

Abstract
Background
Alzheimer’s disease (AD) is characterized by progressive cognitive decline and memory loss. Environmental factors and gene-environment interactions (GXE) may increase AD risk, accelerate cognitive decline, and impair learning and memory. However, there is currently little direct evidence supporting this hypothesis.
Methods
In this study, we assessed for a GXE between lead and ApoE4 on cognitive behavior using transgenic knock-in (KI) mice that express the human Apolipoprotein E4 allele (ApoE4-KI) or Apolipoprotein E3 allele (ApoE3-KI). We exposed 8-week-old male and female ApoE3-KI and ApoE4-KI mice to 0.2% lead acetate via drinking water for 12 weeks and assessed for cognitive behavior deficits during and after the lead exposure. In addition, we exposed a second (cellular) cohort of animals to lead and assessed for changes in adult hippocampal neurogenesis as a potential underlying mechanism for lead-induced learning and memory deficits.
Results
In the behavior cohort, we found that lead reduced contextual fear memory in all animals; however, this decrease was greatest and statistically significant only in lead-treated ApoE4-KI females. Similarly, only lead-treated ApoE4-KI females exhibited a significant decrease in spontaneous alternation in the T-maze. Furthermore, all lead-treated animals developed persistent spatial working memory deficits in the novel object location test, and this deficit manifested earlier in ApoE4-KI mice, with female ApoE4-KI mice exhibiting the earliest deficit onset. In the cellular cohort, we observed that the maturation, differentiation, and dendritic development of adult-born neurons in the hippocampus was selectively impaired in lead-treated female ApoE4-KI mice.
Conclusions
These data suggest that GXE between ApoE4 and lead exposure may contribute to cognitive impairment and that impaired adult hippocampal neurogenesis may contribute to these deficits in cognitive behavior. Together, these data suggest a role for GXE and sex differences in AD risk.
Keywords: Lead, Apolipoprotein E, Cognitive behavior, Adult hippocampal neurogenesis, Learning and memory
Background
Alzheimer’s disease (AD) is the most common age-related neurodegenerative disorder [1]. Autosomal dominant mutations account for less than 5% of all Alzheimer’s cases [2], thus, most AD cases are likely due to a combination of environmental and genetic risk factors [3–5]. A gene-environment interaction (GXE) between genetic risk factors and environmental exposures may perturb hippocampus-dependent learning and memory, accelerate cognitive decline, and contribute to neurodegenerative diseases, including AD. However, there is a paucity of information supporting this GXE hypothesis.
Apolipoprotein E4 allele (ApoE4) is the strongest genetic risk factor for late-onset AD [6]; it increases the frequency and decreases the age at onset of AD in a gene dose-dependent manner [6]. ApoE4 carriers also have a worse prognosis following traumatic brain injury, a higher prevalence of mild cognitive impairment, and accelerated cognitive decline [7–11].
ApoE4 impairs hippocampus-dependent learning and memory in an age- and sex-dependent manner in transgenic knock-in (KI) mouse models [12–14]. In contrast to aging ApoE3-KI mice or male ApoE4-KI mice, female transgenic ApoE4-KI mice exhibit spatial learning and memory deficits starting at 16 months of age [12, 14]. Interestingly, female human ApoE4 carriers have an increased risk of AD compared to males, and female ApoE4 carriers with mild cognitive impairment experience greater hippocampal volume loss and reduced memory performance [15, 16]. Due to the negative effects of ApoE4 on cognition, ApoE4 carriers, particularly female carriers, may be more susceptible to toxicant-induced cognitive impairment compared to ApoE4 non-carriers.
Lead is a ubiquitous environmental contaminant and a significant public health concern in the U.S. and globally [17–19]. This is exemplified by the recent water crisis in Flint, Michigan, where the water supply was contaminated with lead due to lead leaching from old water pipes. The fact that many U.S. cities still have lead service pipes for water distribution underscores the continuing risk of lead exposure in the U.S. [20].
Both animal and epidemiological studies have reported an association between lead exposure and accelerated cognitive decline and/or AD-associated neuropathology in adults [21–25]. Interestingly, among workers occupationally exposed to lead, those with at least one ApoE4 allele experienced accelerated cognitive decline relative to ApoE4 non-carriers [26]. This epidemiological finding suggests that an interaction between lead and ApoE4 may accelerate cognitive decline. However, an experimental model is necessary in order to directly assess for a GXE between ApoE4 and lead regarding cognitive impairment. In this study, we used homozygous male and female ApoE3-KI and ApoE4-KI mice as an experimental model in order to assess the effect of lead exposure and ApoE4 on cognitive behavior and to examine any potential sex differences in susceptibility. We used deficits in hippocampus-dependent learning and memory as a measure of cognitive decline and AD-related memory impairment because the hippocampus is one of the earliest brain regions affected in AD patients and deficits in hippocampus-dependent spatial learning and memory may develop prior to the onset of a clinical diagnosis [27–29].
Interestingly, perturbation of adult hippocampal neurogenesis may cause cognitive behavior deficits, accelerate cognitive decline, and increase AD risk [30]. During adult hippocampal neurogenesis, adult neural precursor cells in the dentate gyrus (DG) of the hippocampus continuously generate adult-born neurons [31] which contribute to hippocampus-dependent learning and memory [32–37]. Various factors have been shown to modulate adult neurogenesis [31, 33, 34, 37, 38], however, little is known about whether toxicants or GXE may impair adult hippocampal neurogenesis. ApoE is expressed in adult neural precursor cells in the DG [6, 31, 39], and in vivo studies using ApoE4-KI mice found that ApoE4 alters adult-born neuron survival and maturation in an age- and sex-dependent fashion [12–14, 40]. Furthermore, we previously reported that lead can impair the proliferation, survival, and differentiation of adult neural precursor cells from the DG of the hippocampus in vitro [41]. No study to date has assessed the effect of adult-only lead exposure on adult hippocampal neurogenesis and cognitive behavior. Thus, we also investigated whether impaired adult hippocampal neurogenesis may contribute to this GXE between ApoE4 and lead exposure regarding cognitive impairment.
Methods
Mice
Humanized Apolipoprotein E3 and E4 knock-in (ApoE3-KI and ApoE4-KI) mice were generated as previously described [42] and provided by Dr. Nobuyo Maeda at the University of North Carolina, Chapel Hill. The human E3 or E4 allele in the ApoE3-KI or ApoE4-KI mice is expressed at physiological levels under control of the endogenous mouse ApoE promoter [13]. ApoE3-KI and ApoE4-KI animals were maintained as homozygous lines and all animals were housed in standard conditions (12 h light/dark cycle) with food and water provided ad libitum.
Reagents
Animal drinking water with 0.2% lead (II) acetate (Cat. 316512, Sigma-Aldrich, St. Louis, MO) was prepared from a stock lead acetate solution and replaced weekly. The preparation, use, and disposal of hazardous agents was carried out according to the Environmental Health and Safety Office at the University of Washington.
Lead exposure
Male and female ApoE3-KI and ApoE4-KI animals were weaned at 28 days and littermates of the same sex were randomly mixed into groups of 3–5 animals per cage. At 8 weeks of age, the mice were either switched to drinking water with 0.2% lead acetate (lead-treated) or kept on normal animal drinking water (control). Body weight was recorded every 1–2 weeks throughout the exposure. Water consumption was monitored for the first week of the exposure period. Lead-treated animals were exposed to lead drinking water for 12 weeks, at which point all the animals were switched to normal drinking water for the remaining duration of the study for the behavior cohort. We used separate cohorts of animals, one for behavior and one for cellular studies, to assess cognitive behavior and adult hippocampal neurogenesis, respectively. The behavior cohorts had n = 8–13 animals per genotype/treatment/sex and the cellular cohorts had n = 6–10 animals per genotype/treatment/sex.
Open field test
The open field test was conducted before and after the lead exposure to assess for the development of any lead-induced locomotor deficits or anxiety. Briefly, each animal was placed into a TruScan Photo Beam Tracking arena (Colbourn Instruments, Whitehall, PA) with Plexiglas walls (10″ X 10″ X 16″) and their movement was monitored with two sets of infrared breams. The animal was allowed to freely explore the arena for 20 min and the data was collected by TruScan 2.0 software (Colbourn Instruments).
Elevated plus maze
We conducted the elevated plus maze test in order to assess the effect of lead exposure on anxiety. The maze (26″ x 26″ x 15.25″; San Diego Instruments, San Diego, CA) consisted of four plastic arms with 7″ walls (two enclosed and two open), and it was placed in the center of the behavior room. Each animal was placed into the center of the maze facing an open arm and allowed to freely explore the maze for 5 min. The open and closed arm ends were defined as the distal 1/3 of the arms. ANYmaze software (San Diego Instruments) was used to collect data on the animal’s movement during the test.
Novel object location test
In order to assess hippocampus-dependent, short-term spatial working memory, we used the 1 h novel object location (NOL) test. This test was performed as previously described with a few modifications [34]. Briefly, each animal was placed into an open field arena (Colbourn Instruments) with two identical objects in two different locations. During training, the animal was allowed to freely explore the two objects for 5 min and then returned to its home cage. After 1 h, the animal was returned to the arena with the same two objects; one object remained in its original location and one object had been moved to a novel location. The time the animal spent actively investigating each object during the training and testing was quantified. Each training and testing session was recorded and scored offline by an experimenter blinded to the animal’s genotype and treatment. We calculated the discrimination index by dividing the difference in exploration time between the novel (C) and familiar (A) locations by the total exploration time.
Cued-contextual fear-conditioning
Contextual fear conditioning is another form of hippocampus-dependent learning and memory [33, 37, 43]. We tested 5–6-month-old animals and used a weak foot shock conditioning paradigm (3 x 0.3 mA, 2 s shocks with 2 min inter-trial intervals) as previously described [33]. For conditioning, the mouse was placed into the foot shock context (10″ x 10″ x 16″ arena with grid shock floor (Colbourn Instruments) and star-shaped wallpaper) and allowed to freely explore the arena for 2 min before the presentation of a 90 dB, 30 s tone (conditioned stimulus, CS). During the last 2 s of the tone presentation, a 0.3 mA foot shock (unconditioned stimulus, US) was delivered via the grid shock floor. This cycle was repeated two more times for a total of three cycles before the animal was returned to its home cage. The CS and US were automated and delivered by TruScan software (Colbourn Instruments). The contextual fear memory test was conducted 24 h after conditioning. The mouse was placed back into the foot shock context for 2 min in the absence of any foot shock. For the cued test (performed 2 h after context test), the animal was placed into a different context (new room; hexagonal Plexiglas arena; cartoon wallpaper) and allowed to freely explore for 2 min followed by the presentation of the CS for 2 min. For the novel context test (performed 2 h after the cued test), the mouse was placed into a novel context (new room; rat cage; striped wallpaper) and allowed to freely explore without any CS or US presentation. In all three tests, persistent freezing behavior (four paws on the ground, no head or body movement besides breathing) was recorded manually every 5 s during the 2 min scoring periods by an experimenter blinded to animal genotype and treatment.
T-maze continuous alternation task
We assessed spontaneous alternation in 12–13- month-old animals using a continuous alternation T-maze protocol with slight modifications [44]. Briefly, the black, plastic T-maze had two goal arms and a start arm (12.2″ x 4.5″ x 8.26″), and was placed on a platform (22.5″) in the center of a room. The test consisted of a first-forced trial followed by 14 free-choice trials. For the first-forced trial, one of the goal arms was (randomly) blocked with a plastic guillotine door. The animal was sequestered in the distal one-third of the start arm for 5 s before the guillotine door was raised and the animal was allowed to enter the unblocked goal arm. Once the animal returned to the start arm, it was sequestered in the start arm for 5 s before the start of the 14 free-choice trials. For each free-choice trial, the mouse was allowed to enter either of the unblocked goal arms; once it entered a goal arm, the other goal arm was blocked with a guillotine door. The animal eventually returned to the start arm and was sequestered for 5 s while all of the goal arms were unblocked. This was repeated for a total of 14 free-choice trials. The alternation percentage was calculated by dividing the number of times the animal entered alternating arms by 14 (free-choice trials). We defined arm entry as the animal’s tail tip entering the arm and defined repetitive arm entries as an animal re-entering the same arm three times in a row (e.g., 5 sequential entries into the same arm is 3 repetitive entries). An experimenter blinded to animal genotype and treatment scored each test.
