

Abstract
Heterotrimeric G proteins signal at a variety of endomembrane locations, in addition to their canonical function at the cytoplasmic surface of the plasma membrane (PM), where they are activated by cell surface G protein-coupled receptors. Here we focus on βγ signaling at the Golgi, where βγ activates a signaling cascade, ultimately resulting in vesicle fission from the trans-Golgi network (TGN). To develop a novel molecular tool for inhibiting endogenous βγ in a spatial-temporal manner, we take advantage of a lipid association mutant of the widely used βγ inhibitor GRK2ct (GRK2ct-KERE) and the FRB/FKBP heterodimerization system. We show that GRK2ct-KERE cannot inhibit βγ function when expressed in cells, but recruitment to a specific membrane location recovers the ability of GRK2ct-KERE to inhibit βγ signaling. PM-recruited GRK2ct-KERE inhibits lysophosphatidic acid-induced phosphorylation of Akt, whereas Golgi-recruited GRK2ct-KERE inhibits cargo transport from the TGN to the PM. Moreover, we show that Golgi-recruited GRK2ct-KERE inhibits model basolaterally targeted but not apically targeted cargo delivery, for both PM-destined and secretory cargo, providing the first evidence of selectivity in terms of cargo transport regulated by βγ. Last, we show that Golgi fragmentation induced by ilimaquinone and nocodazole is blocked by βγ inhibition, demonstrating that βγ is a key regulator of multiple pathways that impact Golgi morphology. Thus, we have developed a new molecular tool, recruitable GRK2ct-KERE, to modulate βγ signaling at specific subcellular locations, and we demonstrate novel cargo selectivity for βγ regulation of TGN to PM transport and a novel role for βγ in mediating Golgi fragmentation.
Keywords: cell biology, G protein, Golgi, intracellular trafficking, protein trafficking (Golgi), signal transduction
Introduction
Heterotrimeric G proteins are molecular switches that activate global downstream signaling cascades within the cell. Canonical G protein activation and signaling occur at the plasma membrane (PM)2 via an activated G protein-coupled receptor (GPCR). The GPCR acts as a guanine-nucleotide exchange factor to promote GDP for GTP exchange, triggering a conformational change resulting in dissociation of Gα and βγ subunits. In addition to Gα and βγ signaling at the PM, there is increasing evidence for signaling at endomembrane locations, where G proteins can bind diverse effectors for discrete signaling pathways (1–3). In one such non-canonical signaling pathway, βγ localizes to the trans-Golgi network (TGN), where it mediates vesicle fission in a protein kinase D (PKD)-dependent manner (4–6).
Golgi-localized βγ stimulates the known effector PLCβ3 to increase the Golgi-localized pool of diacylglycerol (7). This local increase in diacylglycerol at the TGN results in subsequent recruitment of protein kinase Cη (PKCη) and PKD (6, 8). Fully phosphorylated and activated PKD is able to initiate downstream phosphorylation events to activate lipid kinases, such as phosphatidylinositol 4-kinase IIIβ (8–10). A subsequent increase in local phosphatidylinositol 4-phosphate results in recruitment of lipid-modifying enzymes and machinery essential for vesicle fission at the TGN (11, 12).
Proper secretion and transport from the Golgi are essential for cell function; dysregulation of these contributes to diseases (13). In addition to a vital role in secretion, post-TGN vesicles are important in maintaining polarized membrane subdomains and trafficking to endomembrane compartments, regulating cell motility, and proper tissue formation. It is well established that the TGN functions as a sorting center for cargoes known to sort apically or basolaterally in polarized cells, based on sequence identity and known sorting motifs (14). Basolaterally directed transport from the Golgi to the PM has already been shown to be PKD-dependent, whereas the transport of cargo proteins targeted to apical regions occurs independently of PKD (9). Secretion of insulin in response to glucose was found to require PKD signaling at the Golgi (15), and previous results in our laboratory showed that βγ is the upstream effector of PKD at the TGN for regulating Golgi to PM transport of VSV-G and secreted signal sequence horseradish peroxidase (ssHRP), two model basolaterally targeted cargoes (5). Thus, there is a role for βγ in the regulation of basolateral cargo trafficking, but to date, no link has been made between βγ and distribution of cargo toward distinct membrane domains. Specifically, it is not known whether βγ, like PKD, selectively regulates basolaterally targeted, but not apically targeted, cargoes or whether βγ plays a wider role than PKD in regulating distinct cargo transport.
To address this question and to further understand the role of βγ at the Golgi and other subcellular locations, in this report, we describe the development and use of a new molecular tool for spatiotemporal inhibition of endogenous βγ in cells. A C-terminal fragment of GPCR kinase 2 (GRK2ct) binds βγ and has been widely used to inhibit βγ signaling in cells (16). Previous work in our laboratory resulted in targeted inhibition of βγ by fusing GRK2ct to a Golgi-localized mutant KDEL receptor (5). Although this was useful in determining that Golgi-localized βγ was responsible for vesicle fission, it resulted in constitutive inhibition of βγ at the Golgi. Here, we utilize the FRB/FKBP heterodimerization system, which has been extensively characterized, to inducibly recruit a lipid association mutant of GRK2ct (GRK2ct-KERE) either to the PM or Golgi. We show that this mutant GRK2ct only inhibits βγ when it is recruited to a specific membrane, thus providing a tool to inducibly inhibit βγ at specific subcellular locations. We establish the utility of inducibly recruited GRK2ct-KERE by showing that PM recruitment, but not Golgi recruitment, can inhibit LPA-induced phosphorylation of Akt, a βγ-mediated signaling readout; in contrast, Golgi recruitment, but not PM recruitment, of GRK2ct-KERE can inhibit βγ signaling at the Golgi, as assayed by cargo transport from the Golgi to PM. Furthermore, using this inducible inhibition of βγ, here we provide the first direct comparison of the requirement for βγ in PM targeting of basolateral versus apical targeted cargo. Our results indicate that inhibition of βγ fails to prevent Golgi to PM transport of apically targeted cargo, showing cargo selectivity similar to that of PKD.
Last, we used this recruitable GRK2ct-KERE, along with other methods of inhibiting βγ, to investigate a role for βγ in mediating the action of two distinct inducers of Golgi fragmentation. Overexpression of βγ or other key regulators, such as PKD, of the Golgi-localized signaling pathway discussed above can cause fragmentation of the Golgi (5, 6, 17). Although such fragmentation is thought to simply represent overactivation of a pathway that regulates transport vesicle formation at the Golgi, it also begs the question of whether endogenous βγ is involved in any processes that stimulate Golgi fragmentation. It has become increasingly clear that Golgi fragmentation occurs and has important functions in normal cellular function as well as pathophysiologically in infection, neurodegenerative disease, and cancer (18–21). Experimentally, Golgi fragmentation can be induced through distinct mechanisms by several small molecules. Here we focused on ilimaquinone and nocodazole. Ilimaquinone, a compound originally isolated from marine sponges, has been well described to rapidly and reversibly promote Golgi fragmentation of cultured cells (22). Ilimaquinone blocks ADP-ribosylating factor and coat proteins from localizing to Golgi membranes, although they are not direct targets (23). Fragmentation via ilimaquinone is not dependent on microtubule status (24) but does involve activation of PKD and phospholipase D (25) and possibly βγ (4). Nocodazole is widely used to rapidly and reversibly depolymerize microtubules and consequently induce fragmentation of the Golgi. Recently, PKD has been implicated in nocodazole-induced Golgi fragmentation (17), suggesting a signaling process involved in Golgi fragmentation that occurs in response to microtubule disruption. Here we demonstrate a role for βγ in promoting Golgi fragmentation in response to ilimaquinone and nocodazole.
In summary, this report describes the development of a recruitable FRB-GRK2ct-KERE that selectively inhibits βγ signaling at a specific membrane location, such as the Golgi or PM, thus serving as a new molecular tool to modulate subcellular βγ signaling. Here, we use this new method of βγ inhibition to provide novel insight into the cargo selectivity of βγ regulation of Golgi to PM transport and to demonstrate a novel role for βγ in cellular pathways that induce Golgi fragmentation.
Results
A Lipid-binding Region of GRK2 C Terminus Is Necessary for Inhibition of Endogenous βγ Signaling at the Golgi
To be able to differentiate between subcellular pools of βγ responsible for differential signaling pathways, we set out to develop a version of GRK2ct that could inhibit βγ only when targeted to a specific membrane location. We reasoned that a lipid-binding mutant of GRK2ct might serve this purpose, because WT GRK2ct, widely used to inhibit βγ functions, has the drawback that it is able to inhibit βγ at various locations in the cell even in the absence of any specific subcellular targeting. Thus, we tested the ability of a K567E/R578E mutant of GRK2ct (hereafter referred as GRK2ct-KERE) to inhibit βγ in a cargo transport assay. These mutations alter membrane association by disrupting binding of GRK2 to phospholipid headgroups in the PM; full-length GRK2-KERE shows a 90% loss in β2-adrenergic phosphorylation compared with full-length WT GRK2 (26).
Here, we compare WT GRK2ct and GRK2ct-KERE as molecular inhibitors of βγ in VSV-G transport (Fig. 1). We also use GRK2ct-R587Q (RQ), a βγ-binding deficient mutant. WT GRK2ct inhibits βγ-mediated transport of VSV-G from the Golgi to the PM, upon temperature release, as demonstrated previously (5). Less than 10% of WT GRK2ct-positive cells display PM localization of VSV-G. This inhibition of Golgi to PM transport is dependent on the ability of WT GRK2ct to bind and sequester βγ, because GRK2ct-RQ fails to prevent VSV-G from reaching the PM; VSV-G is detected at the PM in >90% of cells expressing GRK2ct-RQ. Similarly, >90% of cells expressing GRK2ct-KERE also show VSV-G at the PM 2 h after temperature release, demonstrating that cytosolic GRK2ct-KERE cannot inhibit βγ signaling, although its βγ-binding surface is not mutationally disrupted.
FIGURE 1.
