,b) Expression of Bak and Bim pro‐apoptotic proteins in healthy controls and MS patients before and at day + 360 post‐AHSCT with CY/rATG conditioning regimen (n = 5 for each group). Protein expression was determined by Western blotting. (c–e) Bcl‐2, Bcl‐xL and cFlip‐L anti‐apoptotic protein expression in healthy controls and MS patients before and at day + 360 post‐AHSCT with CY/rATG conditioning regimen. Statistical analyses were performed by Kruskal–Wallis test followed by Dunn's post‐test. Significance was set at P < 0·05.
Expression of the anti‐apoptotic protein Bcl‐2 was increased in patients before transplantation (median 4·210) when compared with controls (1·389, P = 0·02). At day + 360 post‐AHSCT, Bcl‐2 protein expression was decreased (0·754, P = 0·007) when compared with pretransplant levels (Fig. 6c). At 1 year post‐transplantation, we found that expression levels of Bcl‐2 protein in MS patients were similar to those found in healthy subjects. We found no differences in Bad, Bax and Bid protein expression at baseline and day + 360, compared with controls (data not shown).
Discussion
Mechanistic studies in autoimmune diseases indicate that AHSCT is able to eliminate the immune cell repertoire, promote reconstitution of a new immune system from haematopoietic stem cell precursors (immune resetting) and restore immune tolerance, thereby interrupting inflammatory activity and preventing disease progression 42, 49, 50. To date, more than 800 MS patients have been treated worldwide with AHSCT 44. Currently, BEAM/ATG is the most frequently used AHSCT regimen for MS in Europe, and has also been used by some North American centres 51. Nevertheless, CY/ATG has been adopted by other transplant centres to treat MS and several other autoimmune diseases, with equally positive results 52, 53. Recently, one single North American centre has shown safety and long‐term activity‐free survival in a cohort of relapsing–remitting MS patients treated with CY/ATG conditioning regimen 54. The issue is a matter of debate, and the Italian group still believes that BEAM/ATG may be a more effective scheme for MS patients, leading to longer disease activity‐free survival 55. While a consensus has not yet been achieved, we believe that mechanistic investigations may aid to consolidate one or other regimen in the context of AHSCT for MS patients and perhaps for other autoimmune and inflammatory disorders.
Defective apoptosis affects tolerance processes and deletion of autoreactive cells and may be involved in the development of EAE in mice and MS in humans 10, 23, 24, 25, 27, 28, 29. Therefore, we investigated further the expression profile of apoptosis‐related molecules in MS patients and their modulation for the first 2 years following treatment with AHSCT using BEAM or CY‐based conditioning regimen.
Before transplantation, we detected abnormal expression of genes from the intrinsic apoptosis pathway in MS patients (decreased BAD and BAK, increased A1 and Bcl‐2 protein), suggesting a role of defective apoptosis in disease pathogenesis. Similarly, previous studies have shown alterations in mitochondria‐based apoptosis, such as decreased BAX/BCL‐2 protein ratio in peripheral lymphocytes from MS patients compared to controls 23. Up‐regulated BCL‐2 gene expression has been described in PBMCs from MS patients 56. Moreover, increased Bcl‐xL expression has been correlated with higher resistance to AICD 24.
After AHSCT, we showed that several genes from the intrinsic pathway were modulated in PBMCs, mainly in the BEAM‐conditioned (increased BAK, BIK, BIMEL, A1, BCL‐2, BCL‐XL) compared to the CY‐conditioned group (increased BAX, BID, BCLW and decreased BIK). At 2 years after transplantation, most genes from mitochondria‐based apoptosis reached levels similar to healthy controls, both in the BEAM and CY‐conditioned groups, suggesting that AHSCT plays an essential role in re‐establishing the apoptosis machinery in MS patients early after therapy. To our knowledge, there are no reports in the literature about modulation of apoptosis‐related molecules from the intrinsic pathway after AHSCT.
In the extrinsic apoptosis pathway, we found decreased expression of FASL in PBMCs from our MS patients when compared to baseline. Indeed, studies in mice and humans with autoimmune diseases have demonstrated decreased expression of FAS and FASL in peripheral lymphocytes 57, 58, 59. They also suggest that the breakdown of immune tolerance to self‐antigens could be associated with defective AICD, which is the major mechanism of peripheral tolerance induction, playing an important role in the deletion of autoreactive lymphocytes that have escaped from central tolerance process 58, 60.