Brain and blood lead analysis
We collected blood and brain tissue for lead analysis at sacrifice after the cessation of the lead exposure from a subset of the mice in the cellular cohort (20 weeks old; n = 3–5 per genotype/sex/treatment). The Environmental Health Laboratory at the University of Washington measured blood lead levels using inductively coupled plasma mass spectrometry (ICP-MS; Agilent 7500ce, Agilent Technologies, Santa Clara, CA). The spatial distribution and semi-quantitative measurement of lead and zinc levels (ppm) in 30 μm brain hemisphere sections from 20-week-old ApoE4-KI control or lead-treated females was determined by laser ablation ICP-MS (LA-ICP-MS) using an NWR213 laser ablation system (ESI, Portland, OR) connected to an Agilent 7500ce ICP-MS (Agilent Technologies, Santa Clara, CA). Quantification was achieved using spiked protein matrix as external standards.
TUNEL staining
TUNEL staining was performed using the Trevigen TACS 2 TdT-fluor In situ Apoptosis Detection Kit (Cat. 4812-30-K, Sigma-Aldrich, St. Louis, MO) on 30 μm fresh frozen coronal brain sections according to the manufacturer’s instructions. The cells were permeabilized using Trevigen’s CytoninTM permeabilization buffer and an experimental positive control was included (treated with TACS-NucleaseTM).
Quantification of dentate gyrus volume
The volume of the dentate gyrus was quantified using coronal images (20X magnification) from 15-month-old female ApoE3-KI and ApoE4-KI mice from the behavior cohorts as previously described [45].
BrdU administration
5-bromo-2′-deoxyuridine (BrdU) was from Sigma (Cat. B9285, Sigma-Aldrich, St. Louis, MO) and stored as a 20 mg/ml stock in saline with 0.007% NaOH at -20 °C. After 9 weeks of lead exposure, the mice in one of the cellular cohorts were dosed with 100 mg/kg BrdU by intraperitoneal injection 5 times in 1 day (every 2 h) and sacrificed 3 weeks later (at the end of the 12 week lead exposure) in order to assess the maturation of BrdU-labeled adult-born cells. A second cellular cohort was dosed with BrdU 1 X 100 mg/kg 2 h prior to sacrifice (at the end of the 12 week lead exposure period) in order to assess for proliferative BrdU-labeled cells.
Immunohistochemistry
The primary antibodies and dilutions used in immunohistochemistry were: rat monoclonal anti-BrdU (1:500, Bio-Rad Laboratories AbD Serotec, Raleigh, NC), mouse monoclonal anti-NeuN (1:1000, Millipore, Billerica, MA), mouse monoclonal anti-GAD67 (1:2000, Millipore, Billerica, MA), and donkey polyclonal anti-DCX (1:200, Santa Cruz Biotechnology, Dallas, TX), Goat anti-rat, goat anti-mouse, and donkey anti-rat Alexa Fluor-conjugated secondary antibodies as well as Hoechst 33342 (2.5 μg/ml) were from Invitrogen (Carlsbad, CA). All of the primary and secondary antibodies were diluted into the appropriate blocking buffer (10% donkey or goat serum and 1% BSA).
Mice were anesthetized with ketamine/xylazine and then euthanized by decapitation. One brain hemisphere was freshly frozen for brain lead analysis or other biochemical analysis, while the other hemisphere was post-fixed in 10% neutral-buffered formalin for 3–4 days followed by 30% (w/v) sucrose in phosphate-buffered saline (PBS, pH 7.4) at 4 °C for 3–4 days until saturated. Each post-fixed hemisphere was then stored in cryoprotectant media (30% glycerol; 30% ethylene glycol; 40% PBS) at -80 °C until sectioning. Coronal brain sections (30 μm thick) were used for immunohistochemistry (IHC) using the free-floating antibody staining method as previously described [38].
Quantification and imaging of immunostained cells
Immunostained cells were quantified as previously described [34, 38]. Every eighth serial section (30 μm) was immunostained for each IHC marker (or combination of markers). An experimenter blinded to treatment and genotype quantified marker+ cells in the subgranular zone and granule cell layer of the DG. This number was multiplied by 8 in order to estimate the total number of marker+ cells per DG. Marker colocalization (double-positive cell) was defined as overlapping fluorescent signals in a single cell using a Z-series stack. All the marker+ cells from at least 9 coronal sections per mouse (n = 3–5 mice per genotype/sex/treatment) were quantified for each immuno marker or marker combination. All images were captured with an Olympus Fluoview-1000 laser scanning confocal microscope with the following lenses: numerical aperture (NA) 0.75 10X, NA 0.75 20X, NA1.3 40X (oil), or NA 1.35 60X (oil). Optical Z-sections (1 μm thick) were collected and processed using ImageJ software (NIH, Bethesda, MD). Images were uniformly adjusted for color, brightness, and contrast with Adobe Photoshop CS4 (Adobe Systems Inc., San Jose, CA).
Quantification of dendritic morphology
The dendritic morphology of DCX+ cells in control and lead-treated ApoE3-KI and ApoE4-KI mice (at least 13 individual neurons per genotype/sex/treatment) was assessed by Sholl analysis as previously described [34].
Histology
Kidney and liver sections from control and lead-treated ApoE3-KI and ApoE4-KI males and females (n = 3–4 per genotype/treatment) were fixed in 10% neutral buffered formalin, paraffin embedded, cut into 4- to 5-μm sections, routinely stained with hematoxylin and eosin (H&E), and evaluated by a board-certified veterinary pathologist. Slides were coded to remove information regarding genotype and treatment. Kidneys were scored semi-quantitatively using a 0–4 scale, in which “0” was normal; “1” indicated minimal expansion of the glomerular mesangial matrix and minimal multifocal tubular protein; “2” indicated mild changes of the glomerulus with mild interstitial fibrosis, interstitial nephritis, and tubular protein; “3” indicated moderate multifocal to coalescing changes characterized by more pronounced glomerular, tubular and interstitial changes; and “4” indicated end stage disease with obsolescent glomeruli and severe multifocal to coalescing interstitial nephritis and fibrosis with or without dilation of the renal pelvis (hydronephrosis). Intranuclear inclusion bodies within the proximal renal tubules were noted when present. Livers were assessed using modified criteria from a previous study [46] and scored semi-quantitatively for changes including necrotic foci, cytoplasmic vacuolation, and mononuclear cell infiltration on a 0–4 system in which “0” was normal; “1” indicated minimal changes; “2” indicated mild changes; “3” indicated moderate changes; and “4” indicated severe changes. Images of representative lesions were acquired using NIS-Elements BR 3.2 64-bit and plated in Adobe Photoshop Elements. Image brightness and contrast was adjusted using Auto Smart Fix and Auto White Balance manipulations applied to the entire image.
Statistical analysis
Statistical analyses were conducted using GraphPad Prism software (version 6.0 h for Mac, GraphPad Software Inc., San Diego, CA, USA) and Stata (version 12.0 for Mac, StataCorp LP, College Station, TX, USA). Two-way analysis of variance (ANOVA) with the Holm-Sidak post-test (α = 0.05) was used to analyze the open field, elevated plus maze, contextual fear, and T-maze data in order to account for the main effect of genotype (ApoE3-KI vs. ApoE4-KI), treatment (control vs. lead), or time. Two-tailed t-test (α = 0.05) was used for within-genotype comparisons for the NOL data. Multilevel mixed-effects linear regression (α = 0.05) was used for longitudinal analysis of within-genotype NOL discrimination index data. Two-way analysis of variance (ANOVA) with Fisher’s LSD post-test (α = 0.05) was used to analyze all of the cellular data. All data represent mean ± SEM, n.s. not significant; * p < 0.05; ** p < 0.01; *** p < 0.001.
Results
ApoE3-KI and ApoE4-KI mice do not exhibit weight loss, locomotor deficits, or anxiety following lead exposure
We exposed 8-week-old male and female ApoE3-KI and ApoE4-KI mice to 0.2% lead acetate (lead-treated) or normal drinking water (control) for 12 weeks to model sub-chronic environmental and occupational exposure through ingestion (Fig. 1). We used 0.2% lead acetate in our study because this concentration has been used extensively in the literature to study lead neurotoxicity in mice [47–52]. We recorded body weight every 1–2 weeks during the lead exposure period and did not observe any weight loss in any of the lead-exposed animals at any time during lead exposure (Fig. 1b). Lead-treated ApoE3-KI mice were slightly heavier than their controls at several time points, but this weight gain was not associated with any locomotor deficits. There was no significant difference in body weight between control and lead-treated ApoE4-KI mice.
Fig. 1.
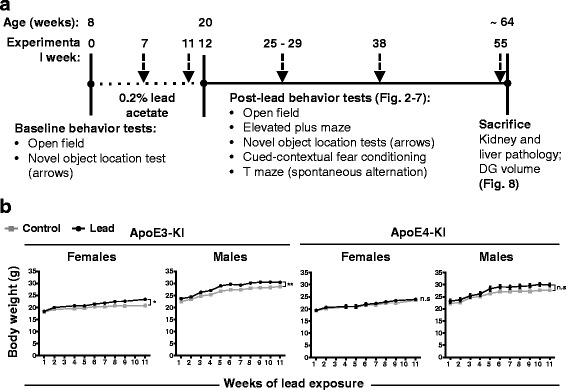
Adult male and female ApoE3-KI and ApoE4-KI mice do not exhibit weight loss upon exposure to 0.2% lead acetate for 12 weeks. a Experimental design and timeline for the behavior cohort. b Body weight was measured every 1–2 weeks during the lead exposure window. The lead-treated mice did not exhibit any weight loss relative to controls of the same sex and genotype. ApoE3-KI mice exposed to lead weighed slightly but significantly more at several time points than control mice during the lead exposure. Data are mean ± SEM with n = 8–13 per genotype/sex/treatment. Two-way ANOVA, significant main effect of treatment: n.s., not significant; * p < 0.05; ** p < 0.01
We conducted the open field test for all of the animals in the behavior cohort before and after the 12-week lead exposure in order to assess locomotor activity and anxiety. The pre-exposure test was performed to exclude any intrinsic differences between the control and lead-treated cohorts that could complicate data interpretation. Although there were some small differences in locomotor activity between the two genotypes, with ApoE4-KI mice exhibiting slightly higher baseline activity than ApoE3-KI mice, there were no significant differences in open field activity between control and lead-treated cohorts prior to the lead exposure of either genotype (data not shown).
After lead exposure, the lead-treated females, both ApoE3-KI and ApoE4-KI, traveled further, spent more time moving, and moved slightly faster than their respective controls (Fig. 2). There was no significant effect of lead on locomotor activity in male mice. It is important to note that there were no significant differences in locomotor activity between lead-treated ApoE3-KI and ApoE4-KI mice of either sex.
Fig. 2.
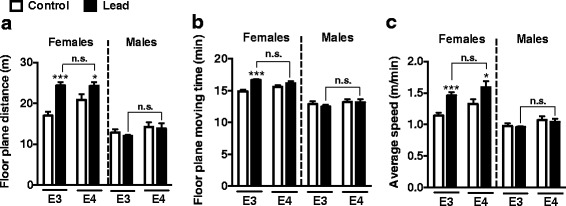
Lead-treated ApoE3-KI and ApoE4-KI male and female mice do not exhibit locomotor deficits in the open field test compared to controls. Mice were exposed to lead as previously described and the open field test was conducted after the cessation of the lead exposure. There was a significant main effect of lead treatment in female animals on floor plane distance, moving time, and average speed (Two-way ANOVA: floor plane distance, F(1,31) = 22.85, p < 0.0001; moving time, F(1,31) = 20.47, p < 0.0001; average speed, F(1,31) = 20.49, p < 0.0001). Post-hoc analyses found that lead-treated ApoE3-KI and/or ApoE4-KI females traveled a (a) greater distance, (b) spent the same or more time moving, and (c) traveled slightly faster than controls. There were no significant differences between lead and control ApoE3-KI and ApoE4-KI males or between lead-treated ApoE3-KI and ApoE4-KI mice (males or females) in any of the locomotor endpoints. Data are mean ± SEM with n = 8–13 per genotype/sex/treatment. Two-way ANOVA with Holm-Sidak post-hoc tests: n.s., not significant; * p < 0.05; *** p < 0.001
We also used the open field test to determine if lead treatment caused anxiety, measured as more time or distance in the margin, less time or distance in the center, or reduced center entries. Lead treatment did not change these parameters in males (Fig. 3a-f). Lead-treated ApoE3-KI females traveled a greater distance in the center of the open field and made more center entries compared to ApoE3-KI control females (Fig. 3d-f). Notably, there were no significant differences in center distance traveled and center entry between lead-treated ApoE3-KI and ApoE4-KI females.