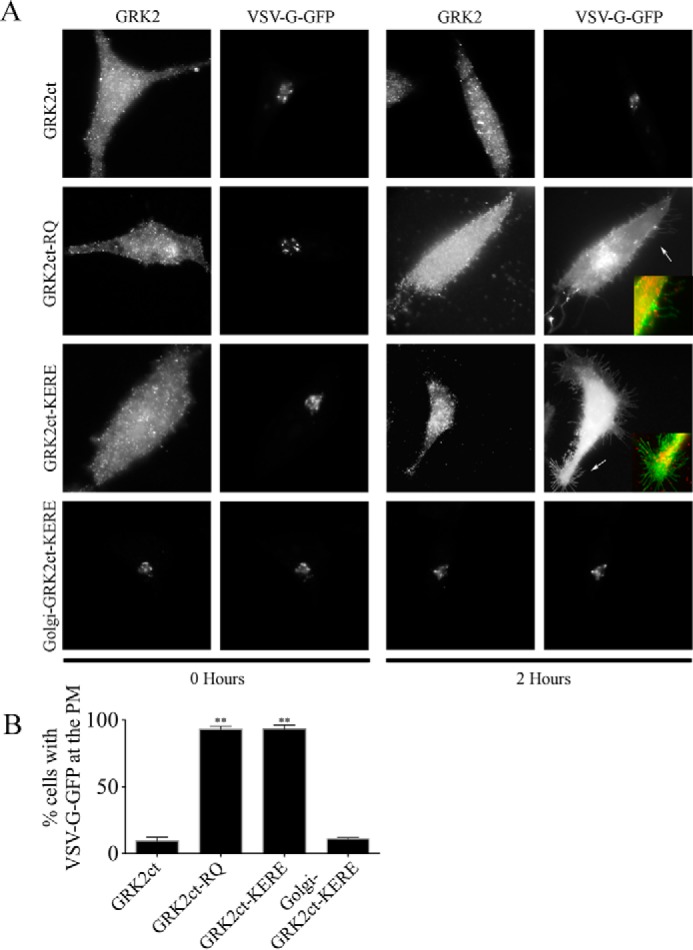
Golgi-GRK2ct-KERE inhibits VSV-G PM transport. HeLa cells were transfected with expression plasmids for temperature-sensitive VSV-G-GFP and GRK2ct, WT or mutant, as indicated. VSV-G-GFP cargo accumulates in the ER, then Golgi, and then transports to the PM when cells are incubated at 39.5, 20, and 32 °C, respectively, as described under “Experimental Procedures.” Cells were fixed at the start of the 32 °C incubation (0 Hours) or 2 h after the shift to 32 °C. A, cells were processed for immunofluorescence microscopy using anti-GRK2 and anti-GFP antibodies to detect the localization of GRK2ct (WT or mutant) and VSV-G-GFP, respectively. Representative images are shown. Inset panels, corresponding to arrows, show dual color images of VSV-G-GFP (green) and GRK2ct-RQ or GRK2ct-KERE (red) to clearly identify PM localization of the VSV-G-GFP cargo. B, percentage of transfected cells with detectable PM localization of VSV-G-GFP after 2 h at 32 °C, as described under “Experimental Procedures.” More than 150 cells were scored for PM localization of VSV-G-GFP in each of n = 5 experiments, and the bars represent the average and S.D. (error bars). Statistical significance compared with WT GRK2ct was tested using an unpaired t test (**, p < 0.0001).
To attempt to “rescue” the ability to inhibit βγ signaling, GRK2ct-KERE was targeted to the cytoplasmic face of Golgi through fusion with a mutant KDEL receptor sequence, described previously (5). When this Golgi-GRK2ct-KERE is co-expressed with VSV-G, there is a dramatic loss of cargo localized at the PM because only 11% of cells display VSV-G at the PM (Fig. 1). Thus, this supports the idea that GRK2ct-KERE can inhibit βγ only when it is specifically targeted to the same membrane location.
Inducible Recruitment of GRK2ct-KERE to the Golgi or Plasma Membrane
The above results suggested the feasibility of using GRK2ct-KERE as an “inducible inhibitor” of βγ by conditional targeting to a specific membrane for spatial and temporal inhibition. To enable this conditional targeting, we employed the FRB/FKBP-inducible heterodimerization system. FRB/FKBP heterodimerization in the presence of rapamycin or rapalog has been well established (27) and allows a spatial and temporal recruitment of proteins of interest. Insertion of GRK2ct-KERE into the FRB vector and of targeting sequences corresponding to the PM or Golgi into a corresponding FKBP vector resulted in a mode of localized membrane recruitment. Fig. 2 shows recruitment of FRB-GRK2ct-KERE and control FRB-GRK2ct-RQ to the PM or Golgi with a 15-min treatment of 1 μm rapamycin. Together, FRB-GRK2ct-(KERE/RQ) and FKBP-(PM/Golgi)-CFP will be referred to as “inducible system components.” These components now provide a tool to inhibit endogenous βγ signaling upon recruitment to specific membranes, and we next sought to validate the utility of this system.
FIGURE 2.
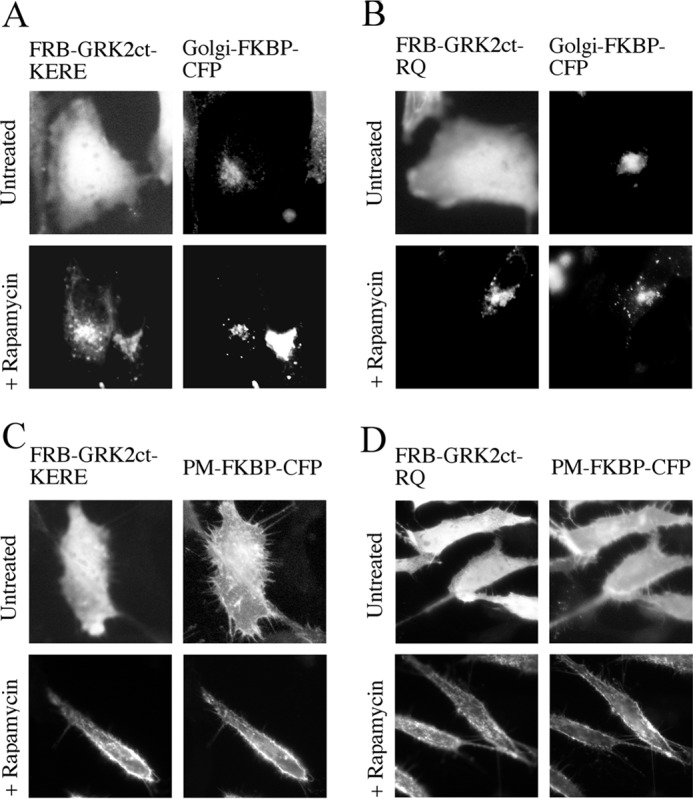
Inducible recruitment of FRB-GRK2ct constructs to FKBP-targeted subcellular locations. A–D, HeLa cells were transiently transfected with expression plasmids for FRB-GRK2ct-KERE and Golgi-FKBP-CFP (A), FRB-GRK2ct-RQ and Golgi-FKBP-CFP (B), FRB-GRK2ct-KERE and PM-FKBP-CFP (C), and FRB-GRK2ct-RQ and PM-FKBP-CFP (D). Cells were incubated with 1 μm rapamycin for 20 min or left untreated, followed by fixation and processing for immunofluorescence microscopy to show that FRB-GRK2ct constructs are appropriately recruited to either the Golgi or PM. Anti-GRK2 antibodies were used to visualize FRB-GRK2ct proteins, and FKBP fusion proteins were visualized via intrinsic CFP fluorescence.
Inducible PM Targeting of GRK2ct-KERE Inhibits LPA-dependent Phosphorylation of Akt
To demonstrate that the inducible system can functionally inhibit βγ at the PM, we assayed phosphorylation of Akt in response to LPA stimulation, a βγ-dependent signaling function in MDA-MB-231 cells (28). LPA agonist treatment promotes rapid phosphorylation of Akt, as measured by immunoblotting with a phospho-specific antibody to detect phosphorylation at serine 473, with maximal intensity ∼10 min post-stimulation (Fig. 3A). Stimulation of pcDNA3 control vector-transfected cells with 10 μm LPA yields a 6-fold increase in phosphorylation of Akt (Fig. 3C). Moreover, pretreatment of pcDNA3-transfected cells with rapamycin shows that there is no effect of short term rapamycin treatment on levels of LPA-stimulated phosphorylation of Akt; this is a critical control because rapamycin inhibits mTORC1, a downstream effector of the Akt pathway. When components of the inducible system are transfected, there is also no appreciable effect on LPA-stimulated phosphorylated Akt levels in the absence of rapamycin treatment. However, upon rapamycin pretreatment and recruitment of GRK2ct-KERE to the PM, a significant decrease in the ability of LPA to promote phosphorylation of Akt is observed; LPA treatment only induces a 2.7-fold increase in Akt phosphorylation (Fig. 3, B and C). Recruitment of GRK2ct-KERE to the Golgi shows no effect on LPA-induced phosphorylation of Akt, demonstrating that only PM-localized βγ is responsible for stimulating phosphorylation of Akt. Additionally, recruitment of GRK2ct-RQ to the PM shows no change in LPA-induced Akt phosphorylation, serving as a negative control for this system (data not shown). These results highlight the utility of this system for inducibly inhibiting canonical signaling by endogenous βγ at the PM.
FIGURE 3.