After transplantation, we showed that genes from the extrinsic pathway were modulated during follow‐up in both groups, conditioned with BEAM (increased FAS, FASL and c‐FLIPL) and CY (increased c‐FLIPL), suggesting the role of these molecules in death of residual autoreactive lymphocytes. In fact, we have demonstrated FAS and FASL up‐regulation previously in type 1 diabetes patients after AHSCT with CY conditioning regimen 22. In the present study, we detected normalization of FAS and FASL expression towards levels similar to controls, both in BEAM and CY groups, at day + 720, suggesting the re‐establishment of peripheral tolerance and AICD. In addition, cytometric analysis showed an increase of CD8+Fas+ T cells at day + 180 and day + 360 when compared to baseline. Previous observations have shown Fas up‐regulation on immune cells after immunosuppression with rabbit ATG 61, 62, 63. Furthermore, Muraro and co‐workers showed increased frequency of CD4+Fas+ and CD8+Fas+ T cells in MS patients treated with AHSCT during the first months of immune reconstitution 64. Based on the immune reconstitution profile of these patients, we suggest that the increase of the CD4+ T cell numbers at day + 720 may account for the observed normalization of the apoptotic gene expression 65. However, this correlation must be confirmed by future experiments. Our results and these reports highlight the possible role of apoptosis modulation in the re‐establishment of immunological tolerance after AHSCT.
Regarding IAP family molecules, we detected up‐regulation of c‐IAP1 in PBMCs from MS patients when compared with healthy counterparts, suggesting deregulated apoptosis in MS. Similarly, Sharief and Semra showed higher expression of IAP proteins (IAP‐1, IAP‐2 and XIAP) in peripheral T lymphocytes from MS patients 66. Hebb and colleagues reported increased mRNA levels of IAP1, IAP2 and XIAP in the peripheral blood of active MS patients 67. In our study, c‐IAP2 and c‐IAP1 were modulated after AHSCT in both BEAM and CY‐conditioned groups, reaching levels similar to controls at 2‐year follow‐up, indicating normalization of apoptosis processes. Such findings have not yet been described in the literature.
In a recent study concerning immune mechanisms after AHSCT in MS patients, we have demonstrated that CD4+ and CD8+ T cells presented decreased FOXP3, FOXO1, PDCD1 and IRF2BP2 genes and increased miR‐16, miR‐155 and miR‐142‐3p before transplantation. At 2 years post‐transplantation the expression of these genes increased, reaching control levels. This study suggests that AHSCT normalizes gene and microRNA expression, thereby improving the immunoregulatory network. These immune mechanisms may be important for disease control in the early periods following AHSCT in MS patients 65.
We have compared previously the clinical aspects of MS patients who underwent AHSCT with two different conditioning regimens, BEAM/hATG and CY/rATG, showing less toxicity in the CY group 47. However, as evidenced by the different baseline EDSS scores in each group, patient characteristics were very dissimilar, having patients with more advanced disease, mainly in the secondary progressive phase, included in the BEAM group 47. Indeed, in the present analysis, we observed increasing EDSS scores after transplantation in the BEAM group, while they stabilized in the CY group. Therefore, due to the clinical heterogeneity between BEAM and CY‐conditioned patients, our study does not allow groups to be compared. We were, however, able to evaluate the effects of AHSCT on the apoptosis‐related gene expression within each of the conditioning regimen groups. Both BEAM and CY regimens were able to modulate expression of apoptosis‐related genes in MS patients throughout follow‐up. In the BEAM‐conditioned patients, we observed that BAD, BAX, BID, BIMEL, NOXA and c‐FLIPL, which are involved in inhibition of the extrinsic apoptosis pathway, did not reach control levels at 2 years post‐transplantation. Deregulation of apoptosis‐related molecules may be involved with the perpetuation of residual memory autoreactive lymphocytes and possibly with disease progression of MS patients.
Normalization of apoptosis‐related molecules during the first 2 years after AHSCT, mainly modulation of FAS and FASL, suggests re‐establishment of peripheral tolerance mechanisms mediated by AICD, possibly increasing apoptosis susceptibility of residual autoreactive lymphocytes. Therefore, we believe that FAS and FASL may be identified as biomarkers for clinical neurological outcomes in MS patients treated with AHSCT. Furthermore, these molecules may become targets for future therapeutic interventions.
Disclosure
The authors report no disclosures.