Fig. 3.
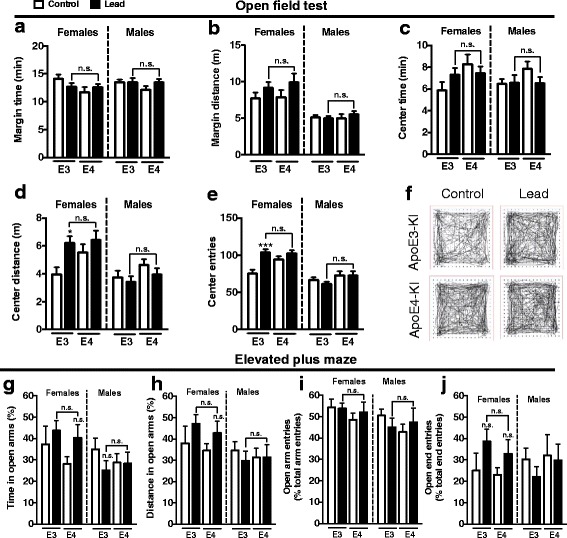
Male and female ApoE3-KI and ApoE4-KI mice exposed to lead do not exhibit overt anxiety in the open field test or elevated plus maze. The open field test was used to determine if lead treatment causes anxiety, measured as more time or distance in the margin, less time or distance in the center, or reduced center entries. There was a significant main effect of lead treatment in female mice on center distance and center entries (Two-way ANOVA: center distance, F(1,31) = 5.936, p = 0.0208; center entries, F(1,31) = 14.76, p = 0.0006). Post-hoc analyses found that there were no significant differences between lead-treated ApoE3-KI and ApoE4-KI mice (males and females) on the (a) time or (b) distance traveled in the margins, the (c) time or (d) distance traveled in the center, or the (e) number of center entries in the open field test. D, ApoE3-KI females treated with lead traveled a slightly greater distance in the center and E, made more center entries than controls. There were no significant differences between control and lead-treated ApoE4-KI females, ApoE3-KI males, or ApoE4-KI males in the open field test. f Representative open field track plots from female control and lead-treated ApoE3-KI and ApoE4-KI mice. In the elevated plus maze, both male and female ApoE3-KI and ApoE4-KI mice exposed to lead spent a (g) similar or greater amount of time and traveled a (h) similar or greater distance in the open arms of the maze compared to controls. Lead-treated males and females did not exhibit reduced (i) open arm or (j) open arm end entries compared to control animals. Data are mean ± SEM with n = 8–13 per genotype/treatment. Two-way ANOVA with Holm-Sidak post-hoc tests: n.s., not significant; * p < 0.05; *** p < 0.001
We also conducted the elevated plus maze test to assess anxiety after the cessation of the lead exposure (Fig. 3g-j). The elevated plus maze is commonly used to assess the anxiogenic and anxiolytic effects of toxicological and pharmacological agents. It relies on a mouse’s natural avoidance of heights and open spaces and it’s preference for dark, sheltered spaces [53]. There was no significant effect of genotype or lead exposure on the percent of total time males or females spent or the total distance they traveled in the open arms by two-way ANOVA (Fig. 3g, h). In addition, there was no significant effect of genotype or treatment on the number of open arm entries or open arm end entries among males or females (Fig. 3i, j).
Thus, subchronic exposure of 8-week-old mice to 0.2% lead acetate did not decrease locomotor activity or cause anxiety in ApoE3-KI or ApoE4-KI mice. Although lead exposure may result in a slight hyperactive phenotype in ApoE3-KI and ApoE4-KI females and a slight anxiolytic phenotype in ApoE3-KI females, there were no significant differences between lead-treated ApoE3-KI and ApoE4-KI mice of either sex.
ApoE4-KI females exposed to lead exhibit impaired contextual fear memory
We also conducted a contextual fear-conditioning test (age of mice: 5–6 months old) to characterize the effect of lead and ApoE4 on hippocampus-dependent learning and memory (Fig. 4a). We used a 0.3 mA x 3 foot shock paradigm because we previously reported that this paradigm is sensitive to changes in contextual fear memory and adult neurogenesis [33]. All mice showed minimal freezing behavior at baseline before foot shock (Fig. 4b, c). We performed the context test 24 h after fear-conditioning in order to assess contextual fear memory. Interestingly, lead-treated ApoE4-KI females had significantly reduced contextual fear memory compared to ApoE4-KI controls (Fig. 4b). Male ApoE4-KI mice and both male and female ApoE3-KI mice exposed to lead exhibited a statistically non-significant reduction in freezing behavior upon lead treatment (Fig. 4b, c). Lead treatment did not cause any statistically significant changes in any animals in auditory-cued fear memory–a form of amygdala-dependent, but hippocampus-independent memory–or in general freezing in a novel context. Furthermore, lead treatment did not affect animals’ shock sensitivity (data not shown). Thus, lead exposure impairs the formation and/or retrieval of contextual fear memory in ApoE4-KI female mice.
Fig. 4.
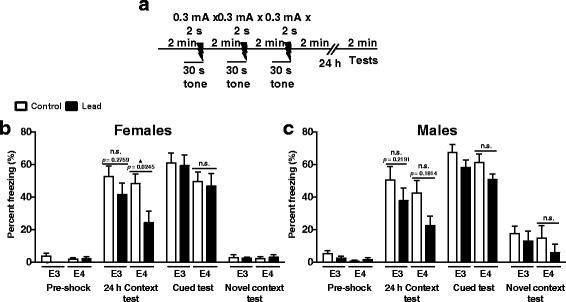
Lead impairs contextual fear memory in lead-treated ApoE4-KI females at 22–24 weeks of age. a Schematic of cued and contextual fear conditioning test performed post-lead exposure. b, c Female and male mice had low baseline freezing behavior (Pre-shock). In the 24 h Context test, there was a significant main effect of lead treatment on freezing behavior in both males and females (Two-way ANOVA: females, F(1,33) = 6.803, p =0.0136; males, F(1,42) = 4.435, p =0.0412). Although lead treatment reduced contextual memory in all animals (manifested as reduced freezing 24 h after fear conditioning) post-hoc tests revealed that this was only statistically significant between control and lead-treated ApoE4-KI females (Holm-Sidak post-hoc test: F(1,33) = 2.648, p = 0.0245). Auditory-cued (hippocampus-independent) fear memory was not affected (Cued test) in any lead-exposed animals. All animals did not freeze when placed into a novel, non-shock context (Novel context test). Data are mean ± SEM with n = 8–13 per sex/genotype/treatment. Two-way ANOVA with Holm-Sidak post-hoc test. n.s. not significant; * p < 0.05
ApoE4-KI females exhibit decreased spontaneous alternation and increased repetitive arm entry in a T-maze
We assessed spontaneous alternation using the T-maze in 12–13-month-old animals. Rodents spontaneously alternate the arms they enter in the T-maze, and this is likely due to a combination of novelty-elicited exploratory behavior as well as spatial working memory [44, 54]. Interestingly, lead-treated ApoE4-KI females exhibited a significant reduction in spontaneous alternation (Fig. 5a) and had a significant increase in repetitive arm entries compared to control ApoE4-KI females (Fig. 5b). These behavior changes were specific to females and were not observed in males. There were no significant differences between control and lead-treated animals on the ratio of left/right arm entries or mean session duration in either females or males (Fig. 5c, d). These data suggest that there may be a GXE interaction between lead and ApoE4 on spontaneous alternation and repetitive entries. Furthermore, lead-treated female ApoE4-KI mice are more sensitive than males. These differences are not due to a bias in arm entries or significant differences in task completion time.
Fig. 5.
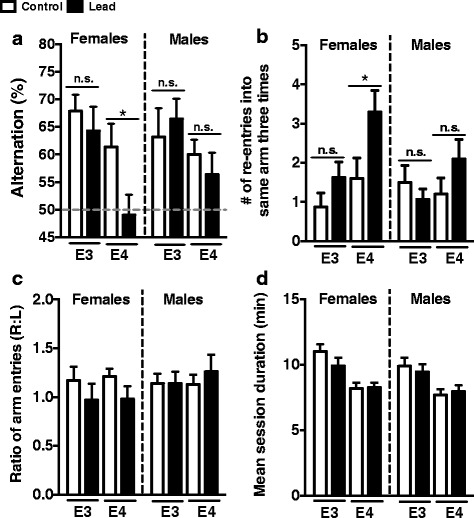
Lead impairs spontaneous alternation and increases repetitive arm entry in lead-treated ApoE4-KI females. Spontaneous alternation was assessed using the T-maze at 12–13 months of age. a There was a significant main effect of genotype on spontaneous alternation in females (Two-way ANOVA: genotype, F(1,32) = 7.666, p = 0.0093). Lead-treated ApoE4-KI female mice exhibited reduced spontaneous alternation compared to female ApoE4-KI control mice (Holm-Sidak post-test: F(1,32) =2.356, p =0.0490). b There was a significant main effect of both genotype and treatment on repetitive arm entries in females (Two-way ANOVA: genotype, F(1,32) = 5.915, p =0.0208; treatment, F(1,32) = 6.164, p =0.0185). ApoE4-KI females exposed to lead exhibited significantly increased repetitive arm entries compared to control ApoE4-KI female mice (Holm-Sidak post-test: F(1,32) = 2.584, p 0.0289). There was no significant difference in spontaneous alternation or arm re-entries in lead-treated ApoE3-KI and ApoE4-KI male mice. c Female and male mice did not exhibit any arm preference. d There was a significant main effect of genotype on mean session duration in both the females and males, with ApoE4-KI mice completing the alternation task slightly faster than ApoE3-KI mice of the same sex (Two-way ANOVA: females, F(1,30) = 19.50, p = 0.0001; males F(1,41) =9.803, p = 0.0032). There was no significant difference in mean session duration between control and lead-treated animals of the same genotype and sex. Data are mean ± SEM with n = 8–13 per sex/genotype/treatment. n.s. not significant; * p < 0.05
ApoE3-KI and ApoE4-KI mice exposed to lead exhibit a GXE interaction and sex differences in the onset of short-term spatial memory deficits in the novel objection location test (NOL)
We conducted a 1 h NOL test to assess hippocampus-dependent spatial working memory before, during, and after the lead exposure. Importantly, at each NOL time point (baseline through 10 months post-lead), none of the mice exhibited a preference for either object or location during the training session, and there was no difference in the total exploration time between lead-treated and control mice (data not shown).
Before lead exposure, all of the ApoE3-KI and ApoE4-KI females and males spent significantly more time exploring the object in the novel vs. familiar (C vs. A) location during the test period, indicating that they remembered the original object location (Fig. 6A). Seven weeks into the lead exposure, all of the mice spent significantly more time exploring the novel location except for lead-treated ApoE4-KI females, which did not discriminate between the old vs. new object locations (Fig. 6B). After 11 weeks of lead exposure, both the lead-treated ApoE4-KI females and males no longer discriminated between the familiar and novel object locations while the lead-treated ApoE3-KI mice continued to spend significantly more time exploring the novel object location (Fig. 6C). Both the lead-treated ApoE4-KI females and males continued to exhibit a deficit in spatial working memory at 3, 6, and 10 months post-lead exposure (Fig. 6D and data not shown). Lead-treated ApoE3-KI mice still spent significantly more time exploring the novel object location at 3, 6, and 10 months post-lead exposure (Fig. 6D and data not shown). However, the differences in exploration time between the new vs. old object locations became smaller over time among the ApoE3-KI mice exposed to lead indicating poorer spatial memory.
Fig. 6.