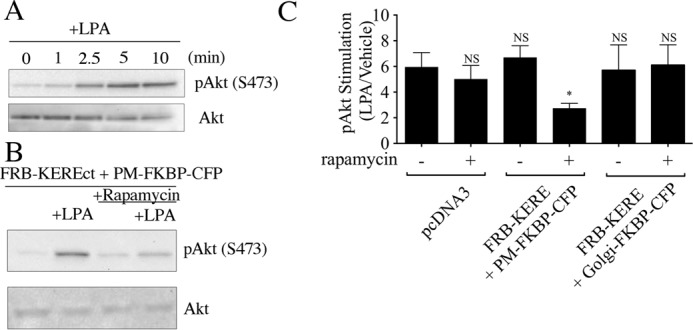
Inducible recruitment of FRB-GRK2ct-KERE to the PM blocks βγ-mediated phosphorylation of Akt. A, non-transfected MDA-MB-231 cells were serum-starved overnight and then stimulated with 10 μm LPA for the time indicated. Cells were lysed and assayed by immunoblot for phospho-Akt and total Akt, as described under “Experimental Procedures,” using antibodies specific for phospho(Ser-473)-Akt (top) and for total Akt (bottom). B, MDA-MB-231 cells were transiently transfected with plasmids encoding the FRB-GRK2ct-KERE and PM-FKBP-CFP and then, 24 h later, serum-starved overnight. Next, cells were pretreated with or without 1 μm rapamycin for 30 min and then stimulated for 10 min with either vehicle or 10 μm LPA. Cell lysates were immunoblotted for phospho(Ser-473)-Akt (top) and for total Akt (bottom). C, quantification of immunoblots (n = 3 experiments), as described under “Experimental Procedures,” is shown for LPA stimulation of Akt phosphorylation in MDA-MB-231 cells transfected with pcDNA3 or with expression plasmids for the indicated inducible system components, including the representative experiment shown in B. Statistical significance compared with the pcDNA3 without rapamycin condition (first bar) was tested using an unpaired t test (*, p < 0.005; NS, no significant difference).
Inducible Golgi Targeting of GRK2ct-KERE Inhibits Transport of Cargo from the TGN to the PM
To demonstrate that the inducible system can functionally inhibit βγ at the Golgi, we examined VSV-G Golgi to PM transport. Inducible system components were expressed together with VSV-G, here referred to as BL-VSV-G to indicate that it is a basolaterally targeted cargo and to differentiate it from an apically targeted form of VSV-G (AP-VSV-G) used below. Rapamycin was added during the final hour of the 2-h 20 °C incubation, the period during which VSV-G moves from the ER to the Golgi and is trapped at the Golgi due to low temperature block, to induce recruitment of FRB-GRK2ct-KERE to either the PM or Golgi. Neither the expression of the inducible system components nor the addition of rapamycin prevented the accumulation of BL-VSV-G at the Golgi during the 20 °C incubation (Fig. 4, A and B, 0 Hours). Moreover, in the absence of rapamycin, FRB-GRK2ct-KERE and Golgi-FKBP-CFP did not prevent the trafficking of BL-VSV-G to the PM as observed after 2 h at the 32 °C permissive temperature (BL-VSV-G at PM in >90% of cells; Fig. 4, A (top row, 2 Hours) and D). The dual color inset shows PM-localized BL-VSV-G clearly distinct from cytoplasmic GRK2ct-KERE (Fig. 4A). Importantly, when rapamycin was added to the cells, Golgi-recruited FRB-GRK2ct-KERE was able to block transport of cargo to the PM (BL-VSV-G at PM in <20% of cells; Fig. 4, A (bottom row, 2 Hours) and D).
FIGURE 4.
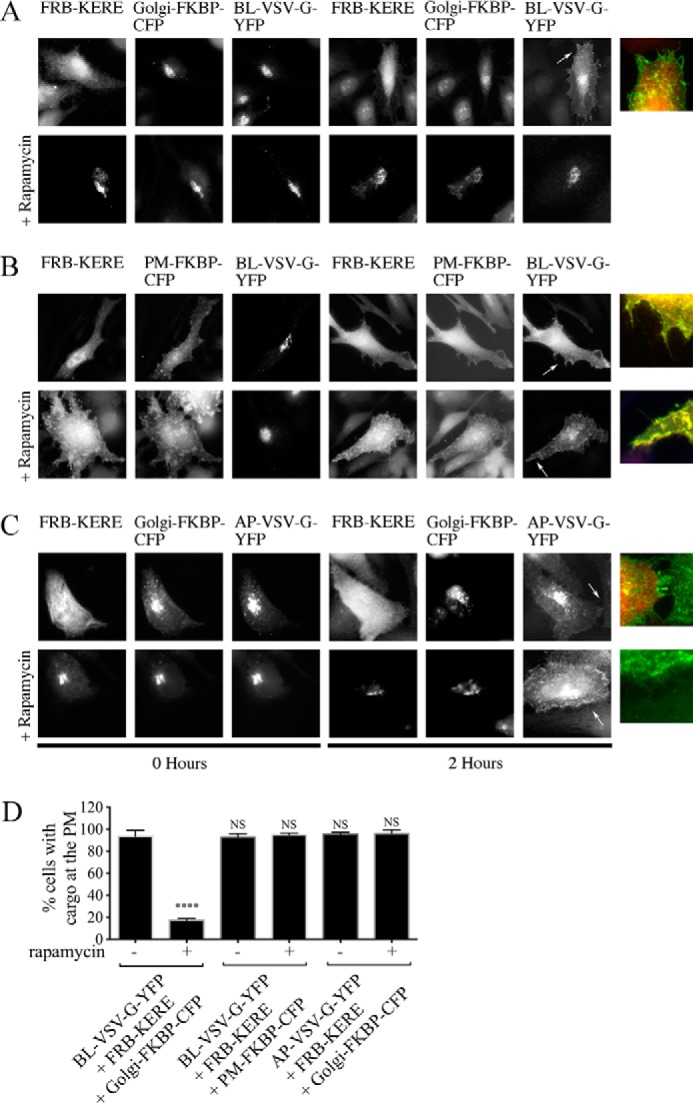
βγ inhibition impairs transport of basolateral but not apical cargo. A–C, HeLa cells were transfected with expression plasmids for BL-VSV-G-YFP (A and B) or AP-VSV-G-YFP (C) together with expression plasmids for the indicated Golgi-targeted (A and C) or PM-targeted (B) inducible system components. Rapamycin was added during the final hour of the 20 °C block where indicated (bottom row of each panel in A–C). Cells were fixed at the start of the 32 °C incubation (0 Hours) or 2 h after the shift to 32 °C, and then cells were processed for immunofluorescence microscopy. Anti-GRK2 antibodies were used to visualize FRB-GRK2ct proteins; FKBP fusion proteins were visualized via intrinsic CFP fluorescence, and VSV-G-YFP proteins were visualized via intrinsic YFP fluorescence. Inset panels, corresponding to arrows, show dual color images of BL-VSV-G-YFP or AP-VSV-G-YFP (green) and FRB-GRK2ct-KERE (red) to clearly identify PM localization of the BL-VSV-G-YFP or AP-VSV-G-YFP cargo. D, percentage of transfected cells with detectable PM localization of BL-VSV-G-YFP or AP-VSV-G-YFP after 2 h at 32 °C, as described under “Experimental Procedures.” More than 150 cells were scored for PM localization of VSV-G-YFP in each of n = 4 experiments, and the bars represent the average and S.D. (error bars). Statistical significance compared with the first bar (BL-VSV-G-YFP + FRB-GRK2ct-KERE + Golgi-FKBP-CFP without rapamycin) was tested using an unpaired t test (****, p < 0.0001; NS, no significant difference).
Interestingly, when FRB-GRK2ct-KERE is recruited to the PM in the presence of rapamycin, BL-VSV-G still traffics out of the TGN to the membrane in >90% of the cells (Fig. 4, B (bottom row) and D). Therefore, inhibition of PM-localized βγ has no effect on cargo delivery; only Golgi-localized βγ is responsible for constitutive cargo delivery from the TGN to the PM. These results demonstrate the utility of this system for inducibly inhibiting endogenous βγ signaling at the Golgi.
βγ Regulates Basolateral but Not Apical Cargo Transport Out of the TGN
Trafficking of model cargo VSV-G and ssHRP is known to be mediated by βγ, PLC, PKD, and PKC, as described earlier. Interestingly, these model cargoes are known to constitutively traffic to basolateral membranes in polarized cell systems (9, 29). Basolateral cargo trafficking can be inhibited by PLC, PKD, and PKC inhibition, but apical cargo remains unaffected (9, 29). Because the role of βγ on basolateral (BL) versus apical (AP) cargoes has not been tested, we sought to determine whether βγ regulates the Golgi to PM transport of apical cargo, in addition to its demonstrated importance for basolateral cargo transport. It has been well established that sorting of apical and basolateral cargoes is conserved in non-polarized systems (30).
Taking advantage of this, components of the inducible system were transfected together with apical cargo, AP-VSV-G, a mutant version of VSV-G shown to traffic to apical domains in polarized cells independently of PKD and also to traffic to the PM independently of PKD in non-polarized cells (9, 30). After incubating transfected cells for 2 h at 20 °C, AP-VSV-G is trapped in the Golgi, similar to BL-VSV-G. After 2 h at the permissive 32 °C incubation, AP-VSV-G is trafficked to the membrane in >95% of cells, with or without rapamycin (Fig. 4, C and D). Although the addition of rapamycin recruits FRB-GRK2ct-KERE to the Golgi and blocks BL-VSV-G cargo delivery (BL-VSV-G at PM in <20% of cells; Fig. 4, A (top row) and D), apical cargo delivery remains unaffected (AP-VSV-G at PM in >95% of cells; Fig. 4, C (bottom row) and D). Further supporting this idea that only basolateral cargoes are affected by βγ inhibition at the Golgi, pretreatment with the pharmacological βγ inhibitor gallein inhibits BL-VSV-G cargo delivery to the PM, as described previously (5), but has no effect on AP-VSV-G delivery to the PM (data not shown).
Two additional model secretory cargoes were used to investigate the role of βγ inhibition. Previous work has shown that secreted alkaline phosphatase (SEAP) and ssHRP are secretory cargoes known to sort in polarized cells apically (31) and basolaterally (29), respectively. Both cargoes are modified so that they are secreted into cell medium, which can then be assayed for enzymatic activity, as described under “Experimental Procedures.” Importantly, treatment with rapamycin alone or expression of inducible system components alone has no effect on either cargo (Fig. 5). Activity of ssHRP in the medium decreases 63 and 50% with βγ inhibition via the pharmacological inhibitor gallein or recruitment of inducible system components to the Golgi, respectively (Fig. 5A). Contrastingly, activity of SEAP in the medium is unchanged with gallein treatment or inducible system recruitment to the Golgi (Fig. 5B). Additionally, recruitment of FRB-GRK2ct-RQ to the Golgi has no effect on either cargo, indicating that this block of ssHRP delivery is due to inhibition of βγ. Thus, consistent with the results with constitutive PM-targeted BL-VSV-G and AP-VSV-G (Fig. 4), βγ inhibition blocks basolaterally targeted, but not apically targeted, secretory cargo delivery (Fig. 5).