Author contributions
G. L. V. de O. participated in all aspects of the study, experimental conception, sample collection, PCR and protein extraction standardization, experimental procedures, data acquisition, data analysis, interpretation and manuscript writing. A. F. F. and E. P. L. G. helped with PCR and WB standardizations; C. R. T., A. A. B. and D. G. B. were responsible for all clinical aspects involving MS patients. S. K. and D. T. C. provided administrative and financial support. J. C. V. contributed to conception and study design. M. C. O., F. A. de C. and K. C. R. M. participated in experimental conception, interpretation and drafting/revising the manuscript.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Table S1. Gene expression levels of apoptotic genes of patients at pretransplant period BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimens] and controls.
Table S2. A1 gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S3. BCL2 gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S4. BCL‐W gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S5. BCL‐XL gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S6. MCL‐1 gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S7. C‐FLIP gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S8. clAP‐1 gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S9. clAP‐2 gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S10. BAD gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S11. BAK gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S12. BAX gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S13. BID gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S14. BIK gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S15. BIMEL gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S16. BOK gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S17. NOXA gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S18. FAS gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S19. FASL gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Acknowledgements
The study was supported by Brazilian governmental agencies, as follows: Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Foundation for the Support of Research in the State of São Paulo; Processes numbers 2008/58387 and 2008/57877‐3), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Coordination of the Advancement of Higher Education) and Fundação Hemocentro de Ribeirão Preto (FUNDHERP, Ribeirao Preto Blood Bank Foundation).
References
- 1. Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest 2012; 122:1180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol 2015; 15:545–58. [DOI] [PubMed] [Google Scholar]
- 3. Stys PK, Zamponi GW, Van Minnen J, Geurts JJ. Will the real multiple sclerosis please stand up? Nat Rev Neurosci 2012; 13:507–14. [DOI] [PubMed] [Google Scholar]
- 4. Hafler DA, Compston A, Sawcer S et al Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007; 357:851–62. [DOI] [PubMed] [Google Scholar]
- 5. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann Neurol 2007; 61:288–99. [DOI] [PubMed] [Google Scholar]
- 6. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann Neurol 2007; 61:504–13. [DOI] [PubMed] [Google Scholar]
- 7. Gatzka M, Walsh CM. Apoptotic signal transduction and T cell tolerance. Autoimmunity 2007; 40:442–52. [DOI] [PubMed] [Google Scholar]
- 8. Kinnunen T, Chamberlain N, Morbach H et al Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest 2013; 123:2737–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol 2010; 10:849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Comi C, Fleetwood T, Dianzani U. The role of T cell apoptosis in nervous system autoimmunity. Autoimmun Rev 2012; 12:150–6. [DOI] [PubMed] [Google Scholar]
- 11. Opferman JT. Apoptosis in the development of the immune system. Cell Death Differ 2008; 15:234–42. [DOI] [PubMed] [Google Scholar]
- 12. Tischner D, Woess C, Ottina E, Villunger A. Bcl‐2‐regulated cell death signalling in the prevention of autoimmunity. Cell Death Dis 2010; 1:e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol 2007; 7:532–42. [DOI] [PubMed] [Google Scholar]
- 14. Maniati E, Potter P, Rogers NJ, Morley BJ. Control of apoptosis in autoimmunity. J Pathol 2008; 214:190–8. [DOI] [PubMed] [Google Scholar]