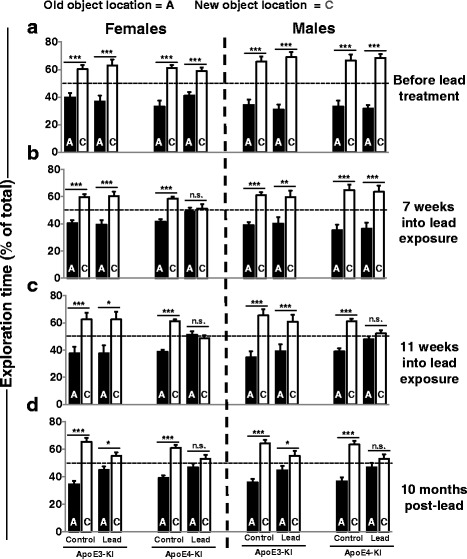
The effect of lead on short-term spatial memory in the NOL test. The NOL test was performed before, during, and after the lead exposure to assess for spatial working memory deficits. The time animals spent investigating the object in the old (location A) vs. new (location C) locations was quantified. More time spent exploring the object in the novel vs. old location indicates memory for the old location. ( A ) All the animals had intact spatial memory prior to the lead exposure. ( B ) At 7 weeks into the lead exposure, only the lead-treated ApoE4-KI females did not discriminate between the old vs. new object locations (Two-tailed t-test (A vs. C): p = 1.000). ( C ) At 11 weeks, both the lead-treated ApoE4-KI females and males no longer discriminated between the object locations (females, p = 0.4042; males, p = 0.1959) while the ApoE3-KI females and males spent significantly more time exploring the novel object location (females, p = 0.0107; males, p = 0.0098). ( D ) Lead-treated ApoE4-KI females and males continued to exhibit a deficit in spatial working memory at 10 months post-lead exposure (females, p = 0.0961; males, p = 0.1855). Lead-treated ApoE3-KI mice still spent statistically significantly more time exploring the novel object location at 10 months post-lead exposure (females, p = 0.0186; males, p = 0.0320). Data are mean ± SEM with n = 8–13 per genotype/treatment. Two-tailed t–test: n.s. not significant, * p < 0.05; ** p < 0.01; *** p < 0.001
We calculated the discrimination index for each NOL test, and the discrimination indices at each time point are summarized by genotype and sex in Fig. 7. There was an effect of lead exposure on the discrimination index in all lead-exposed animals (Multiple mixed-effects linear regression: ApoE3-KI females, p = 0.001; ApoE4-KI females, p < 0.0001; ApoE3-KI males, p = 0.019; ApoE4-KI males, p < 0.0001). During the lead exposure period, there was no difference in the discrimination index between control and lead-treated ApoE3-KI females (Fig. 7a). However, lead-treated ApoE3-KI females had a significantly lower discrimination index at 3 and 10 months post-lead exposure compared to controls.
Fig. 7.
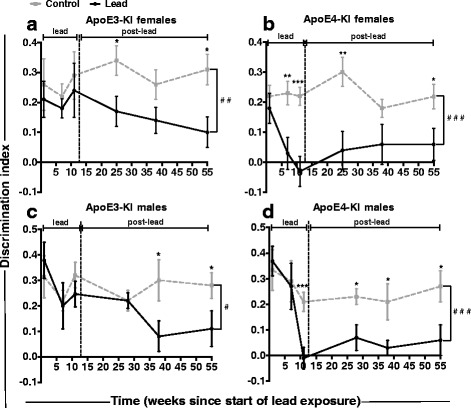
Genotype and sex differences in lead-induced reduction of discrimination index over time. The discrimination index in the NOL test was calculated by dividing the difference in exploration time between the novel (c) and familiar (a) locations by the total exploration time, and used to compare changes of spatial memory over time. There was a main effect of lead exposure on the discrimination index in all lead-exposed animals (Multiple mixed-effects linear regression: ApoE3-KI females, p = 0.001; ApoE4-KI females, p < 0.0001; ApoE3-KI males, p = 0.019; ApoE4-KI males, p < 0.0001). a There was no difference in the discrimination index between control and lead-treated ApoE3-KI females during the lead exposure (Two-tailed t-test: 7 week, p = 0.3046; 11 week, p = 0.9977). Lead-treated ApoE3-KI females had a significantly lower discrimination index at 3 and 10 months post-lead exposure (3 months, p = 0.0268; 10 months, p = 0.0156) and a non-significant decrease 6 months post-lead (p = 0.0901) compared to controls. b Lead-treated ApoE4-KI females had a significantly lower discrimination index than ApoE4-KI control mice starting at 7 weeks into the lead exposure and this persisted through 10 months post-lead (7 weeks, p = 0.0016; 11 weeks, p = 0.0002; 3 months, p = 0.0030; 10 months, p = 0.0269). The lead-treated ApoE4-KI discrimination index was lower than controls at 6 months post-lead but not statistically significant (p = 0.0713). c Lead-treated ApoE3-KI males had a significantly lower discrimination index at 6 and 10 months post-lead compared to controls (6 months, p = 0.0356; 10 months, p = 0.0418). d In contrast, lead-treated ApoE4-KI males had a significantly lower discrimination index than controls starting at 11 weeks into the lead exposure and this effect persisted through 10 months post-lead exposure (11 weeks, p = 0.0064; 4 months, p = 0.0433; 6 months, p = 0.0382; 10 months, p = 0.0275). Data are mean ± SEM with n = 8–13 per sex/genotype/treatment. Multi-level mixed-effects linear regression; significant effect of treatment: # p < 0.05; ## p < 0.01; ### p < 0.001. Two-tailed t-test; significant difference between control and lead: n.s., not significant; * p < 0.05; ** p < 0.01; *** p < 0.001
Interestingly, the lead-treated ApoE4-KI females had a significantly lower discrimination index than control ApoE4-KI females starting at 7 weeks into the lead exposure, and this decrease persisted through 10 months post-lead (Fig. 7b). Thus, lead-treated ApoE3-KI and ApoE4-KI females exhibited persistent deficits in short-term spatial memory, with ApoE4-KI females exhibiting a significant decrease in the discrimination index starting at 7 weeks into the lead exposure, much earlier than lead-treated ApoE3-KI females at 3 months post-lead exposure.
Lead-treated ApoE3-KI males had a significantly lower discrimination index at 6 and 10 months post-lead compared to controls (Fig. 7c), suggesting that spatial memory deficits can develop long after lead exposure. In contrast, lead-treated ApoE4-KI males had a significantly lower discrimination index than controls starting at 11 weeks into the lead exposure and this effect persisted through 10 months post-lead exposure (Fig. 7d). Thus, both ApoE3-KI and ApoE4-KI males exposed to lead exhibited persistent deficits in short-term spatial memory, but the lead-treated ApoE4-KI males showed these deficits much earlier (starting at 11 weeks into the lead exposure vs. 6 months post-lead).
Together, data in Figs. 6 and 7 demonstrate that ApoE4-KI mice exposed to lead developed impaired spatial working memory much earlier than lead-treated ApoE3-KI mice, in both males and females. When we compare males and females of the same genotype, lead-induced spatial memory deficits manifested earlier in females than in males for both the ApoE3-KI and ApoE4-KI mice. These data suggest a GXE between lead exposure and ApoE4 on cognitive impairment as well as sex differences in the onset of cognitive deficits.
Lead-treated ApoE3-KI and ApoE4-KI mice do not exhibit significant pathology in the DG and other tissues
In order to determine if lead exposure causes systemic toxicity, we assessed for pathological changes in the liver and kidney of 64-week-old (~15-month-old) male and female ApoE3-KI and ApoE4-KI mice after various behavioral testing. There were no significant differences in kidney or liver pathology scores between control and lead-treated mice of either sex or genotype (Fig. 8a-d and data not shown). Furthermore, there were no changes in the volume of the DG of the hippocampus in ApoE3-KI or ApoE4-KI females upon lead exposure (Fig. 8e). In addition, we did not observe an increase in TUNEL+ cells in the hippocampus or cortex of lead-treated animals (Fig. 9). Thus, the lead exposure regimen used in this study did not result in gross pathological changes in the DG, kidney, or liver.
Fig. 8.
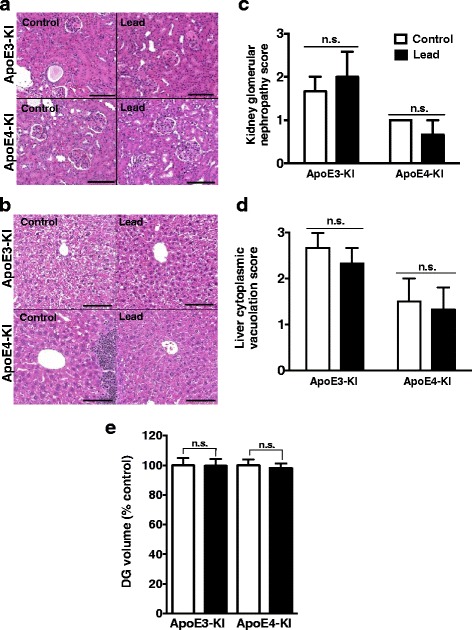
Adult lead does not cause significant liver or kidney toxicity or DG volume loss in aged ApoE3-KI and ApoE4-KI mice. 8-week-old female ApoE3-KI and ApoE4-KI mice were exposed to 0.2% lead acetate for 12 weeks, then switched to normal drinking water and sacrificed 43–45 weeks post-lead exposure (14.5–15 months old). H&E staining of a kidney and b liver sections and quantification of c kidney glomerular nephropathy and d liver cytoplasmic vacuolation in female ApoE3-KI and ApoE4-KI mice. e DG volume was measured in female ApoE3-KI and ApoE4-KI mice using ImageJ analysis of confocal images of coronal sections from one brain hemisphere. Data are mean ± SEM. n = 3–5 per genotype/treatment. Two-way ANOVA with Fisher’s LSD post-test: n.s., not significant. Scale bars, 100 μm
Fig. 9.
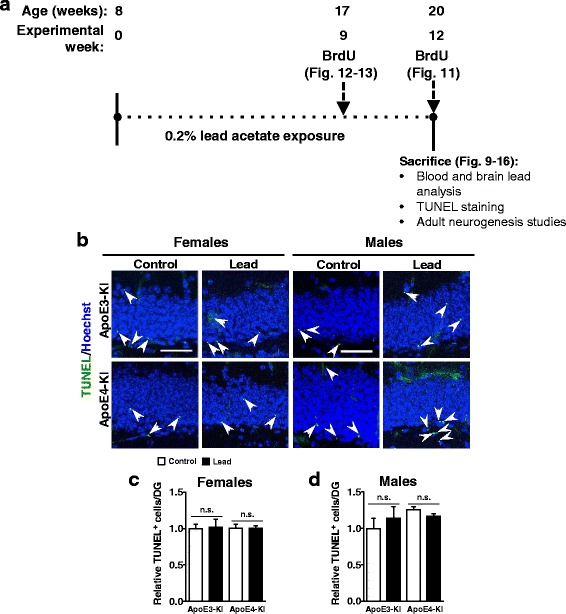
Adult-only lead exposure does not increase the number of TUNEL+ apoptotic cells in the DG of the hippocampus. a Experimental design and timeline for cellular studies (BrdU dosing) for Figs. 9, 10, 11, 12, 13, 14, 15 and 16. b Representative images of TUNEL+ (green) cells in the DG of control and lead-treated ApoE3-KI and ApoE4-KI males and females. Quantification of TUNEL+ cells per DG in ApoE3-KI and ApoE4-KI c females and d males. Data are mean ± SEM with n = 3–4 per genotype/sex/treatment. Two-way ANOVA with Fisher’s LSD post-test: n.s., not significant. Scale bar, 50 μm
Adult lead exposure results in elevated blood lead levels and lead deposition in the brain
We collected blood and brain tissue for lead analysis from the cellular cohort at sacrifice after the 12-week lead exposure. Lead exposure significantly raised blood lead levels in all animals relative to controls at the end of the 12 week exposure, and lead levels were higher in exposed females than males of the same genotype (Fig. 10a). Lead-exposed animals also had significantly higher brain lead levels relative to their controls at the end of the 12-week exposure, with the highest levels in ApoE4-KI female brains (Fig. 10b). Low-resolution LA-ICP-MS of sections from a control and a lead-treated female ApoE4-KI mouse illustrate that adult-only lead exposure is sufficient to cause increased lead deposition throughout the brain (Fig. 10c-f), while levels of the essential metal zinc (an internal control for the imaging and exposure) were not significantly different between control and lead-treated mice (data not shown).
Fig. 10.