FIGURE 5.
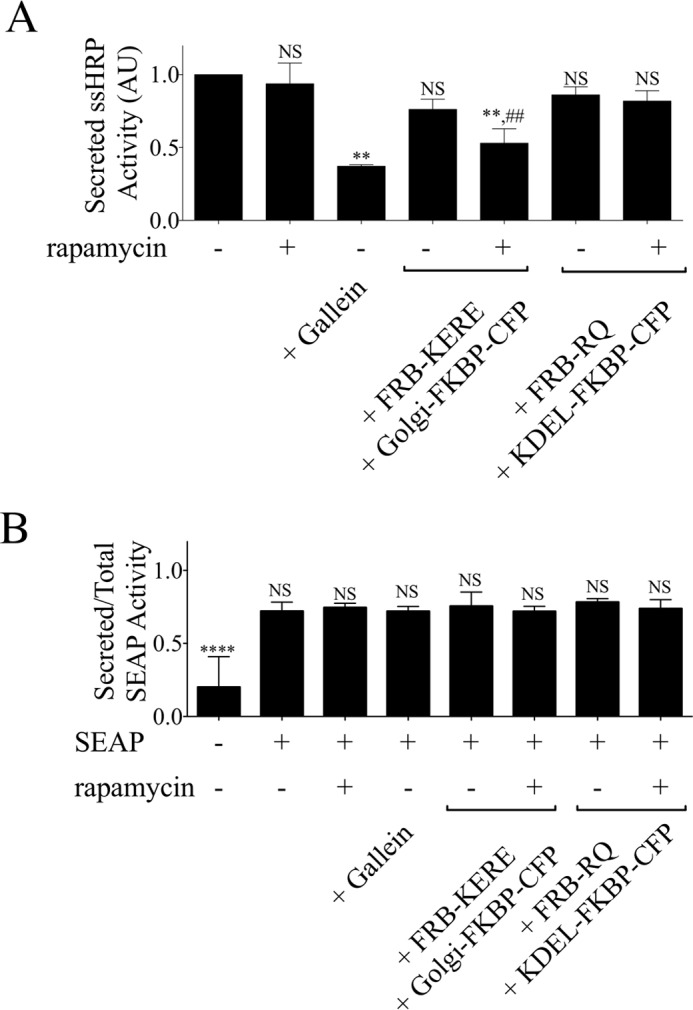
βγ inhibition impairs transport of constitutive basolateral but not apical secretory cargo. A, ssHRP assay. HeLa cells were transfected with expression plasmids for ssHRP cargo and either pcDNA3 or inducible system components, as indicated. Cells were washed and pretreated for 30 min with 10 μm gallein (βγ inhibitor), 1 μm rapamycin, or serum-free medium, and then medium (with or without pharmacological compounds) was replenished and harvested after a 5-h incubation. Medium was assayed as described under “Experimental Procedures” for luminescence as a readout of activity of secreted horseradish peroxidase. Activity secreted into the medium from cells transfected with ssHRP only (first bar) was set to 1 arbitrary unit (AU). In each case, bars represent the average and S.D. (error bars) for triplicate samples from each of n = 3 experiments. Statistical significance was tested using paired t test. **, statistically significant difference compared with ssHRP only (first bar) (p < 0.05; NS, no significant difference). ##, statistically significant difference compared with ssHRP + FRB-KERE + Golgi-FKBP-CFP without rapamycin (fourth bar) (p < 0.05; NS, no significant difference). B, SEAP assay. HeLa cells were transfected with expression plasmids for SEAP cargo and either pcDNA3 or inducible system components, as indicated. Cells were washed and pretreated for 30 min with 10 μm gallein, 1 μm rapamycin, or serum free-medium, and then medium (with or without pharmacological compounds) was replenished and harvested after a 5-h incubation. Medium and cell lysates were assayed for luminescence produced by secreted placental alkaline phosphatase activity, as described under “Experimental Procedures.” Activity is presented as a ratio of secreted to total activity for each condition. In each case, bars represent the average and S.D. (error bars) for triplicate samples from each of n = 3 experiments. Statistical significance compared with the SEAP only condition (second bar) was tested using an unpaired t test (****, p < 0.0001; NS, no significant difference).
βγ Regulates Ilimaquinone- and Nocodazole-stimulated Golgi Fragmentation
Next, we examined whether βγ functions in pathways that regulate the structure of the Golgi and lead to Golgi fragmentation, in addition to its function in regulating cargo trafficking. Several small molecules, as well as normal cellular and disease processes, induce fragmentation of the Golgi, but the mechanisms that lead to Golgi fragmentation are not well defined. Here, we investigated a role for βγ in Golgi fragmentation induced by ilimaquinone or nocodazole. Ilimaquinone treatment of cells has been demonstrated to cause Golgi fragmentation in a PKD-dependent manner, although the direct targets of ilimaquinone remain elusive (32). An early report also suggested that ilimaquinone-induced Golgi fragmentation might also require βγ, and thus we sought to characterize this using the inducible βγ inhibitor (23). Nocodazole is widely used to depolymerize microtubules in cells, and it has been well described that nocodazole-induced microtubule disruption also causes Golgi fragmentation (33). Surprisingly, a report indicated that nocodazole-induced Golgi fragmentation is not simply a mechanical result of loss of microtubules, but requires activation of PKD (17). This novel dependence on PKD prompted us to examine whether βγ might also be involved in nocodazole-induced Golgi fragmentation.
Initially, we used the βγ inhibitor gallein to test for a role for βγ in Golgi fragmentation. Treatment of HeLa or MDCK cells with 30 μm ilimaquinone or 5 μg/ml nocodazole for 2 h caused the expected dramatic fragmentation of the Golgi (Fig. 6). However, cells pretreated with gallein retained a compact Golgi and did not display fragmented Golgi after ilimaquinone or nocodazole treatment. These results provide the first indication that not only is βγ required for ilimaquinone-induced Golgi fragmentation, but βγ also surprisingly is required to connect nocodazole-induced microtubule depolymerization to Golgi fragmentation. To further demonstrate a role for βγ, we used siRNA-mediated depletion. Gβ1 and Gβ2, the two major Gβ isoforms in HeLa cells (34), were depleted with siRNA, as used previously (Fig. 7C). In addition, siRNAs targeting PKD1/PKD2 were used to confirm the importance of PKD. siRNA knockdown of either Gβ1/β2 or PKD1/PKD2 inhibited ilimaquinone- and nocodazole-induced Golgi fragmentation (Fig. 7, A and B). As described previously, ilimaquinone treatment typically fragments the Golgi into a diffuse pattern of smaller, harder to identify vesicles (35), whereas nocodazole treatment fragments the Golgi into small vesicles that are more readily observed throughout the cytosol. Thus, for two distinct compounds known to fragment the Golgi, there is a role for βγ and PKD.
FIGURE 6.

Pharmacological inhibition of βγ can block ilimaquinone- or nocodazole-induced Golgi fragmentation in HeLa and MDCK cells. HeLa cells (A) or Madin-Darby MDCK II cells (B) were seeded onto coverslips for 48 h and then pretreated with medium or 1 μm gallein for 30 min. Cells were then treated with 30 μm ilimaquinone or 5 μg/ml nocodazole for 2 h. Cells pretreated with gallein continued to have gallein in the medium during the subsequent ilimaquinone or nocodazole treatment. Cells were processed for immunofluorescence microscopy using an anti-GM130 antibody to visualize Golgi structure.
FIGURE 7.
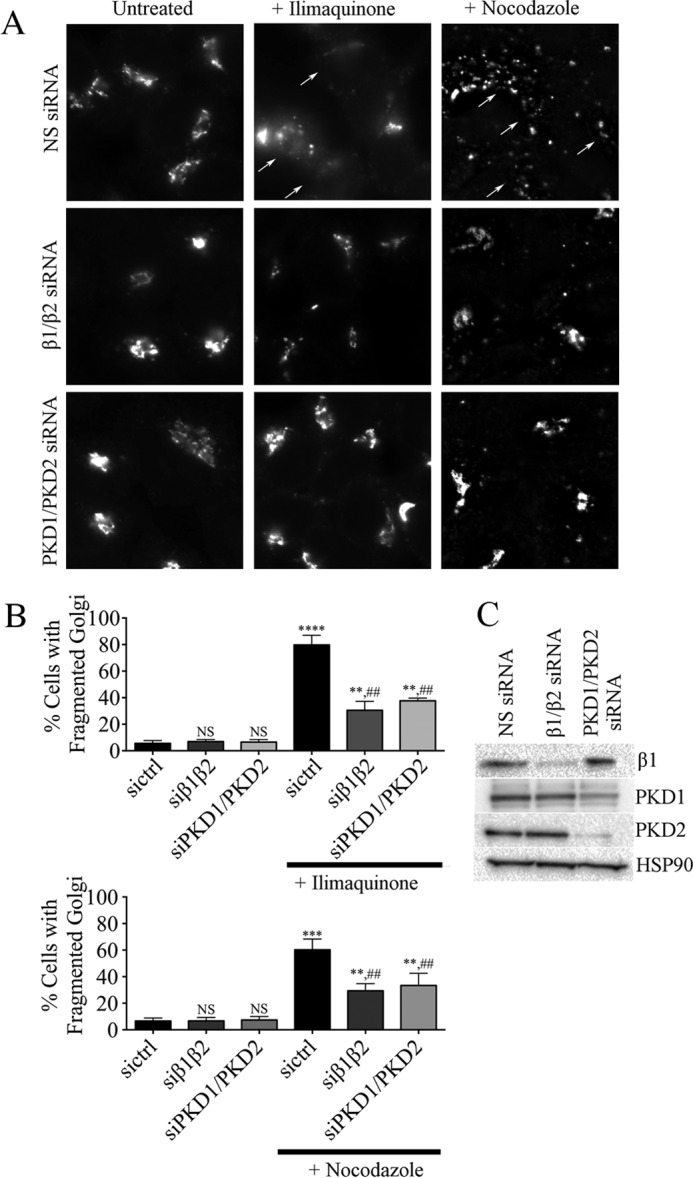
Gβ1/β2 or PKD1/2 siRNA knockdown blocks ilimaquinone- or nocodazole-induced Golgi fragmentation. HeLa cells were transfected with nonspecific control siRNA (NS siRNA) or siRNA targeting Gβ1/β2 or PKD1/2, as described under “Experimental Procedures.” After 48 h, cells were treated for 1 h with 30 μm ilimaquinone or 5 μg/ml nocodazole. A, cells were stained with anti-TGN46 to visualize Golgi structure. Representative images show ilimaquinone- or nocodazole-induced Golgi fragmentation (arrows). B, the percentage of cells with fragmented Golgi before and after ilimaquinone (top graph) or nocodazole (bottom graph) was scored, as described under “Experimental Procedures.” More than 200 cells were scored for fragmented Golgi in each of n = 3 experiments, and the bars represent the average and S.D. (error bars). Statistical significance of each condition compared with nonspecific control siRNA-transfected cells (sictrl) in the absence of ilimaquinone or nocodazole treatment (****, p < 0.0001; ***, p < 0.0005; **, p < 0.01; NS, no significant difference) was tested using an unpaired t test. In addition, statistical significance of ilimaquinone-treated (top graph) or nocodazole-treated (bottom graph) cells compared with the corresponding untreated condition is indicated (##, p < 0.01). C, cell lysates from each siRNA knockdown were immunoblotted using antibodies against Gβ1, PKD1, PKD2, or HSP90 (loading control), as indicated.