- 15. Bouillet P, O'Reilly LA. CD95, BIM and T cell homeostasis. Nat Rev Immunol 2009; 9:514–9. [DOI] [PubMed] [Google Scholar]
- 16. Liphaus BL, Kiss MHB, Carrasco S, Goldenstein‐Schainberg C. Increased Fas and Bcl‐2 expression on peripheral mononuclear cells from patients with active juvenile‐onset systemic lupus erythematosus. J Rheumatol 2007; 34:1580–4. [PubMed] [Google Scholar]
- 17. Munoz LE, Van Bavel C, Franz S, Berden J, Herrmann M, Van der Vlag J. Apoptosis in the pathogenesis of systemic lupus erythematosus. Lupus 2008; 17:371–5. [DOI] [PubMed] [Google Scholar]
- 18. Pope RM. Apoptosis as a therapeutic tool in rheumatoid arthritis. Nat Rev Immunol 2002; 2:527–35. [DOI] [PubMed] [Google Scholar]
- 19. Liu H, Pope RM. The role of apoptosis in rheumatoid arthritis. Curr Opin Pharmacol 2003; 3:317–22. [DOI] [PubMed] [Google Scholar]
- 20. Hayashi T, Faustman DL. Implications of altered apoptosis in diabetes mellitus and autoimmune disease. Apoptosis 2001; 6:31–45. [DOI] [PubMed] [Google Scholar]
- 21. Gronski M, Weinem M. Death pathways in T cell homeostasis and their role in autoimmune diabetes. Rev Diabet Stud 2006; 3:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Oliveira GLV, Malmegrim KC, Ferreira AF et al Up‐regulation of fas and fasL pro‐apoptotic genes expression in type 1 diabetes patients after autologous haematopoietic stem cell transplantation. Clin Exp Immunol 2012; 168:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sharief MK, Douglas M, Noori M, Semra YK. The expression of pro‐ and antiapoptosis Bcl‐2 family proteins in lymphocytes from patients with multiple sclerosis. J Neuroimmunol 2002; 125:155–62. [DOI] [PubMed] [Google Scholar]
- 24. Waiczies S, Weber A, Lünemann JD, Aktas O, Zschenderlein R, Zipp F. Elevated Bcl‐X(L) levels correlate with T cell survival in multiple sclerosis. J Neuroimmunol 2002; 126:213–20. [DOI] [PubMed] [Google Scholar]
- 25. Sharief MK, Matthews H, Noori MA. Expression ratios of the Bcl‐2 family proteins and disease activity in multiple sclerosis. J Neuroimmunol 2003; 134:158–65. [DOI] [PubMed] [Google Scholar]
- 26. Eguchi K. Apoptosis in autoimmune diseases. Intern Med 2001; 40:275–84. [DOI] [PubMed] [Google Scholar]
- 27. Okuda Y, Okuda M, Bernard CC. The suppression of T cell apoptosis influences the severity of disease during the chronic phase but not the recovery from the acute phase of experimental autoimmune encephalomyelitis in mice. J Neuroimmunol 2002; 131:115–25. [DOI] [PubMed] [Google Scholar]
- 28. Lev N, Barhum Y, Melamed E, Offen D. Bax‐ablation attenuates experimental autoimmune encephalomyelitis in mice. Neurosci Lett 2004; 359:139–42. [DOI] [PubMed] [Google Scholar]
- 29. Tseveleki V, Bauer J, Taoufik E et al Cellular FLIP (long isoform) overexpression in T cells drives Th2 effector responses and promotes immunoregulation in experimental autoimmune encephalomyelitis. J Immunol 2004; 173:6619–26. [DOI] [PubMed] [Google Scholar]
- 30. Reichardt HM, Lühder F. The ambivalent role of apoptosis in experimental autoimmune encephalomyelitis and multiple sclerosis. Curr Pharm Des 2012; 18:4453–64. [DOI] [PubMed] [Google Scholar]
- 31. Achiron A, Feldman A, Mandel M, Gurevich M. Impaired expression of peripheral blood apoptotic‐related gene transcripts in acute multiple sclerosis relapse. Ann N Y Acad Sci 2007; 1107:155–67. [DOI] [PubMed] [Google Scholar]
- 32. Rinta S, Kuusisto H, Raunio M et al Apoptosis‐related molecules in blood in multiple sclerosis. J Neuroimmunol 2008; 205:135–41. [DOI] [PubMed] [Google Scholar]
- 33. Gomes AC, Jönsson G, Mjörnheim S, Olsson T, Hillert J, Grandien A. Upregulation of the apoptosis regulators cFLIP, CD95 and CD95 ligand in peripheral blood mononuclear cells in relapsing‐remitting multiple sclerosis. J Neuroimmunol 2003; 135:126–34. [DOI] [PubMed] [Google Scholar]
- 34. Satoh J, Nakanishi M, Koike F et al Microarray analysis identifies an aberrant expression of apoptosis and DNA damage‐regulatory genes in multiple sclerosis. Neurobiol Dis 2005; 18:537–50. [DOI] [PubMed] [Google Scholar]
- 35. Okuda Y, Apatoff BR, Posnett DN. Apoptosis of T cells in peripheral blood and cerebrospinal fluid is associated with disease activity of multiple sclerosis. J Neuroimmunol 2006; 171:163–70. [DOI] [PubMed] [Google Scholar]