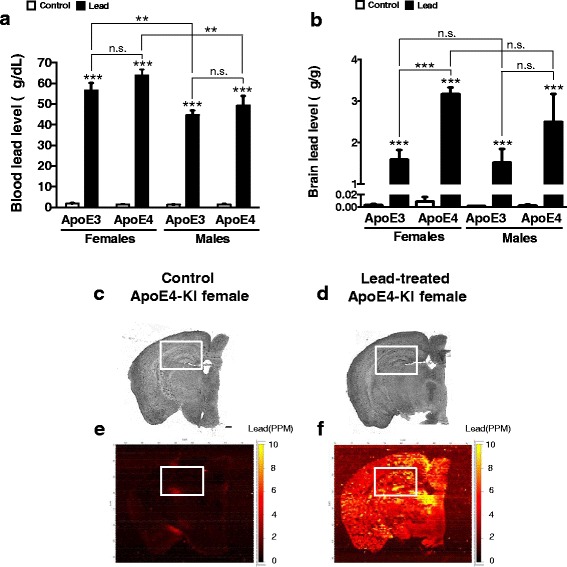
Adult-only lead exposure results in elevated blood lead levels and lead deposition in the brain. 8-week-old ApoE3-KI and ApoE4-KI male and female mice were exposed to 0.2% lead acetate for 12 weeks and then sacrificed. Blood and one brain hemisphere were collected at sacrifice and (a) blood lead and (b) brain lead levels were measured using ICP-MS. Brightfield images of one brain hemisphere from a female (c) control and (d) lead-treated ApoE4-KI mouse after the 12 week lead exposure. Semi-quantitative measurement of lead in the brain of a (e) control and (f) lead-treated ApoE4-KI mouse using LA-ICP-MS. Two-way ANOVA with Fisher’s LSD post-test: n.s., not significant; ** p < 0.01; *** p < 0.001. Scale bars, 100 μm
Lead decreases proliferation of adult-born cells in the hippocampus in all lead-exposed mice
Adult hippocampal neurogenesis is the process whereby adult neural stem cells in the DG of the hippocampus generate adult-born neurons [31]. These adult-born neurons can influence hippocampus-dependent learning and memory [33, 34, 37, 55]. Interestingly, ApoE4 alters adult-born neuron differentiation and maturation in the DG of female ApoE4-KI mice [40]. Thus, we examined whether adult lead exposure inhibits adult neurogenesis in the hippocampus and whether there is a GXE between lead exposure and ApoE4 on this process using a second cohort (the cellular cohort) of mice.
To determine if adult-only lead exposure is sufficient to impair cell proliferation during adult neurogenesis, we treated half of the cellular cohort of mice with BrdU 2 h prior to sacrifice at the end of the 12-week lead exposure in order to label actively proliferating cells in S-phase of the cell cycle (Fig. 11). We found that lead significantly reduced the total number of BrdU+ cells in all lead-treated animals compared to their respective controls of the same genotype and sex (Fig. 11a-d). The percent change in BrdU+ cells in lead-treated mice compared to control mice was not statistically significantly different between ApoE3-KI and ApoE4-KI mice, in either sex (Fig. 11e-f). These data suggest that adult-only lead exposure for 12 weeks is sufficient to significantly decrease adult-born cell proliferation in the DG of the hippocampus of all mice. Although this cellular mechanism does not explain the behavior differences observed between ApoE3-KI vs. ApoE4-KI mice, or between males and females, it may contribute to the eventual memory loss in all lead-exposed mice, such as that observed in the novel object location test.
Fig. 11.
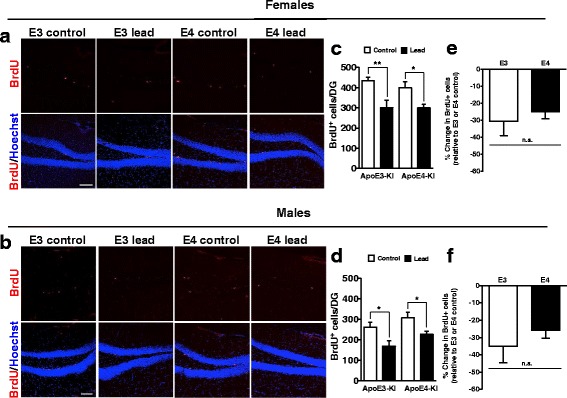
Adult lead exposure decreases adult-born cell proliferation in the DG of the hippocampus of all mice. 8-week-old male and female ApoE3- and ApoE4-KI mice were exposed to 0.2% lead acetate for 12 weeks and then sacrificed. BrdU was administered 2 h prior to sacrifice (1 x 100 mg/kg). Representative images of BrdU (red) immunostaining in the DG of 20-week-old (a) female and (b) male ApoE3-KI and ApoE4-KI control and lead-treated mice. Quantification of the total BrdU+ cells per DG in (c) females and (d) males. Quantification of the percent change in total BrdU+ cells in lead-treated mice relative to ApoE3-KI or ApoE4-KI control (e) females and (f) males. Data are mean ± SEM with n = 3–5 per genotype/sex/treatment. Two-way ANOVA with Fisher’s LSD post-test: n.s., not significant; * p < 0.05; ** p < 0.01. Scale bars, 100 μm
Lead impairs adult-born neuron differentiation and decreases the dendritic complexity of immature neurons in the DG of female ApoE4-KI mice
We examined the effect of lead on neuronal differentiation of adult-born cells in the DG, using immunostaining for DCX and NeuN as markers of immature and mature neurons, respectively (Figs. 12a and 13a). BrdU was administered to the remaining half of the cellular cohort of mice at 9 weeks into the lead exposure (3 weeks prior to sacrifice) in order to label 3-week-old adult-born cells. Lead treatment significantly reduced the proportion of adult-born cells that express DCX (BrdU+DCX+/total BrdU+) or NeuN (BrdU+NeuN+/total BrdU+) in female ApoE4-KI mice, but not in other lead-treated mice (Fig. 12b-c, and Fig. 13b-c), suggesting that lead inhibits neuronal differentiation of adult-born neurons in the hippocampus and that female ApoE4-KI mice are more sensitive. There was also a statistically significant decrease in the number of total adult-born mature neurons (BrdU+NeuN+) in both male and female ApoE4-KI mice, although the effect was smaller in males than in females (Fig. 13d-e).
Fig. 12.
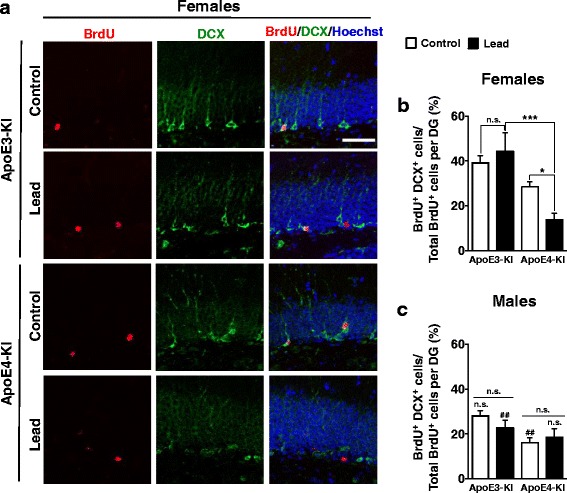
Lead decreases adult-born immature neuron maturation in the DG of ApoE4-KI female mice. 8-week-old male and female ApoE3- and ApoE4-KI mice were exposed to 0.2% lead acetate for 12 weeks and then sacrificed. 100 mg/kg BrdU was administered 5 times in 1 day (every 2 h) 3 weeks prior to sacrifice (i.e., at 9 weeks into lead exposure). a Representative images of BrdU (red) and DCX (green) immunostaining in the DG of control and lead-treated female ApoE3-KI and ApoE4-KI animals. Quantification of the percent of total BrdU+ cells that are BrdU+DCX+ per DG in females (b) and males (c) respectively. Data are mean ± SEM with n = 4–5 per genotype/sex/treatment. Two-way ANOVA with Fisher’s LSD post-test: Comparison between control and lead-treated: n.s., not significant; * p < 0.05; *** p < 0.001. Comparison between males and females of the same genotype and treatment: n.s., not significant; ##, p < 0.01. Scale bar, 50 μm
Fig. 13.
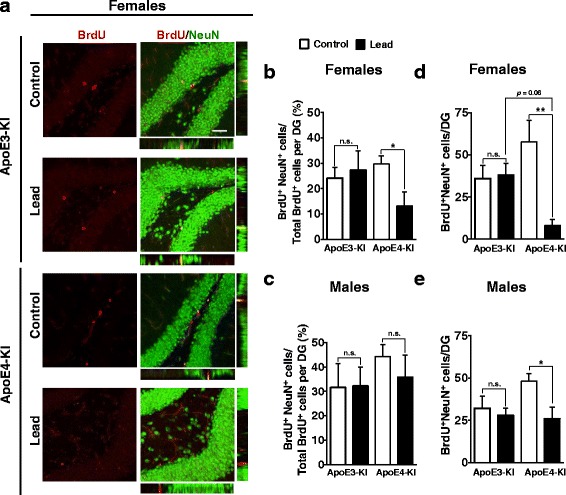
Lead decreases adult-born neuron differentiation in the DG of ApoE4-KI female mice. 8-week-old male and female ApoE3-KI and ApoE4-KI mice were exposed to 0.2% lead acetate for 12 weeks and then sacrificed. 100 mg/kg BrdU was administered 5 times in 1 day (every 2 h) 3 weeks prior to sacrifice (i.e., at 9 weeks into lead exposure). a Representative images of BrdU (red) and NeuN (green) immunostaining in the DG of control and lead-treated female ApoE3-KI and ApoE4-KI animals. Quantification of the percent of total BrdU+ cells that are BrdU + NeuN+ per DG in b females and c males, respectively. Quantification of the total number of BrdU + NeuN+ cells in d females and e males, respectively. Data are mean ± SEM with n = 4–5 per genotype/sex/treatment. Two-way ANOVA with Fisher’s LSD post-test: n.s., not significant; * p < 0.05; ** p < 0.01. Scale bar, 50 μm
We also assessed the dendritic morphology, a measure of neuronal maturation, of adult-born neurons in this same cohort of mice. Because the acid treatment necessary for BrdU staining damages neuronal processes, we used DCX immunostaining alone to visualize neuronal processes of newly generated neurons (Fig. 14a-b, 15a-b). Neurite tracing and Sholl analysis showed that lead treatment significantly decreased the total dendritic length (Fig. 14c) and the number of dendritic crossings (Fig. 14e) in female ApoE4-KI mice. This effect was not seen in female ApoE3-KI mice (Fig. 14d) or any of the male mice (Fig. 15). These data suggest that ApoE4-KI females are more sensitive to the effects of lead on neuronal differentiation and maturation.
Fig. 14.
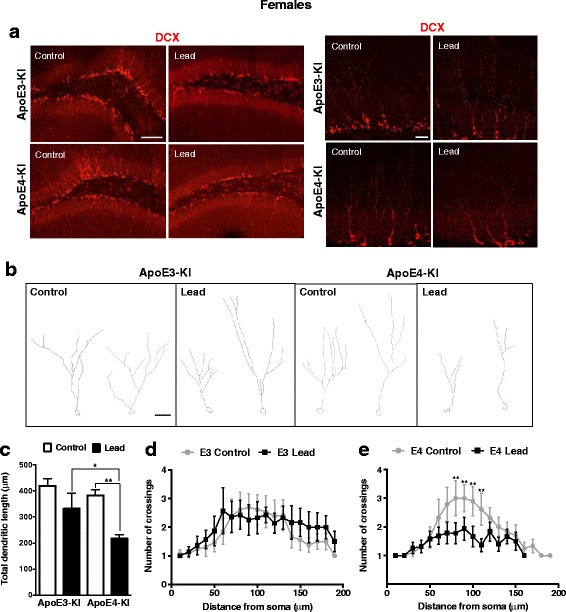
Lead decreases the dendritic complexity of immature neurons in the DG of ApoE4-KI female mice. a, Representative confocal images of DCX (red) immunostaining in the DG of 20-week-old female ApoE3-KI and ApoE4-KI control and lead-treated mice (left scale bar, 100 μm; right scale bar, 25 μm). b, Representative examples of DCX+ neurons from female ApoE3-KI and ApoE4-KI control and lead-treated mice traced using the ImageJ Simple Neurite Tracer plug-in (scale bar, 25 μm). c, Quantification of the total dendritic length of DCX+ neurons in the DG. Sholl analysis of DCX+ neurons from (d) ApoE3-KI and (e) ApoE4-KI female mice. Data are mean ± SEM with n = 4–5 per genotype/treatment. Two-way ANOVA with Fisher’s LSD post-test for analysis of total dendritic length; two-tailed t-test for within genotype comparisons of the number of crossings in control vs. lead-treated mice: * p < 0.05; ** p < 0.01
Fig. 15.