To investigate which subcellular pool of βγ mediates Golgi fragmentation, we used our inducible system to inhibit PM- or Golgi-localized βγ. Components of the inducible system were expressed in HeLa cells pretreated with or without rapamycin for 30 min and then stimulated with 30 μm ilimaquinone for 40 min (Fig. 8). Basally, only 10% of HeLa cells display a fragmented Golgi phenotype. Simply recruiting FRB-GRK2ct-KERE to the Golgi or PM via the addition of rapamycin does not increase Golgi fragmentation above this basal level (Fig. 8, A and B, second row). As described above, Golgi fragmentation is dramatically increased with ilimaquinone treatment; 70–80% of cells display diffuse fragmented Golgi (Fig. 8, A and B (third row) and C). Interestingly, both PM-recruited and Golgi-recruited FRB-GRK2ct-KERE block this fragmentation, as observed by ilimaquinone-induced Golgi fragmentation in only 40 and 20% of cells, respectively. This suggests, surprisingly, that both PM-localized and Golgi-localized βγ can contribute to a pathway leading to disrupted Golgi morphology in response to ilimaquinone.
FIGURE 8.
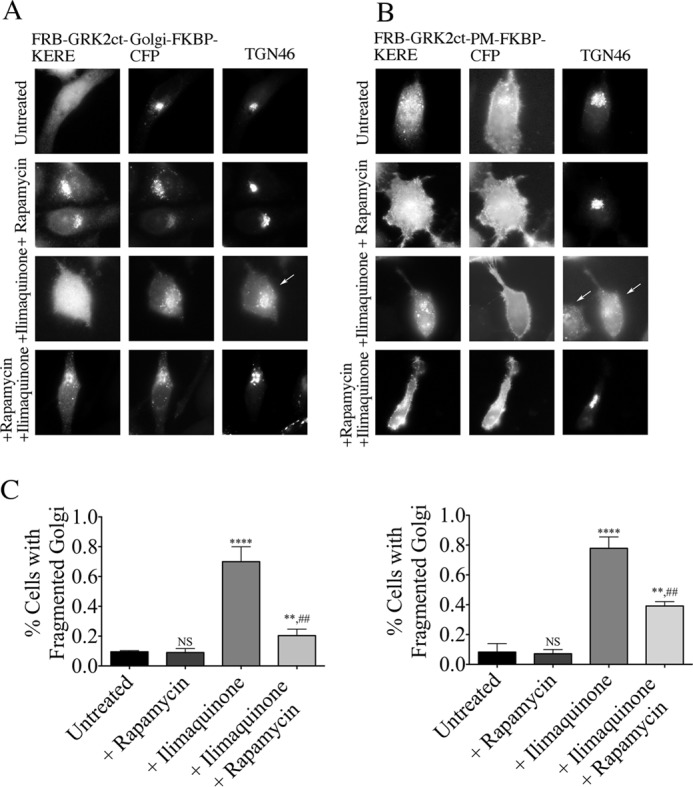
Inhibition of βγ at the Golgi or PM blocks ilimaquinone-induced Golgi fragmentation. HeLa cells were transfected with expression plasmids for either Golgi-targeted (A) or PM-targeted (B) inducible system components. Cells were pretreated with 1 μm rapamycin or washed with medium for 30 min, followed by 30 μm ilimaquinone or vehicle treatment for 40 min. Cells were then fixed and processed for immunofluorescence microscopy. Anti-GRK2 and anti-TGN46 antibodies were used to visualize FRB-GRK2ct-KERE proteins and the Golgi, respectively; FKBP fusion proteins were visualized via intrinsic CFP fluorescence. Representative images show ilimaquinone-induced Golgi vesiculation (arrows). C, the percentage of cells with fragmented Golgi in each condition in which cells expressed the Golgi-targeted (left graph) or PM-targeted (right graph) inducible system components was scored. More than 200 cells were scored for fragmented Golgi in each of n = 3 experiments, and the bars represent the average and S.D. (error bars). Statistical significance compared with the untreated condition (****, p < 0.0001; **, p < 0.05; NS, no significant difference) was tested using an unpaired t test. In addition, statistical significance of the ilimaquinone + rapamycin condition compared with the +ilimaquinone condition is indicated (##, p < 0.005).
The inducible system was similarly used to inhibit PM- or Golgi-localized βγ after nocodazole treatment (Fig. 9). At basal conditions, <20% of HeLa cells display Golgi fragmentation (representative images in the top row of Fig. 9, A and B; quantification in Fig. 9C), and rapamycin treatment alone has no effect on Golgi fragmentation (row 2 of Fig. 9, A and B). Upon treatment with nocodazole but in the absence of rapamycin, >80% of cells expressing the inducible system components display a fragmented Golgi (Fig. 9, A and B, third row). When FRB-GRK2ct-KERE is recruited to the Golgi (upon rapamycin addition), only 44% of cells display a fragmented Golgi phenotype (Fig. 9A, bottom row). In contrast, when FRB-GRK2ct-KERE is recruited to the PM, there is no reduction in the percentage of cells (>80%) that display a fragmented Golgi phenotype (Fig. 9, B (bottom row) and C). Thus, there is a striking difference in the subcellular pools of Gβγ regulating ilimaquinone- versus nocodazole-induced Golgi fragmentation.
FIGURE 9.
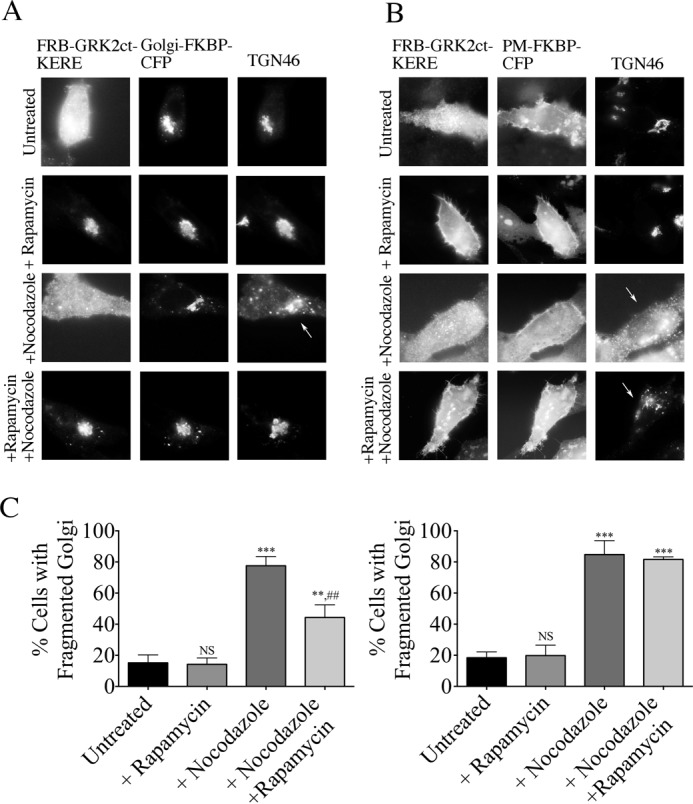
Inhibition of βγ at the Golgi blocks nocodazole-induced Golgi fragmentation. HeLa cells were transfected with expression plasmids for either Golgi-targeted (A) or PM-targeted (B) inducible system components. Cells were pretreated with 1 μm rapamycin or washed with medium for 30 min, followed by 5 μg/ml nocodazole or vehicle treatment for 1 h. Cells were then fixed and processed for immunofluorescence microscopy. Anti-GRK2 and anti-TGN46 antibodies were used to visualize FRB-GRK2ct-KERE proteins and the Golgi, respectively; FKBP fusion proteins were visualized via intrinsic CFP fluorescence. Representative images show nocodazole-induced Golgi vesiculation (arrows). C, the percentage of cells with fragmented Golgi in each condition in which cells expressed the Golgi-targeted (left) or PM-targeted (right) inducible system components was scored. More than 200 cells were scored for fragmented Golgi in each of n = 3 experiments, and the bars represent the average and S.D. (error bars). Statistical significance compared with the untreated condition (***, p < 0.0005; **, p < 0.01; NS, no significant difference) was tested using an unpaired t test. In addition, statistical significance of the nocodazole + rapamycin condition compared with the +nocodazole condition is indicated (##, p < 0.005).