- 36. Lim SY, Constantinescu CS. Current and future disease‐modifying therapies in multiple sclerosis. Int J Clin Pract 2010; 64:637–50. [DOI] [PubMed] [Google Scholar]
- 37. Tramacere I, Del Giovane C, Salanti G, D'Amico R, Filippini G. Immunomodulators and immunosuppressants for relapsing‐remitting multiple sclerosis: a network meta‐analysis. Cochrane Database Svst Rev 2015; 9:CD011381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Farjam M, Zhang GX, Ciric B, Rostami A. Emerging immunopharmacological targets in multiple sclerosis. J Neurol Sci 2015; 358:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fassas A, Anagnostopoulos A, Kazis A et al Peripheral blood stem cell transplantation in the treatment of progressive multiple sclerosis: first results of a pilot study. Bone Marrow Transplant 1997; 20:631–8. [DOI] [PubMed] [Google Scholar]
- 40. Bowen JD, Kraft GH, Wundes A et al Autologous hematopoietic cell transplantation following high‐dose immunosuppressive therapy for advanced multiple sclerosis: long‐term results. Bone Marrow Transplant 2012; 47:946–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mancardi GL, Sormani MP, Di Gioia M et al Autologous haematopoietic stem cell transplantation with an intermediate intensity conditioning regimen in multiple sclerosis: the Italian multi‐centre experience. Mult Scler 2012; 18:835–42. [DOI] [PubMed] [Google Scholar]
- 42. Abrahamsson SV, Angelini DF, Dubinsky AN et al Non‐myeloablative autologouhaematopoietic stem cell transplantation expands regulatory cells and depletes IL‐17 producing mucosal‐associated invariant T cells in multiple sclerosis. Brain 2013; 136:2888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burt RK, Balabanov R, Voltarelli JC, Barreira A, Burman J. Autologous hematopoietic stem cell transplantation for multiple sclerosis – if confused or hesitant, remember: ‘treat with standard immune suppressive drugs and if no inflammation, no response’. Mult Scler 2012; 18:772–5. [DOI] [PubMed] [Google Scholar]
- 44. Mancardi GL, Sormani MP, Gualandi F et al Autologous hematopoietic stem cell transplantation in multiple sclerosis: a phase II trial. Neurology 2015; 84:981–8. [DOI] [PubMed] [Google Scholar]
- 45. Muraro RK. Andiamo! Moving forward with autologous hematopoietic transplantation for highly active MS. Neurology 2015; 84:968–9. [DOI] [PubMed] [Google Scholar]
- 46. Poser CM, Paty DW, Scheinberg L et al New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol 1983; 13:227–31. [DOI] [PubMed] [Google Scholar]
- 47. Hamerschlak N, Rodrigues M, Moraes DA et al Brazilian experience with two conditioning regimens in patients with multiple sclerosis: BEAM/horse ATG and CY/rabbit ATG. Bone Marrow Transplant 2010; 45:239–48. [DOI] [PubMed] [Google Scholar]
- 48. Albesiano E, Messmer BT, Damle RN, Allen SL, Rai KR, Chiorazzi N. Activation‐induced cytidine deaminase in chronic lymphocytic leukemia B cells: expression as multiple forms in a dynamic, variably sized fraction of the clone. Blood 2003; 102:3333–9. [DOI] [PubMed] [Google Scholar]
- 49. Abrahamsson S, Muraro PA. Immune re‐education following autologous hematopoietic stem cell transplantation. Autoimmunity 2008; 41:577–84. [DOI] [PubMed] [Google Scholar]
- 50. Muraro PA, Abrahamsson SV. Resetting autoimmunity in the nervous system: the role of hematopoietic stem cell transplantation. Curr Opin Invest Drugs 2010; 11:1265–75. [PubMed] [Google Scholar]
- 51. Saccardi R, Freedman MS, Sormani MP et al A prospective, randomized, controlled trial of autologous haematopoietic stem cell transplantation for aggressive multiple sclerosis: a position paper. Mult Scler 2012; 18:825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burt RK, Loh Y, Cohen B et al Autologous non‐myeloablative haemopoietic stem cell transplantation in relapsing‐remitting multiple sclerosis: a phase I/II study. Lancet Neurol 2009; 8:244–53. [DOI] [PubMed] [Google Scholar]
- 53. Snowden JA, Saccardi R, Allez M et al Haematopoietic SCT in severe autoimune diseases: updated guidelines of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant 2012; 47:770–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burt RK, Balabanov R, Han X et al Association of nonmyeloablative hematopoietic stem cell transplantation with neurological disability in patients with relapsing‐remitting multiple sclerosis. JAMA 2015; 313:275–84. [DOI] [PubMed] [Google Scholar]