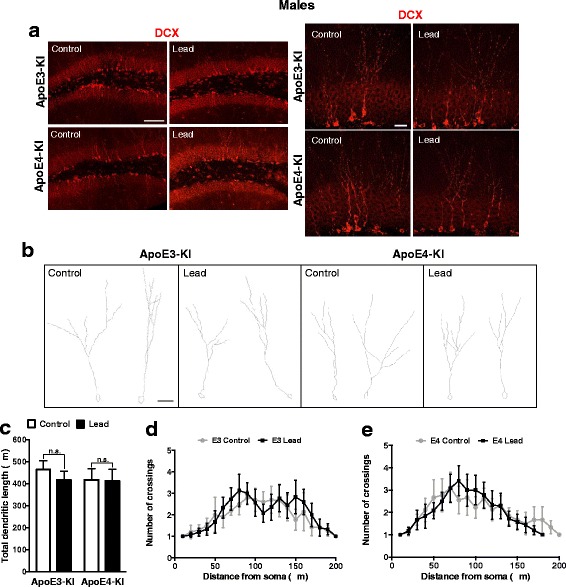
Lead does not impair the dendritic complexity of immature neurons in the DG of male mice. a, Representative confocal images of DCX (red) immunostaining in the DG of 20-week-old male ApoE3-KI and ApoE4-KI control and lead-treated mice (left scale bar, 100 μm; right scale bar, 25 μm). b, Representative examples of DCX+ neurons from male ApoE3-KI and ApoE4-KI control and lead-treated mice traced using the ImageJ Simple Neurite Tracer plug-in (scale bar, 25 μm). c, Quantification of the total dendritic length of DCX+ neurons in the DG. Sholl analysis of DCX+ neurons from d ApoE3-KI and e ApoE4-KI male mice. Data are mean ± SEM with n = 4–5 per genotype/treatment. Two-way ANOVA with Fisher’s LSD post-test: n.s., not significant
Lead reduces the number of GABAergic interneurons in the DG of ApoE4-KI female mice
GABAergic interneurons play an important role in adult hippocampal neurogenesis [56]. Previous studies report that ApoE4-KI female mice exhibit an age-dependent decrease in the number of GABAergic interneurons in the DG of the hippocampus, starting at 6 months of age [12, 14, 40]. Thus, we examined the effect of lead on the number of GABAergic neurons at the end of the 12-week lead exposure (20-weeks old) using GAD67 staining as a marker (Fig. 16a). There was no significant difference in the total number of GABAergic neurons between control ApoE3-KI and ApoE4-KI mice in either sex (Fig. 16b). However, lead treatment significantly decreased the number of GAD67+ cells in the DG of ApoE4-KI females but not in other mice.
Fig. 16.
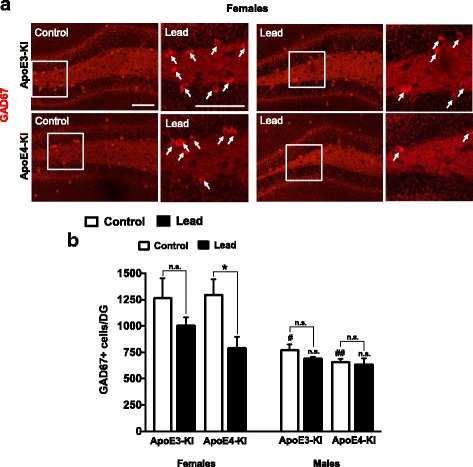
Lead decreases the total number of GAD67+ GABAergic interneurons in 20-week-old female ApoE4-KI mice. 8-week-old male and female ApoE3-KI and ApoE4-KI mice were exposed to 0.2% lead acetate for 12 weeks and sacrificed. a Representative images of GAD67 (red) immunostaining in the DG of 20-week-old female ApoE3-KI and ApoE4-KI control and lead-treated mice. b Quantification of the total number of GAD67+ cells in the DG of 20-week-old ApoE3-KI and ApoE4-KI females and males. Data are mean ± SEM with n = 3–5 per genotype/treatment. Two-way ANOVA with Fisher’s LSD post-test. Comparison between control and lead-treated: n.s., not significant; * p < 0.05. Comparison between males and females of the same genotype and treatment: n.s., not significant; #, p < 0.05; ##, p < 0.01. Scale bars, 100 μm
Discussion
AD currently affects nearly 35 million people and is projected to affect 117 million people worldwide by 2050 [57]. AD and cognitive decline are associated with significant societal, financial, and health care costs. Yet, there is a paucity of research on the role of environmental exposures and GXE on neurodegenerative disease risk and AD pathogenesis. Thus, additional investigation into how environmental factors and GXE may influence AD risk or accelerate cognitive decline is of immense public health importance.
Functional impairment in cognition is often used as a proxy to estimate the presence and degree of AD-associated neuropathology [58, 59]. Here, we used humanized transgenic knock-in mice as an animal model of human ApoE4 carriers to assess whether there is a GXE between lead and ApoE4 on cognitive behavior, and potentially on AD risk. ApoE3-KI and ApoE4-KI female mice develop spontaneous learning and memory deficits starting at 15–18 months [12–14]. Consequently, we assessed learning and memory prior to 15 months of age to determine if lead can accelerate the onset of cognitive behavior deficits. We defined a GXE as a different effect of lead exposure on cognitive behavior in mice expressing the human ApoE4 allele from those expressing the human ApoE3 allele. Based on this definition, we concluded that lead interacts with ApoE4 because we observed statistically significant deficits in contextual fear memory and spontaneous alternation in lead-treated ApoE4-KI female mice but not in other animals. In addition, lead causes an earlier onset of spatial working memory deficits in the NOL test in lead-treated ApoE4-KI mice compared to ApoE3-KI mice, for both males and females. Together, these data suggest that the effect of lead exposure on cognitive behavior is different depending on ApoE genotype. More specifically, a GXE between lead and ApoE4 may accelerate cognitive decline, contribute to a more rapid progression from cognitive aging to mild cognitive impairment, and potentially contribute to AD. Furthermore, females may be more susceptible to this GXE.
We found that lead exposure decreased spontaneous alternation in the T-maze in female ApoE4-KI animals. While alternation is not strictly hippocampus-dependent, alternation performance is very sensitive to hippocampal lesions [44, 54, 60]. More importantly, impaired alternation and/or repetitive arm entry has been reported in several different AD-mouse models, including models with presenilin and amyloid precursor protein mutations [61–64].
Lead exposure also impaired hippocampus-dependent spatial memory in the NOL test in all lead-treated animals. Interestingly, lead-exposed ApoE3-KI male mice developed memory loss in the NOL test at 6 months post-lead exposure. Moreover, the memory loss in all lead-exposed animals was persistent through 10 months post-lead. These data suggest that sub-chronic adult lead exposure alone is sufficient to cause long lasting or irreversible memory impairment, and that this behavior phenotype can occur long after exposure has ended. These findings are pertinent to human health because irreversible loss of memory formation is characteristic of AD, senile dementia, and up to 50% of Parkinson’s disease cases [65]. Importantly, the aging population in the U.S. experienced high lead exposure in early life [66]. Thus, it is possible that lead exposure may lead to cognitive impairment later in life and contribute to cognitive decline associated with aging and neurodegeneration, such as late-onset AD.
Epidemiological reports suggest an increased risk of late-onset AD and increased cognitive decline with mild cognitive impairment in women [15, 16]. We observed that lead impaired contextual fear memory and working memory in the T-maze in female but not male ApoE4-KI mice. Furthermore, lead-induced spatial memory impairment in the NOL test manifested earlier in females than males for both ApoE3-KI and ApoE4-KI mice. These data provide strong evidence for sex differences in susceptibility to lead-induced cognitive decline in an animal model and suggest that women may be more susceptible to cognitive impairment and/or AD following exposure to toxicants such as lead.
We report average peak blood lead levels of 44.4–63.4 μg/dL, similar to previous animal studies and comparable to some occupational exposure settings in the 20th century [49, 67–69]. Elevated blood lead levels (≥10 μg/dL for adults) can still be encountered among U.S. workers, and Occupational Safety & Health Administration regulations do not require U.S. workers to be removed from their job sites until they have a blood lead level ≥ 50 μg/dL. [70]. Importantly, high levels of lead exposure from environmental sources and in occupational settings are still a major concern in developing countries [18, 71]. Therefore, 0.2% lead acetate serves as an experimental model for occupational exposures globally, for environmental exposure in developing countries, and for the US population who were exposed to high levels of environmental lead prior to the ban of leaded gasoline and paint.
Brain and blood lead levels were highest in female ApoE4-KI mice. There were no differences in water consumption during the first week but water consumption thereafter was not monitored. Although we cannot formally exclude the possibility that ApoE4-KI females drank more water than other animals after the first week of lead exposure, there was no statistically significant difference between blood lead levels of female ApoE3-KI and ApoE4-KI mice. Thus, it seems unlikely that higher water consumption alone led to more lead deposition in the brain of female ApoE4-KI.
It is possible that the ApoE4-KI enhances lead deposition in the brain and differences in brain lead disposition may contribute to the GXE between ApoE4 and lead. Interestingly, ApoE4-KI mice exhibit decreased basement membrane thickness, reduced collagen and tight junction protein expression, as well as increased matrix metalloproteinase 9 activity in the blood-brain barrier, suggesting that more lead may accumulate in the brain of ApoE4-KI mice due to reduced blood-brain barrier integrity [72–74]. Furthermore, human ApoE4 carriers with AD also exhibit a thinner basement membrane, increased fibrin extravasion, and pericyte loss in the blood-brain barrier compared to ApoE3-KI carriers with AD or non-AD controls [75, 76]. Thus, effects of E4 on the blood-brain barrier may increase the accumulation of lead in the brain in ApoE4-KI mice or maybe even in humans, and contribute to more severe or earlier onset of learning and memory impairments.
We previously found that lead can impair the survival, proliferation, and differentiation of adult neural precursor cells in vitro [41]. Thus, we sought to determine whether lead can interact with ApoE4 to impair adult hippocampal neurogenesis in vivo and whether this could be an underlying mechanism for the observed cognitive deficits. We exposed mice to lead starting at 8 weeks of age in order to avoid the cumulative effects of lead on both the developing and adult brain [77]. Unlike previous reports using the ApoE3-KI and ApoE4-KI models, we did not see a significant effect of ApoE4 alone on adult-born cell proliferation and maturation. This may be due to the fact that the effects of ApoE4 are age-dependent and we analyzed adult neurogenesis 1.5–2.5 months earlier (in 4.6-month-old vs. 6–7-month-old mice) than previous studies [12, 14, 40]. Data presented here demonstrate that adult lead exposure impairs adult-born cell proliferation in all lead-treated animals; this mechanism may contribute to lead impairment of spatial memory in the novel object location test that eventually develop in all lead-treated mice. Furthermore, lead exposure significantly impaired adult-born neuron differentiation and decreased dendritic complexity specifically in ApoE4-KI females. These cellular changes correlate with the fact that lead impairment of cognition is most pronounced in ApoE4-KI females. There was also a statistically significant reduction in the number of adult-born mature neurons (BrdU+NeuN+ cells) in male ApoE4-KI mice, although the reduction is smaller than that in females. This cellular change occurs at the end of the 12-week lead exposure and correlates with the onset of NOL deficits in the lead-treated ApoE4-KI males in the behavior cohort. Together, these data suggest that lead and ApoE4 may interact to perturb neuronal differentiation and maturation during adult hippocampal neurogenesis, and that this mechanism may contribute to the lead and ApoE4 interaction on accelerated cognitive impairment in ApoE4 mice and the heightened sensitivity of ApoE4 females.
The survival and integration of adult-born neurons is activity-dependent and occurs at approximately three weeks after neuronal birth [78, 79]. Several different cell types and inputs facilitate the survival and maturation of adult-born cells during this window, including GABAergic interneurons that may exert trophic effects on adult-born immature neuron survival and maturation [56, 80, 81]. Previous studies found that there are basal sex differences in the number of GAD67+ interneurons in ApoE3-KI and ApoE4-KI mice, with female mice having significantly more GAD67+ interneurons, and that female ApoE4-KI mice have significantly fewer GAD67+ cells than ApoE3-KI mice starting at 6–7 months of age [40]. We found that lead significantly decreased the total number of GAD67+ cells in 20-week-old, lead-treated ApoE4-KI females. This cellular effect correlates closely with our observation of significantly reduced adult-born neuron maturation and dendritic complexity in ApoE4-KI females, suggesting that an interaction between lead and ApoE4 may contribute to earlier impairment of GABAergic input and contribute to the observed impairment in immature neuron maturation in ApoE4-KI females.
Adult-born, immature neurons play a critical role in synaptic plasticity within the DG [79, 82, 83]. Notably, the increased responsiveness and excitability of newborn granule cells may give these adult-born cells a specific and significant role in learning and memory [55, 84]. In rats, a greater proportion of young, adult-born neurons are activated during spatial exploration compared to existing granule cells [85, 86]. Similarly, the selective loss of immature neurons impairs long-term spatial memory retention and contextual fear extinction [36]. Thus, the disruption of adult-born immature neuron maturation in lead-treated ApoE4-KI females may be associated with the impaired hippocampus-dependent learning and memory in these mice.