Discussion
Here we report the development of a novel molecular tool for spatial and temporal inhibition of endogenous βγ, and we have used this tool to expand our knowledge of the role of βγ in regulating Golgi to PM transport and Golgi morphology. Specifically, the results show that 1) a Golgi pool, but not a PM pool, of βγ regulates Golgi to PM transport; 2) Golgi-localized βγ selectively regulates Golgi to PM transport of basolateral but not apical cargo; 3) βγ is required for Golgi fragmentation induced by ilimaquinone, and both Golgi and PM pools of βγ play a role in this Golgi fragmentation; and 4) βγ is required for Golgi fragmentation induced by nocodazole, but only Golgi-localized βγ plays a role in this Golgi fragmentation.
The βγ molecular inhibitor described here takes advantage of a lipid-binding mutant of GRK2 to generate a GRK2ct that can only inhibit βγ when recruited to a specific subcellular membrane location. Previous work with full-length GRK2 showed that the mutations K567E/R578E (KERE) prevented the stimulation of GPCR phosphorylation by GRK2 in response to negative phospholipids and prevented agonist-stimulated phosphorylation of GPCRs in cells; however, GRK2-K567E/R578E retained the ability to interact with βγ (26). The studies reported here now provide the first test of the K567E/R578E mutations in the context of GRK2ct, the C-terminal pleckstrin homology domain of GRK2 that has been widely used as an inhibitor of βγ function in cells. GRK2ct-KERE failed to inhibit βγ-dependent Golgi to PM transport of the model cargo VSV-G, but GRK2ct-KERE strongly inhibited such transport when constitutively targeted to the Golgi. This is in contrast to WT GRK2ct, which is able to inhibit this Golgi to PM transport regardless of whether it is constitutively recruited to Golgi membranes. These findings suggest that not only is binding to negatively charged membranes essential for the ability of full-length GRK2 to phosphorylate activated GPCRs, but binding to negatively charged membranes by GRK2ct is also necessary for efficient interaction with βγ.
The identification of GRK2ct-KERE as a conditional inhibitor of βγ in cells (i.e. only when recruited to membranes) allowed us to use the rapamycin-inducible heterodimerization system to develop GRK2ct-KERE as spatial and temporal inhibitor of βγ. Thus, FRB-GRK2ct-KERE can be recruited in a rapamycin-dependent manner to PM or Golgi in our studies when the cognate PM-FKBP or Golgi-FKBP is also expressed. Indeed, we show that FRB-GRK2ct-KERE can inhibit βγ-dependent activation of Akt in response to LPA only when inducibly recruited to the PM and not when recruited to the Golgi or in the absence of rapamycin-induced recruitment. Likewise, FRB-GRK2ct-KERE only inhibited Golgi to PM transport when specifically recruited to the Golgi. We anticipate that this membrane-recruitable βγ inhibitor system can be adapted to investigate the role of βγ signaling in response to a variety of stimuli at diverse subcellular locations.
The inducible βγ inhibitor developed here complements a recent study in which an optogenetic system was used to recruit GRK2ct to the PM in single cells (36). In that study, GRK2ct was recruited to the PM specifically on one side of a migrating cell to generate a gradient of βγ signaling. Optogenetic targeting of a G protein inhibitor, such as GRK2ct, to a specific region of the PM is a powerful approach for interrogating signaling. Optogenetic targeting has the advantage that a signaling molecule or inhibitor can be precisely targeted to a defined region in a single cell, whereas the rapamycin-inducible system is well suited to use in a population of cells. In the study mentioned above, GRK2ct was only targeted to the PM, but additional targeting partners, as used in our studies, could allow optogenetic targeting to other organelles, such as the Golgi. Interestingly, in the optogenetic studies, WT GRK2ct was used, and thus it is possible that a basal level of βγ inhibition might be present even in the absence of GRK2ct being optogenetically targeted to the PM. We predict that the use of GRK2ct-KERE in other recruitable systems, such as optogenetic systems, could further improve the systems.
By inhibiting βγ at distinct subcellular locations, our studies confirmed that Golgi-localized βγ regulates constitutive cargo delivery but that PM-localized βγ has little to no contribution to trafficking of cargo. Additionally, we asked whether different types of constitutive cargoes could be regulated by βγ. In a polarized cell system, there are a multitude of post-TGN pathways. Cargo can be directed to the apical or basolateral membranes or go through endosome intermediates on the way to their final destinations. Typically, this is through cargo sorting sequences interacting with adaptor proteins at the Golgi, endosome, and destination membranes (37). Importantly, it has been shown that machinery is conserved between polarized and non-polarized cells (38), so that although some of the intermediate compartments may not exist in non-polarized cells, the protein components and sorting pathways remain. This allowed us to use traditionally basolateral (BL-VSV-G and ssHRP) and apical (AP-VSV-G and SEAP) cargoes in simple trafficking assays. Previous work, using these and other cargoes, made the important finding that PKD was required for Golgi to PM transport of basolaterally targeted cargo but not apically targeted cargo (9), yet no studies until now had addressed whether βγ shows similar selectively as PKD in terms of cargo or whether βγ has a much broader role compared with PKD in regulating generation of transport carriers containing a wide variety of cargo. Utilizing our inducible system, we observed that only basolateral cargoes were affected by inhibiting βγ signaling. Apical cargo remained unaffected by the recruitment of FRB-GRK2ct-KERE to the Golgi. Our results show that inhibition of βγ does not result in block of all Golgi to PM transport. Instead, these results provide the first support for the idea that βγ regulates only a subset of cargo delivery pathways, involving activation and recruitment of PKD.
In addition to PKD being involved in cargo transport, it has also been implicated in overactivation of this pathway, leading to Golgi fragmentation, similar to our previous results with overexpression of βγ (5, 17). Although this is often considered simply an experimental read-out of overactivation of the βγ signaling pathway at the Golgi, there is an increasing body of evidence supporting Golgi fragmentation as a physiological process, such as occurs preceding mitosis (39). In addition to defining further the role of βγ in Golgi to PM transport, we chose to examine a role for βγ in ilimaquinone-induced or nocodazole-induced Golgi fragmentation.
Here we showed that inhibition of endogenous βγ using the inducible system, as well as using the pharmacological inhibitor gallein and siRNA depletion, decreased the ability of ilimaquinone to cause Golgi fragmentation. These results support early studies in which GDP-bound Gα subunits added to permeabilized cells attenuated ilimaquinone-stimulated Golgi fragmentation, thereby first suggesting a role for βγ in mediating the effect of ilimaquinone (23). Surprisingly, we found that ilimaquinone-stimulated Golgi fragmentation could be decreased by inhibition of both PM- and Golgi-localized βγ, indicating that G proteins from both locations contribute to ilimaquinone-induced Golgi fragmentation (Fig. 8). One hypothesis is that ilimaquinone is functioning, directly or indirectly, through a GPCR located at the PM, whereas another possibility is that βγ translocates from the PM to the Golgi to induce Golgi fragmentation after ilimaquinone treatment. Ilimaquinone promotes several cellular activities, in addition to Golgi fragmentation, but the cellular targets of ilimaquinone that mediate its effects on cells remain to be definitively identified (20, 40).
Fragmentation of the Golgi via nocodazole treatment was once thought to be purely a mechanical event resulting from depolymerization of microtubules. Recent work, however, provided evidence for the importance of a signaling event, in which nocodazole treatment causes activation of PKD and nocodazole-induced Golgi fragmentation requires PKD (17). Here we show the surprising finding that, in addition to PKD, βγ is necessary for nocodazole-induced Golgi fragmentation (Figs. 6, 7, and 9). Distinct from ilimaquinone-mediated Golgi fragmentation, use of the inducible system showed that only Golgi-localized βγ contributed to nocodazole-induced Golgi fragmentation. This hints at a distinction between how these pharmacological agents are activating βγ. Nocodazole only utilizes Golgi-localized βγ, similar to the cargo transport pathway. This points to a potential role for MT-associated proteins or free tubulin to serve as local activators of βγ as a result of microtubule destabilization. βγ is already known to associate with tubulin, with γ-tubulin association not dependent on microtubule status (41). Further work is needed to see how this could regulate cytoskeleton-associated processes, like cell division. Together, our studies using the inducible system reveal an interesting difference in the Golgi functions of βγ; both PM- and Golgi-localized βγ can mediate ilimaquinone-induced Golgi fragmentation, but only Golgi-localized βγ regulates constitutive cargo transport or nocodazole-induced Golgi fragmentation in our studies.
Although it is accepted that βγ signals at the Golgi, there remain gaps in knowledge to fully understand this pathway. It remains to be seen whether translocation of βγ from the PM to Golgi is always necessary for βγ signaling at the Golgi, or if there is an “activatable” pool of βγ already resident at the Golgi. Recent studies have addressed the role of βγ in mediating regulated Golgi to PM transport in response to GPCR activation. βγ was shown to be required for insulin secretion in response to agonist activation of the M3 muscarinic acetylcholine receptor and for protease-activated receptor-2 (PAR-2) transport from the Golgi to PM in response to agonist activation of the cell surface pool of PAR-2 (42, 43); in both studies, observations of GPCR activation-induced translocation of overexpressed βγ from the PM to Golgi were correlated with the effects on regulated cargo transport. Constitutive cargoes, such as those examined in our studies, may rely on a pool of Golgi-localized βγ and/or may require constitutive translocation of βγ from the PM to Golgi. The system of inducible βγ inhibition developed here, along with additional molecular tools for understanding and visualizing βγ signaling, may help to better define how agonist-activated and constitutive trafficking of G proteins regulates their intracellular functions.
Experimental Procedures
Reagents
The Argent regulated heterodimerization kit was provided by ARIAD pharmaceuticals (Cambridge, MA). Gallein was obtained from Tocris Bioscience. Rapamycin, HEPES, LPA, ilimaquinone, nocodazole, and cycloheximide were from Sigma-Aldrich.