- 55. Curro D, Vuolo L, Gualandi F et al Low intensity lympho‐ablative regimen followed by autologous hematopoietic stem cell transplantation in severe forms of multiple sclerosis: a MRI‐based clinical study. Mult Scler 2015; 21:1423–30. [DOI] [PubMed] [Google Scholar]
- 56. Bomprezzi R, Rignér M, Kim S et al Gene expression profile in multiple sclerosis patients and healthy controls: identifying pathways relevant to disease. Hum Mol Genet 2003; 12:2191–9. [DOI] [PubMed] [Google Scholar]
- 57. Giordano C, De Maria R, Stassi G et al Defective expression of the apoptosis‐inducing CD95 (Fas/APO‐1) molecule on T and B cells in IDDM. Diabetologia 1995; 38:1449–54. [DOI] [PubMed] [Google Scholar]
- 58. Decallonne B, Van Etten E, Giulietti A et al Defect in activation induced cell death in non‐obese diabetic (NOD) T lymphocytes. J Autoimmun 2003; 20:219–26. [DOI] [PubMed] [Google Scholar]
- 59. Zucchelli S, Holler P, Yamagata T, Roy M, Benoist C, Mathis D. Defective central tolerance induction in NOD mice: genomics and genetics. Immunity 2005; 22:385–96. [DOI] [PubMed] [Google Scholar]
- 60. Su X, Hu Q, Kristan JM et al Significant role for Fas in the pathogenesis of autoimmune diabetes. J Immunol 2000; 164:2523–32. [DOI] [PubMed] [Google Scholar]
- 61. Genestier L, Fournel S, Flacher M, Assossou O, Revillard JP, Bonnefoy‐Berard N. Induction of Fas (Apo‐1, CD95)‐mediated apoptosis of activated lymphocytes by polyclonal antithymocyte globulins. Blood 1998; 91:2360–8. [PubMed] [Google Scholar]
- 62. Dubey S, Nityanand S. Involvement of Fas and TNF pathways in the induction of apoptosis of T cells by antithymocyte globulin. Ann Hematol 2003; 82:496–9. [DOI] [PubMed] [Google Scholar]
- 63. Grüllich C, Ziegler C, Finke J. Rabbit anti T‐lymphocyte globulin induces apoptosis in peripheral blood mononuclear cell compartments and leukemia cells, while hematopoetic stem cells are apoptosis resistant. Biol. Blood Marrow Transplant 2009; 15:173–82. [DOI] [PubMed] [Google Scholar]
- 64. Muraro PA, Douek DC, Packer A et al Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med 2005; 201:805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Arruda LC, Lorenzi JC, Sousa AP et al Autologous hematopoietic SCT normalizes miR‐16, ‐155 and ‐142‐3p expression in multiple sclerosis patients. Bone Marrow Transplant 2015; 50:380–9. [DOI] [PubMed] [Google Scholar]
- 66. Sharief MK, Semra YK. Upregulation of the inhibitor of apoptosis proteins in activated T lymphocytes from patients with multiple sclerosis. J Neuroimmunol 2001; 119:350–7. [DOI] [PubMed] [Google Scholar]
- 67. Hebb AL, Moore CS, Bhan V et al Expression of the inhibitor of apoptosis protein family in multiple sclerosis reveals a potential immunomodulatory role during autoimmune mediated demyelination. Mult Scler 2008; 14:577–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Table S1. Gene expression levels of apoptotic genes of patients at pretransplant period BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimens] and controls.
Table S2. A1 gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S3. BCL2 gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S4. BCL‐W gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S5. BCL‐XL gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S6. MCL‐1 gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S7. C‐FLIP gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S8. clAP‐1 gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S9. clAP‐2 gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S10. BAD gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S11. BAK gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S12. BAX gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S13. BID gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S14. BIK gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S15. BIMEL gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S16. BOK gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S17. NOXA gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S18. FAS gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.
Table S19. FASL gene expression levels of gene in patients at pre‐ and post‐transplantation periods BCNU, Etoposide, AraC and Melphalan (BEAM) or cyclophosphamide conditioning regimen groups] and controls.