Although lead was deposited in the mouse brain broadly including the hippocampus and no regional specific deposition was found, we did not observe overt motor impairment. In humans, high levels of lead exposure can cause fine and gross motor impairments. However, lead exposure during development, which interferes with the proper development of the nervous system, likely plays a significant role in these cases. In the present study, mice were exposed to lead beginning at 8 weeks of age, eliminating the effect of lead on the developing nervous system. We only measured gross motor function (open field test), so we cannot rule out if lead exposure caused fine motor dysfunction. We did not observe a significant increase in TUNEL+ cells in the hippocampus or cortex of lead-treated animals. However, lead impaired adult neurogenesis in the hippocampus, with the greatest effect seen in female ApoE4-KI mice. Thus, newborn cells and neurons in the hippocampus, especially those in mice expressing human ApoE4 allele, may be more vulnerable than neurons in other regions of the brain to lead toxicity in the adult mice.
Similar to other studies that have reported basal sex differences in adult neurogenesis in these mice [14], we report baseline differences in the total number of adult-born cells (total BrdU+ cells), and the total number of GAD67+ cells in the DG between control males and females. However, we do not think these differences alter the conclusions of this study. For example, we found that all control mice performed equally well in tests for contextual fear memory, spontaneous alternation in the T-maze, and memory for novel object location (NOL). In addition, while ApoE3-KI and ApoE4-KI male mice had a lower number of BrdU+ and GAD67+ cells, they did not show statistically significant memory deficits after lead exposure when tested for contextual fear memory or spontaneous alternation in the T-maze. They also exhibited a later onset of deficits in the NOL test than their respective females of the same genotype. These data suggest that the lower basal adult neurogenesis in males does not affect their basal shot-term spatial and contextual memory, nor lead to higher sensitivity to lead impairment of cognition.
Conclusions
Although it has been hypothesized that exposure to environmental factors and a GXE interaction may increase AD risk, there is little direct evidence supporting this hypothesis. In this study, we found that adult lead exposure is sufficient to impair hippocampus-dependent learning and memory and that these deficits are more severe or occur earlier in female ApoE4-KI mice. These findings provide some of the first direct evidence for a GXE interaction between lead and ApoE4 on cognitive impairment. Furthermore, impaired adult hippocampal neurogenesis may be a potential mechanism underlying this GXE interaction. These results also provide new insight into how environmental toxicants and genetic risk factors may interact to contribute to cognitive decline and neurodegeneration in non-AD patients.
Acknowledgements
We thank Mike Paulsen, Dr. Christopher Simpson, and the Environmental Health Laboratory at the University of Washington for providing LA-ICP-MS services. We thank Dr. Daniel Storm and members of the Xia laboratory for critically reading the manuscript and scientific discussions.
Funding
This work was supported by the National Institutes of Health (R01 MH95840 and ES026591 to Z.X., T32ES015459 to A.E.).
Availability of data and material
The datasets generated during and/or analyzed during the current study are not publicly available due to the large amount of raw data generated for each of the behavior and cellular studies for all 8 treatment groups but are available from the corresponding author on reasonable request.
Authors’ contributions
A.E. and Z.X. designed the research; A.E. performed the behavior and cellular experiments; J.S. performed histology and pathology experiments; N.M. contributed unpublished reagents/analytic tools; A.E. and Z.X. analyzed data; A.E., J.S., and Z.X. wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The University of Washington Institutional Animal Care and Use Committee approved all animal protocols.
Abbreviations
- AD
Alzheimer’s disease
- ApoE3-KI and ApoE4-KI
Apolipoprotein E3 or E4 knock-in mouse model
- BrdU
5-bromo-2′-deoxyuridine
- DG
Dentate gyrus
- GXE
Gene-environment interaction
- H&E
Hematoxylin and eosin
- ICP-MS
Inductively coupled mass spectrometry
- NOL
Novel Object Location
Contributor Information
Anna K. Engstrom, Email: anna28@uw.edu
Jessica M. Snyder, Email: snyderjm@uw.edu
Nobuyo Maeda, Email: nobuyo@med.unc.edu.
Zhengui Xia, Phone: 1- 206 616 9433, Email: zxia@u.washington.edu.
References
- 1.Ott A, Breteler MM, van Harskamp F, Claus JJ, van der Cammen TJ, Grobbee DE, Hofman A. Prevalence of Alzheimer’s disease and vascular dementia: association with education. The Rotterdam study. BMJ. 1995;310(6985):970–973. doi: 10.1136/bmj.310.6985.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu JF, Basha R, Zawia NH. The environment, epigenetics and amyloidogenesis. Journal of Molecular Neuroscience. 2008;34(1):1–7. doi: 10.1007/s12031-007-0009-4. [DOI] [PubMed] [Google Scholar]
- 3.Gatz M, Fratiglioni L, Johansson B, Berg S, Mortimer JA, Reynolds CA, Fiske A, Pedersen NL. Complete ascertainment of dementia in the Swedish Twin Registry: the HARMONY study. Neurobiol Aging. 2005;26(4):439–447. doi: 10.1016/j.neurobiolaging.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 4.Raiha I, Kaprio J, Koskenvuo M, Rajala T, Sourander L. Alzheimer’s disease in twins. Biomed Pharmacother. 1997;51(3):101–104. doi: 10.1016/S0753-3322(97)86906-5. [DOI] [PubMed] [Google Scholar]
- 5.Ketelaar ME, Hofstra EM, Hayden MR. What monozygotic twins discordant for phenotype illustrate about mechanisms influencing genetic forms of neurodegeneration. Clin Genet. 2012;81(4):325–333. doi: 10.1111/j.1399-0004.2011.01795.x. [DOI] [PubMed] [Google Scholar]
- 6.Liu CC, Kanekiyo T, Xu HX, Bu GJ. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nature Reviews Neurology. 2013;9(2):106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mortensen EL, Hogh P. A gender difference in the association between APOE genotype and age-related cognitive decline. Neurology. 2001;57(1):89–95. doi: 10.1212/WNL.57.1.89. [DOI] [PubMed] [Google Scholar]
- 8.Cosentino S, Scarmeas N, Helzner E, Glymour MM, Brandt J, Albert M, Blacker D, Stern Y. APOE epsilon 4 allele predicts faster cognitive decline in mild Alzheimer disease. Neurology. 2008;70(19 Pt 2):1842–1849. doi: 10.1212/01.wnl.0000304038.37421.cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitehair DC, Sherzai A, Emond J, Raman R, Aisen PS, Petersen RC, Fleisher AS, Alzheimer’s Disease Cooperative S Influence of apolipoprotein E varepsilon4 on rates of cognitive and functional decline in mild cognitive impairment. Alzheimers Dement. 2010;6(5):412–419. doi: 10.1016/j.jalz.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beydoun MA, Boueiz A, Abougergi MS, Kitner-Triolo MH, Beydoun HA, Resnick SM, O’Brien R, Zonderman AB. Sex differences in the association of the apolipoprotein E epsilon 4 allele with incidence of dementia, cognitive impairment, and decline. Neurobiol Aging. 2012;33(4):720–731. doi: 10.1016/j.neurobiolaging.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blair CK, Folsom AR, Knopman DS, Bray MS, Mosley TH, Boerwinkle E, Atherosclerosis Risk in Communities Study I APOE genotype and cognitive decline in a middle-aged cohort. Neurology. 2005;64(2):268–276. doi: 10.1212/01.WNL.0000149643.91367.8A. [DOI] [PubMed] [Google Scholar]
- 12.Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen LG, Huang YD. Apolipoprotein E4 Causes Age- and Tau-Dependent Impairment of GABAergic Interneurons, Leading to Learning and Memory Deficits in Mice. Journal Of Neuroscience. 2010;30(41):13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bour A, Grootendorst J, Vogel E, Kelche C, Dodart JC, Bales K, Moreau PH, Sullivan PM, Mathis C. Middle-aged human apoE4 targeted-replacement mice show retention deficits on a wide range of spatial memory tasks. Behav Brain Res. 2008;193(2):174–182. doi: 10.1016/j.bbr.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Leung L, Andrews-Zwilling Y, Yoon SY, Jain S, Ring K, Dai J, Wang MM, Tong L, Walker D, Huang YD. Apolipoprotein E4 Causes Age- and Sex-Dependent Impairments of Hilar GABAergic Interneurons and Learning and Memory Deficits in Mice. Plos One. 2012;7(12):18. doi: 10.1371/journal.pone.0053569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleisher A, Grundman M, Jack CR, Jr, Petersen RC, Taylor C, Kim HT, Schiller DH, Bagwell V, Sencakova D, Weiner MF, et al. Sex, apolipoprotein E epsilon 4 status, and hippocampal volume in mild cognitive impairment. Arch Neurol. 2005;62(6):953–957. doi: 10.1001/archneur.62.6.953. [DOI] [PubMed] [Google Scholar]
- 16.Bretsky PM, Buckwalter JG, Seeman TE, Miller CA, Poirier J, Schellenberg GD, Finch CE, Henderson VW. Evidence for an interaction between apolipoprotein E genotype, gender, and Alzheimer disease. Alzheimer Dis Assoc Disord. 1999;13(4):216–221. doi: 10.1097/00002093-199910000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Cox DD, Gary; O; Haver, Robert; Salatino, Brendon: American Healthy Homes Survey: Lead and Arsenic Findings. In.: U.S. Department of Housing and Urban Development, Office of Healthy Homes and Lead Hazard Control; 2011.
- 18.Brosche S, Denney V, Weinberg J, Calonzo M, Withanage H, Clark S. IPEN. 2014. Asia Regional Paint Report; pp. 3–34. [Google Scholar]
- 19.Willmsen C, Kamb Lewis, Mayo J. Loaded with lead. In: Seattle Times. Seattle, WA: Seattle Times; 2014.