Cell Culture, Plasmids, and Transfections
All cell culture experiments were performed with HeLa, MDA-MB-231, or MDCK II cell lines, as indicated. All plasmid transfections were performed with Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific), and all siRNA transfections were performed with HiPerFect transfection reagent (Qiagen). GRK2ct and GRK2ct-RQ constructs were described previously (5). To construct GRK2ct-KERE, full-length GRK2-KERE (provided by Dr. Jeff Benovic) served as the template, with amino acids 495–689 amplified by PCR and digested with HindIII and EcoRI. The KERE fragment was purified and inserted into GRK2ct in a pcDNA3 backbone. To construct Golgi-GRK2ct-KERE, GRK2ct-KERE was cut with EcoNI and EcoRI and then purified and inserted into the previously described KDEL-GRK2ct backbone (5).
To construct components of the inducible system, GRK2ct-RQ and GRK2ct-KERE were amplified with SpeI and BamHI restriction sites. Then they were purified, digested with enzymes, and subcloned into the pC4-RHE vector from Ariad Pharmaceuticals. This results in an N-terminal fusion of FRB-GRK2ct-RQ and FRB-GRK2ct-KERE. Golgi-FKBP-CFP (KDEL-FKBP-CFP) and PM-FKBP-CFP constructs are as described (5).
VSV-G-GFP is a temperature-sensitive cargo that traffics from the Golgi as a basolateral cargo, as described previously (5). AP-VSV-G-YFP (VSV-G3-YFP) is an apically targeted cargo, whereas BL-VSV-G-YFP (VSV-G3-SP-YFP) is a basolaterally targeted cargo (both gifts from Dr. Kai Simons), as seen in polarized cells (30). Secretory cargo ssHRP is a basolateral secretory cargo, as described previously (5), and SEAP (Addgene plasmid 24595) is a secreted apical cargo.
β1/β2 siRNA treatment includes one siRNA targeting β1 and one targeting β2, obtained from Thermo Scientific, as described previously (5). Nonspecific siRNA control corresponds to a pool of four nonspecific RNAs, from Dharmacon. PKD1/PKD2 siRNA corresponds to one pool of four siRNAs against PKD1 and one pool of four siRNAs against PKD2, from Dharmacon, as described previously (39).
Immunofluorescence
Transfected cells were fixed with 4% formaldehyde in phosphate-buffered saline (PBS) for 15 min and permeabilized by incubation in blocking buffer (2.5% nonfat milk in 1% Triton X-100 in Tris-buffered saline) for 20 min. Cells were then incubated with the indicated primary antibodies in blocking buffer for 1 h. Primary antibodies used included anti-TGN46 antibody (Abcam), anti-GFP antibody (Rockland), anti-GRK2 antibody (Sigma), and anti-GM130 (BD Biosciences). The cells were washed with blocking buffer and incubated in a 1:200 dilution of one or more of the following secondary antibodies (Invitrogen) for 1 h: chicken anti-rabbit antibody conjugated with Alexa 594, donkey anti-goat antibody conjugated with Alexa 488, goat anti-mouse Alexa 488, goat anti-mouse Alexa 594, or goat anti-rabbit Alexa 594. The coverslips were then washed with 1% Triton X-100 in Tris-buffered saline, rinsed in distilled water, and mounted on glass slides with Prolong anti-fade reagent (Invitrogen). Images were acquired using an Olympus BX-61 upright microscope with a ×60 1.4 numerical aperture or a ×100 1.3 numerical aperture oil immersion objective and an ORCA-ER cooled charge-coupled device camera (Hamamatsu, Bridgewater, NJ) controlled by Slidebook version 4.0 (Intelligent Imaging Innovations, Denver, CO).
LPA-stimulated Phosphorylation of Akt
MDA-MB-231 cells were seeded into 6-cm dishes. Cells were transfected with 1 μg of DNA with Lipofectamine 2000. Medium was exchanged 24 h later. Cells were then serum-starved overnight. Agonist and vehicle (PBS + 0.1% (w/v) bovine serum albumin) were diluted in serum-free medium, with cells stimulated with 10 μm LPA or control vehicle for the times indicated. Cells were immediately lysed in 1× Sample Buffer, boiled for 5 min, and then stored at −20 °C. Lysates were run on a 12% SDS-polyacrylamide gel and then transferred onto a polyvinylidene difluoride membrane. The membrane was blocked for 1 h in TBS-Tween + 5% milk. Membranes were then immunostained for anti-phospho(Ser-473)-Akt (Cell Signaling Technology, D9E) and anti-Akt (Cell Signaling Technology) overnight at 4 °C. Membranes were washed and then incubated in horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Luminescent bands were imaged by Quantity One software (Bio-Rad) using the Bio-Rad Chemi-Doc. Densitometry from these bands was measured via ImageJ for three separate experiments. First, a ratio of phosphorylated Akt to Akt was calculated for each condition. Reported is the -fold change of that ratio for LPA stimulation to vehicle for each condition.
Cargo Transport Assays
VSV-G Transport Assay
HeLa cells were co-transfected with GRK2ct or inducible system plasmids and VSV-G plasmids and incubated for 24 h at 37 °C. Cells were then washed with fresh medium, moved to 39.5 °C, and incubated overnight at this non-permissive temperature, where accumulation of VSV-G occurred in the ER. Then medium was changed to serum-free medium, and cells were incubated for 2 h at 20 °C, allowing the VSV-G protein to fold properly and be transported from the ER to the Golgi. 10 μg/ml cycloheximide was added during the last hour of incubation at 20 °C. After the 20 °C incubation, one set of cells were fixed as the 0 h time point. A second set of cells were incubated in serum-free medium for 2 h at the permissive 32 °C temperature and then fixed to be evaluated by immunofluorescence microscopy. For experiments in which cells were treated with rapamycin, the dimerization agent was added after a 1-h incubation at 20 °C. Cells were scored as having VSV-G delivered to the PM or not and are shown as a percentage of total cells. Any cell with observable PM localization of VSV-G was scored as “PM”; cells without observable VSV-G at the PM were scored as “not PM.” 150 cells were counted in each of at least four different experiments.
ssHRP Secretion Assay
HeLa cells were co-transfected with the inducible system components and ssHRP plasmid. 48 h after transfection, cells were washed and incubated with fresh medium for 5 h. Gallein or rapamycin was added for a 30-min pretreatment before washing with fresh, serum-free medium and maintained in the serum-free medium during the subsequent 5 h. To assay for secreted HRP, 5 μl of the culture medium from a 35-mm dish was harvested and added to 50 μl of SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). The linear range of luminescence was determined based on both the maximal luminescence of ssHRP and the linear range of ssHRP secretion over time. HRP activity was then measured using a Lumat LB 9507 luminometer. A background reading, from medium of cells transfected with pcDNA3, was subtracted as a baseline luminescence for each sample.
SEAP Assay
HeLa cells were co-transfected with the inducible system components and SEAP plasmid. 48 h after SEAP transfection, cells were washed and incubated with fresh medium for 5 h. For experiments in which cells were treated with rapamycin, dimerization agent was added 30 min before washing with fresh medium and maintained in the medium during the 5-h incubation. To assay for SEAP activity, medium was collected at 5 h, and cells were lysed according to the Phospha-LightTM system protocol provided by Applied Biosystems. The linear range of luminescence was determined, based on both the maximal luminescence of SEAP and the linear range of SEAP secretion over time. Luminescence was measured for both media and cell lysates, to determine the proportion of total SEAP secreted into the medium, and shown as a ratio of secreted/total activity.
Golgi Fragmentation
HeLa cells were transfected with components of the inducible system or siRNA. Cells were washed 24 h later. For conditions with the inducible system, 48 h post-transfection, HeLa cells were pretreated for 30 min with 1 μm rapamycin and then washed and maintained with rapamycin for each treatment when indicated. For ilimaquinone-induced Golgi fragmentation, cells were treated with 30 μm ilimaquinone or DMSO control for 40 min and then fixed in 4% formaldehyde. For nocodazole-induced Golgi fragmentation, cells were treated with 5 μg/ml nocodazole or DMSO control for 1 h and then fixed in 4% formaldehyde and processed for immunofluorescence microscopy. Cells were scored as displaying a fragmented Golgi phenotype and are shown as a percentage of total cells. We define a fragmented Golgi phenotype as a loss of compact Golgi structure. Specifically, for cells treated with ilimaquinone, the Golgi fragments into a diffuse pattern of cytoplasmic puncta. For cells treated with nocodazole, the Golgi is broken down into smaller vesicle-type structures. 150 cells were counted in each of at least three different experiments.
Author Contributions
L. M. K. and P. B. W. designed the study and wrote the paper. L. M. K. performed and analyzed the experiments and prepared the figures. L. M. K. and P. B. W. reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank J. Benovic, F. Paumet, and R. Irannejad for critically reading the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM56444 (to P. B. W.). This work was also supported by American Heart Association Grant 14PRE19980023 (to L. M. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PM
- plasma membrane
- GPCR
- G protein-coupled receptor
- TGN
- trans-Golgi network
- GRK2ct
- GPCR kinase 2 C terminus
- FRB
- FKBP-rapamycin binding domain of FRAP
- FKBP
- FK506-binding protein
- LPA
- lysophosphatidic acid
- Akt
- protein kinase B
- PKD
- protein kinase D
- PLCβ3
- phospholipase C β3
- PKCη
- protein kinase Cη
- VSV-G
- vesicular stomatitis virus-G
- ssHRP
- signal sequence horseradish peroxidase
- AP
- apical cargo
- BL
- basolateral cargo
- SEAP
- secreted alkaline phosphatase
- ER
- endoplasmic reticulum
- MDCK
- Madin-Darby canine kidney.