- 20.Rabin R. The lead industry and lead water pipes “A Modest Campaign”. Am J Public Health. 2008;98(9):1584–1592. doi: 10.2105/AJPH.2007.113555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005;25(4):823–829. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart WF, Schwartz BS, Davatzikos C, Shen D, Liu D, Wu X, Todd AC, Shi W, Bassett S, Youssem D. Past adult lead exposure is linked to neurodegeneration measured by brain MRI. Neurology. 2006;66(10):1476–1484. doi: 10.1212/01.wnl.0000216138.69777.15. [DOI] [PubMed] [Google Scholar]
- 23.Weisskopf MG, Wright RO, Schwartz J, Spiro A, 3rd, Sparrow D, Aro A, Hu H. Cumulative lead exposure and prospective change in cognition among elderly men: the VA Normative Aging Study. Am J Epidemiol. 2004;160(12):1184–1193. doi: 10.1093/aje/kwh333. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, et al. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 2008;28(1):3–9. doi: 10.1523/JNEUROSCI.4405-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bihaqi SW, Zawia NH. Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to lead (Pb) Neurotoxicology. 2013;39:95–101. doi: 10.1016/j.neuro.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart WF, Schwartz BS, Simon D, Kelsey K, Todd AC. ApoE genotype, past adult lead exposure, and neurobehavioral function. Environ Health Perspect. 2002;110(5):501–505. doi: 10.1289/ehp.02110501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buccione I, Perri R, Carlesimo GA, Fadda L, Serra L, Scalmana S, Caltagirone C. Cognitive and behavioural predictors of progression rates in Alzheimer’s disease. Eur J Neurol. 2007;14(4):440–446. doi: 10.1111/j.1468-1331.2007.01693.x. [DOI] [PubMed] [Google Scholar]
- 28.Johnson DK, Storandt M, Morris JC, Galvin JE. Longitudinal study of the transition from healthy aging to Alzheimer disease. Arch Neurol. 2009;66(10):1254–1259. doi: 10.1001/archneurol.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weintraub S, Wicklund AH, Salmon DP. The neuropsychological profile of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(4):a006171. doi: 10.1101/cshperspect.a006171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazarov O, Marr RA. Neurogenesis and Alzheimer’s disease: at the crossroads. Exp Neurol. 2010;223(2):267–281. doi: 10.1016/j.expneurol.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garthe A, Behr J, Kempermann G. Adult-generated hippocampal neurons allow the flexible use of spatially precise learning strategies. PLoS One. 2009;4:e5464. doi: 10.1371/journal.pone.0005464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan YW, Chan GCK, Kuo CT, Storm DR, Xia Z. Inhibition of Adult Neurogenesis by Inducible and Targeted Deletion of ERK5 Mitogen-Activated Protein Kinase Specifically in Adult Neurogenic Regions Impairs Contextual Fear Extinction and Remote Fear Memory. J Neurosci. 2012;32:6444–6455. doi: 10.1523/JNEUROSCI.6076-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang W, Pan YW, Zou J, Li T, Abel GM, Palmiter RD, Storm DR, Xia Z. Genetic Activation of ERK5 MAP Kinase Enhances Adult Neurogenesis and Extends Hippocampus-Dependent Long-Term Memory. J Neurosci. 2014;34(6):2130–2147. doi: 10.1523/JNEUROSCI.3324-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clelland CD, Choi M, Romberg C, Clemenson GD, Jr, Fragniere A, Tyers P, Jessberger S, Saksida LM, Barker RA, Gage FH, et al. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 2009;325(5937):210–213. doi: 10.1126/science.1173215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng W, Saxe MD, Gallina IS, Gage FH. Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci. 2009;29:13532–13542. doi: 10.1523/JNEUROSCI.3362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan YW, Storm DR, Xia Z. The maintenance of established remote contextual fear memory requires ERK5 MAP kinase and ongoing adult neurogenesis in the hippocampus. PLoS One. 2012;7(11):e50455. doi: 10.1371/journal.pone.0050455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan YW, Zou J, Wang W, Sakagami H, Garelick MG, Abel G, Kuo CT, Storm DR, Xia Z: Inducible and conditional deletion of extracellular signal-regulated kinase 5 disrupts adult hippocampal neurogenesis. J Biol Chem 2012, (in press). [DOI] [PMC free article] [PubMed]
- 39.Yang CP, Gilley JA, Zhang G, Kernie SG. ApoE is required for maintenance of the dentate gyrus neural progenitor pool. Development. 2011;138(20):4351–4362. doi: 10.1242/dev.065540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li G, Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Ring K, Halabisky B, Deng CH, Mahley RW, Huang YD. GABAergic Interneuron Dysfunction Impairs Hippocampal Neurogenesis in Adult Apolipoprotein E4 Knockin Mice. Cell Stem Cell. 2009;5(6):634–645. doi: 10.1016/j.stem.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engstrom A, Wang H, Xia Z. Lead decreases cell survival, proliferation, and neuronal differentiation of primary cultured adult neural precursor cells through activation of the JNK and p38 MAP kinases. Toxicol In Vitro. 2015;29(5):1146–1155. doi: 10.1016/j.tiv.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu PT, Schmechel D, Rothrock-Christian T, Burkhart DS, Qiu HL, Popko B, Sullivan P, Maeda N, Saunders AM, Roses AD, et al. Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol Dis. 1996;3(3):229–245. doi: 10.1006/nbdi.1996.0023. [DOI] [PubMed] [Google Scholar]
- 43.Shan Q, Chan GC, Storm DR. Type 1 adenylyl cyclase is essential for maintenance of remote contextual fear memory. J Neurosci. 2008;28(48):12864–12867. doi: 10.1523/JNEUROSCI.2413-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spowart-Manning L, van der Staay FJ. The T-maze continuous alternation task for assessing the effects of putative cognition enhancers in the mouse. Behav Brain Res. 2004;151(1–2):37–46. doi: 10.1016/j.bbr.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Sahay A, Scobie KN, Hill AS, O’Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, Hen R. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011;472:466–470. doi: 10.1038/nature09817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mabrouk A, Bel Hadj Salah I, Chaieb W, Ben Cheikh H. Protective effect of thymoquinone against lead-induced hepatic toxicity in rats. Environmental science and pollution research international. 2016;23(12):12206–12215. doi: 10.1007/s11356-016-6419-5. [DOI] [PubMed] [Google Scholar]
- 47.Bihaqi SW, Bahmani A, Subaiea GM, Zawia NH. Infantile exposure to lead and late-age cognitive decline: relevance to AD. Alzheimers Dement. 2014;10(2):187–195. doi: 10.1016/j.jalz.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masoud AM, Bihaqi SW, Machan JT, Zawia NH, Renehan WE. Early-Life Exposure to Lead (Pb) Alters the Expression of microRNA that Target Proteins Associated with Alzheimer’s Disease. J Alzheimers Dis. 2016;51(4):1257–64. doi: 10.3233/JAD-151018. [DOI] [PubMed] [Google Scholar]
- 49.Dosunmu R, Alashwal H, Zawia NH. Genome-wide expression and methylation profiling in the aged rodent brain due to early-life Pb exposure and its relevance to aging. Mechanisms of ageing and development. 2012;133(6):435–443. doi: 10.1016/j.mad.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patkova J, Vojtisek M, Tuma J, Vozeh F, Knotkova J, Santorova P, Wilhelm J. Evaluation of lipofuscin-like pigments as an index of lead-induced oxidative damage in the brain. Exp Toxicol Pathol. 2012;64(1–2):51–56. doi: 10.1016/j.etp.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Yu H, Li T, Cui Y, Liao Y, Wang G, Gao L, Zhao F, Jin Y. Effects of lead exposure on D-serine metabolism in the hippocampus of mice at the early developmental stages. Toxicology. 2014;325:189–199. doi: 10.1016/j.tox.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 52.Liu F, Xue Z, Li N, Huang H, Ying Y, Li J, Wang L, Li W. Effects of lead exposure on the expression of amyloid beta and phosphorylated tau proteins in the C57BL/6 mouse hippocampus at different life stages. Journal of trace elements in medicine and biology : organ of the Society for Minerals and Trace Elements. 2014;28(2):227–232. doi: 10.1016/j.jtemb.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 53.Walf AA, Frye CA. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc. 2007;2(2):322–328. doi: 10.1038/nprot.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerlai R. A new continuous alternation task in T-maze detects hippocampal dysfunction in mice. A strain comparison and lesion study. Behav Brain Res. 1998;95(1):91–101. doi: 10.1016/S0166-4328(97)00214-3. [DOI] [PubMed] [Google Scholar]
- 55.Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11:339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tozuka Y, Fukuda S, Namba T, Seki T, Hisatsune T. GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron. 2005;47:803–815. doi: 10.1016/j.neuron.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 57.Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11(2):111–128. doi: 10.31887/DCNS.2009.11.2/cqiu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones RN, Manly J, Glymour MM, Rentz DM, Jefferson AL, Stern Y. Conceptual and measurement challenges in research on cognitive reserve. J Int Neuropsychol Soc. 2011;17(4):593–601. doi: 10.1017/S1355617710001748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yassa MA, Lacy JW, Stark SM, Albert MS, Gallagher M, Stark CE. Pattern separation deficits associated with increased hippocampal CA3 and dentate gyrus activity in nondemented older adults. Hippocampus. 2011;21(9):968–979. doi: 10.1002/hipo.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dudchenko PA. An overview of the tasks used to test working memory in rodents. Neurosci Biobehav Rev. 2004;28(7):699–709. doi: 10.1016/j.neubiorev.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 61.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4(1):97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 62.Arendash GW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE, Diamond DM. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 2001;891(1–2):42–53. doi: 10.1016/S0006-8993(00)03186-3. [DOI] [PubMed] [Google Scholar]
- 63.Laursen B, Mork A, Plath N, Kristiansen U, Bastlund JF. Impaired hippocampal acetylcholine release parallels spatial memory deficits in Tg2576 mice subjected to basal forebrain cholinergic degeneration. Brain Res. 2014;1543:253–262. doi: 10.1016/j.brainres.2013.10.055. [DOI] [PubMed] [Google Scholar]
- 64.Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29(3):177–185. doi: 10.1023/A:1021691918517. [DOI] [PubMed] [Google Scholar]
- 65.Arnaldi D, Campus C, Ferrara M, Fama F, Picco A, De Carli F, Accardo J, Brugnolo A, Sambuceti G, Morbelli S, et al. What predicts cognitive decline in de novo Parkinson’s disease? Neurobiol Aging. 2012;33(6):1127. doi: 10.1016/j.neurobiolaging.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 66.ATSDR . Toxicological profile for lead. Atlanta, GA, US: U.S. Department of Health and Human Services, Public Health Serivce, Agency for Toxic Substances and Disease Registry; 2007. [Google Scholar]
- 67.Mantere P, Hanninen H, Hernberg S. Subclinical neurotoxic lead effects: two-year follow-up studies with psychological test methods. Neurobehav Toxicol Teratol. 1982;4(6):725–727. [PubMed] [Google Scholar]
- 68.Williamson AM, Teo RK. Neurobehavioural effects of occupational exposure to lead. British journal of industrial medicine. 1986;43(6):374–380. doi: 10.1136/oem.43.6.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lucchini R, Albini E, Cortesi I, Placidi D, Bergamaschi E, Traversa F, Alessio L. Assessment of neurobehavioral performance as a function of current and cumulative occupational lead exposure. Neurotoxicology. 2000;21(5):805–811. [PubMed] [Google Scholar]
- 70.OSHA: 2005d. Toxic and Hazardous Substances. Occupational safety and health standards. Washington, DC: Occupational Safety and Health Administration. Code of Federal Regulations. 29 CFR 1910.1025. In.; March 25, 2005.
- 71.Ilyas F. DAWNcom. 2014. City air heavily polluted with lead, cadmium, and CO. [Google Scholar]
- 72.Tai LM, Thomas R, Marottoli FM, Koster KP, Kanekiyo T, Morris AW, Bu G. The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 2016;131(5):709–23. doi: 10.1007/s00401-016-1547-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alata W, Ye Y, St-Amour I, Vandal M, Calon F. Human apolipoprotein E varepsilon4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J Cereb Blood Flow Metab. 2015;35(1):86–94. doi: 10.1038/jcbfm.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Salloway S, Gur T, Berzin T, Tavares R, Zipser B, Correia S, Hovanesian V, Fallon J, Kuo-Leblanc V, Glass D, et al. Effect of APOE genotype on microvascular basement membrane in Alzheimer’s disease. J Neurol Sci. 2002;203–204:183–187. doi: 10.1016/S0022-510X(02)00288-5. [DOI] [PubMed] [Google Scholar]
- 76.Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, Zlokovic BV. Accelerated pericyte degeneration and blood–brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. Journal of Cerebral Blood Flow and Metabolism. 2016;36(1):216–227. doi: 10.1038/jcbfm.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14(1):83–144. [PubMed] [Google Scholar]
- 78.Tashiro A, Sandler VM, Toni N, Zhao C, Gage FH. NMDA-receptor-mediated, cell-specific integration of new neurons in adult dentate gyrus. Nature. 2006;442:929–933. doi: 10.1038/nature05028. [DOI] [PubMed] [Google Scholar]
- 79.Marin-Burgin A, Schinder AF. Requirement of adult-born neurons for hippocampus-dependent learning. Behav Brain Res. 2012;227(2):391–399. doi: 10.1016/j.bbr.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 80.Ge S, Goh EL, Sailor KA, Kitabatake Y, Ming GL, Song H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;439:589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim WR, Christian K, Ming GL, Song H. Time-dependent involvement of adult-born dentate granule cells in behavior. Behav Brain Res. 2012;227(2):470–479. doi: 10.1016/j.bbr.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Snyder JS, Kee N, Wojtowicz JM. Effects of adult neurogenesis on synaptic plasticity in the rat dentate gyrus. J Neurophysiol. 2001;85:2423–2431. doi: 10.1152/jn.2001.85.6.2423. [DOI] [PubMed] [Google Scholar]
- 83.Esposito MS, Piatti VC, Laplagne DA, Morgenstern NA, Ferrari CC, Pitossi FJ, Schinder AF. Neuronal differentiation in the adult hippocampus recapitulates embryonic development. J Neurosci. 2005;25:10074–10086. doi: 10.1523/JNEUROSCI.3114-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pan YW, Storm DR, Xia Z. Role of adult neurogenesis in hippocampus-dependent memory, contextual fear extinction and remote contextual memory: new insights from ERK5 MAP kinase. Neurobiol Learn Mem. 2013;105:81–92. doi: 10.1016/j.nlm.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ramirez-Amaya V, Marrone DF, Gage FH, Worley PF, Barnes CA. Integration of new neurons into functional neural networks. J Neurosci. 2006;26:12237–12241. doi: 10.1523/JNEUROSCI.2195-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sandoval CJ, Martinez-Claros M, Bello-Medina PC, Perez O, Ramirez-Amaya V. When are new hippocampal neurons, born in the adult brain, integrated into the network that processes spatial information? PLoS One. 2011;6(3):e17689. doi: 10.1371/journal.pone.0017689. [DOI] [PMC free article] [PubMed] [Google Scholar]