References
- 1.Hewavitharana T., and Wedegaertner P. B. (2012) Non-canonical signaling and localizations of heterotrimeric G proteins. Cell. Signal. 24, 25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saini D. K., Chisari M., and Gautam N. (2009) Shuttling and translocation of heterotrimeric G proteins and Ras. Trends Pharmacol. Sci. 30, 278–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giannotta M., Ruggiero C., Grossi M., Cancino J., Capitani M., Pulvirenti T., Consoli G. M., Geraci C., Fanelli F., Luini A., and Sallese M. (2012) The KDEL receptor couples to Gα(q/11) to activate Src kinases and regulate transport through the Golgi. EMBO J. 31, 2869–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jamora C., Yamanouye N., Van Lint J., Laudenslager J., Vandenheede J. R., Faulkner D. J., and Malhotra V. (1999) Gβγ-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell 98, 59–68 [DOI] [PubMed] [Google Scholar]
- 5.Irannejad R., and Wedegaertner P. B. (2010) Regulation of constitutive cargo transport from the trans-Golgi network to plasma membrane by Golgi-localized G protein βγ subunits. J. Biol. Chem. 285, 32393–32404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Díaz Añel A. M., and Malhotra V. (2005) PKCeta is required for β1γ2/β3γ2- and PKD-mediated transport to the cell surface and the organization of the Golgi apparatus. J. Cell Biol. 169, 83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Díaz and Añel A. M. (2007) Phospholipase C β3 is a key component in the Gβγ/PKCη/PKD-mediated regulation of trans-Golgi network to plasma membrane transport. Biochem. J. 406, 157–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liljedahl M., Maeda Y., Colanzi A., Ayala I., Van Lint J., and Malhotra V. (2001) Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell 104, 409–420 [DOI] [PubMed] [Google Scholar]
- 9.Yeaman C., Ayala M. I., Wright J. R., Bard F., Bossard C., Ang A., Maeda Y., Seufferlein T., Mellman I., Nelson W. J., and Malhotra V. (2004) Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat. Cell Biol. 6, 106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hausser A., Storz P., Märtens S., Link G., Toker A., and Pfizenmaier K. (2005) Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIβ at the Golgi complex. Nat. Cell Biol. 7, 880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine T. P., and Munro S. (2002) Targeting of Golgi-specific pleckstrin homology domains involves both PtdIns 4-kinase-dependent and -independent components. Curr. Biol. 12, 695–704 [DOI] [PubMed] [Google Scholar]
- 12.Godi A., Di Campli A., Konstantakopoulos A., Di Tullio G., Alessi D. R., Kular G. S., Daniele T., Marra P., Lucocq J. M., and De Matteis M. A. (2004) FAPPs control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat. Cell Biol. 6, 393–404 [DOI] [PubMed] [Google Scholar]
- 13.De Matteis M. A., and Luini A. (2011) Mendelian disorders of membrane trafficking. N. Engl. J. Med. 365, 927–938 [DOI] [PubMed] [Google Scholar]
- 14.Pandey K. N. (2010) Small peptide recognition sequence for intracellular sorting. Curr. Opin. Biotechnol. 21, 611–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sumara G., Formentini I., Collins S., Sumara I., Windak R., Bodenmiller B., Ramracheya R., Caille D., Jiang H., Platt K. A., Meda P., Aebersold R., Rorsman P., and Ricci R. (2009) Regulation of PKD by the MAPK p38δ in insulin secretion and glucose homeostasis. Cell 136, 235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koch W. J., Inglese J., Stone W. C., and Lefkowitz R. J. (1993) The binding site for the βγ subunits of heterotrimeric G proteins on the β-adrenergic receptor kinase. J. Biol. Chem. 268, 8256–8260 [PubMed] [Google Scholar]
- 17.Fuchs Y. F., Eisler S. A., Link G., Schlicker O., Bunt G., Pfizenmaier K., and Hausser A. (2009) A Golgi PKD activity reporter reveals a crucial role of PKD in nocodazole-induced Golgi dispersal. Traffic 10, 858–867 [DOI] [PubMed] [Google Scholar]
- 18.Haase G., and Rabouille C. (2015) Golgi fragmentation in ALS motor neurons: new mechanisms targeting microtubules, tethers, and transport vesicles. Front. Neurosci. 9, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farber-Katz S. E., Dippold H. C., Buschman M. D., Peterman M. C., Xing M., Noakes C. J., Tat J., Ng M. M., Rahajeng J., Cowan D. M., Fuchs G. J., Zhou H., and Field S. J. (2014) DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell 156, 413–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu P. H., Chueh S. C., Kung F. L., Pan S. L., Shen Y. C., and Guh J. H. (2007) Ilimaquinone, a marine sponge metabolite, displays anticancer activity via GADD153-mediated pathway. Eur. J. Pharmacol. 556, 45–54 [DOI] [PubMed] [Google Scholar]
- 21.Heuer D., Rejman Lipinski A., Machuy N., Karlas A., Wehrens A., Siedler F., Brinkmann V., and Meyer T. F. (2009) Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature 457, 731–735 [DOI] [PubMed] [Google Scholar]
- 22.Takizawa P. A., Yucel J. K., Veit B., Faulkner D. J., Deerinck T., Soto G., Ellisman M., and Malhotra V. (1993) Complete vesiculation of Golgi membranes and inhibition of protein transport by a novel sea sponge metabolite, ilimaquinone. Cell 73, 1079–1090 [DOI] [PubMed] [Google Scholar]
- 23.Jamora C., Takizawa P. A., Zaarour R. F., Denesvre C., Faulkner D. J., and Malhotra V. (1997) Regulation of Golgi structure through heterotrimeric G proteins. Cell 91, 617–626 [DOI] [PubMed] [Google Scholar]
- 24.Veit B., Yucel J. K., and Malhotra V. (1993) Microtubule independent vesiculation of Golgi membranes and the reassembly of vesicles into Golgi stacks. J. Cell Biol. 122, 1197–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sonoda H., Okada T., Jahangeer S., and Nakamura S. (2007) Requirement of phospholipase D for ilimaquinone-induced Golgi membrane fragmentation. J. Biol. Chem. 282, 34085–34092 [DOI] [PubMed] [Google Scholar]
- 26.Carman C. V., Barak L. S., Chen C., Liu-Chen L. Y., Onorato J. J., Kennedy S. P., Caron M. G., and Benovic J. L. (2000) Mutational analysis of Gβγ and phospholipid interaction with G protein-coupled receptor kinase 2. J. Biol. Chem. 275, 10443–10452 [DOI] [PubMed] [Google Scholar]
- 27.Belshaw P. J., Ho S. N., Crabtree G. R., and Schreiber S. L. (1996) Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc. Natl. Acad. Sci. U.S.A. 93, 4604–4607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riaz A., Huang Y., and Johansson S. (2016) G-protein-coupled lysophosphatidic acid receptors and their regulation of AKT signaling. Int. J. Mol. Sci. 17, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bossard C., Bresson D., Polishchuk R. S., and Malhotra V. (2007) Dimeric PKD regulates membrane fission to form transport carriers at the TGN. J. Cell Biol. 179, 1123–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keller P., Toomre D., Díaz E., White J., and Simons K. (2001) Multicolour imaging of post-Golgi sorting and trafficking in live cells. Nat. Cell Biol. 3, 140–149 [DOI] [PubMed] [Google Scholar]
- 31.Paladino S., Sarnataro D., Pillich R., Tivodar S., Nitsch L., and Zurzolo C. (2004) Protein oligomerization modulates raft partitioning and apical sorting of GPI-anchored proteins. J. Cell Biol. 167, 699–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharlow E. R., Giridhar K. V., LaValle C. R., Chen J., Leimgruber S., Barrett R., Bravo-Altamirano K., Wipf P., Lazo J. S., and Wang Q. J. (2008) Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J. Biol. Chem. 283, 33516–33526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dinter A., and Berger E. G. (1998) Golgi-disturbing agents. Histochem. Cell Biol. 109, 571–590 [DOI] [PubMed] [Google Scholar]
- 34.Krumins A. M., and Gilman A. G. (2006) Targeted knockdown of G protein subunits selectively prevents receptor-mediated modulation of effectors and reveals complex changes in non-targeted signaling proteins. J. Biol. Chem. 281, 10250–10262 [DOI] [PubMed] [Google Scholar]
- 35.Axelsson M. A., and Warren G. (2004) Rapid, endoplasmic reticulum-independent diffusion of the mitotic Golgi haze. Mol. Biol. Cell 15, 1843–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Neill P. R., and Gautam N. (2014) Subcellular optogenetic inhibition of G proteins generates signaling gradients and cell migration. Mol. Biol. Cell 25, 2305–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mellman I., and Nelson W. J. (2008) Coordinated protein sorting, targeting and distribution in polarized cells. Nat. Rev. Mol. Cell Biol. 9, 833–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Müsch A., Xu H., Shields D., and Rodriguez-Boulan E. (1996) Transport of vesicular stomatitis virus G protein to the cell surface is signal mediated in polarized and nonpolarized cells. J. Cell Biol. 133, 543–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kienzle C., Eisler S. A., Villeneuve J., Brummer T., Olayioye M. A., and Hausser A. (2013) PKD controls mitotic Golgi complex fragmentation through a Raf-MEK1 pathway. Mol. Biol. Cell 24, 222–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee H. Y., Chung K. J., Hwang I. H., Gwak J., Park S., Ju B. G., Yun E., Kim D. E., Chung Y. H., Na M., Song G. Y., and Oh S. (2015) Activation of p53 with ilimaquinone and ethylsmenoquinone, marine sponge metabolites, induces apoptosis and autophagy in colon cancer cells. Mar. Drugs. 13, 543–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montoya V., Gutierrez C., Najera O., Leony D., Varela-Ramirez A., Popova J., Rasenick M. M., Das S., and Roychowdhury S. (2007) G protein βγ subunits interact with αβ- and γ-tubulin and play a role in microtubule assembly in PC12 cells. Cell Motil. Cytoskeleton 64, 936–950 [DOI] [PubMed] [Google Scholar]
- 42.Saini D. K., Karunarathne W. K., Angaswamy N., Saini D., Cho J. H., Kalyanaraman V., and Gautam N. (2010) Regulation of Golgi structure and secretion by receptor-induced G protein βγ complex translocation. Proc. Natl. Acad. Sci. U.S.A. 107, 11417–11422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jensen D. D., Zhao P., Jimenez-Vargas N. N., Lieu T., Gerges M., Yeatman H. R., Canals M., Vanner S. J., Poole D. P., and Bunnett N. W. (2016) Protein kinase D and Gβγ subunits mediate agonist-evoked translocation of protease-activated receptor-2 from the Golgi apparatus to the plasma membrane. J. Biol. Chem. 291, 11285–11299 [DOI] [PMC free article] [PubMed] [Google Scholar]