

Abstract
Oxidative stress plays a key role in the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Glutathione is the major anti-oxidant involved in cellular oxidative defense, however there are currently no simple non-invasive methods for assessing hepatic glutathione metabolism in patients with NAFLD. As a primary source of plasma glutathione, liver plays an important role in interorgan glutathione homeostasis. In this study, we have tested the hypothesis that measurements of plasma glutathione turnover could be used to assess the hepatic glutathione metabolism in LDLR−/− mice, a mouse model of diet-induced NAFLD. Mice were fed a standard low fat diet (LFD) or a high fat diet containing cholesterol (a Western type diet (WD)). The kinetics of hepatic and plasma glutathione were quantified using the 2H2O metabolic labeling approach. Our results show that a WD leads to reduced fractional synthesis rates (FSR) of hepatic (25%/h in LFD vs. 18%/h in WD, P < 0.05) and plasma glutathione (43%/h in LFD vs. 21%/h in WD, P <0.05), without any significant effect on their absolute production rates (PRs). WD-induced concordant changes in both hepatic and plasma glutathione turnover suggest that the plasma glutathione turnover measurements could be used to assess hepatic glutathione metabolism. The safety, simplicity, and low cost of the 2H2O-based glutathione turnover approach suggest that this method has the potential for non-invasive probing of hepatic glutathione metabolism in patients with NAFLD and other diseases.
Keywords: Glutathione, Oxidative stress, Heavy water, Flux, Mass spectrometer, Western Diet, NAFLD, NASH
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease that ranges from benign steatosis (triglyceride accumulation in the hepatocytes) to nonalcoholic steatohepatitis (NASH), fibrosis, cirrhosis and can develop to hepatocarcinoma [1]. Insulin resistance and obesity are the most common features of NAFLD. The loss of insulin's ability to control adipose tissue lipolysis combined with the increased hepatic de novo lipogenesis stimulates triglyceride synthesis in the liver that leads to hepatic steatosis, the early stage of NAFLD. Excessive hepatic fatty acid load in NAFLD can also results in increased mitochondrial, peroxisomal and microsomal hepatic fatty acid oxidation, all associated with the generation of reactive oxygen species (ROS) [2–4]. According to the current paradigm, hepatic oxidative stress is considered a “second hit” which initiates inflammatory process involved in the disease progression from steatosis to NASH [5–8]. Recently an elegant approach utilizing non-invasive nuclear magnetic resonance (NMR) in combination with [2-13C]glycine administration was applied to measure glutathione synthesis in human and rat livers as an oxidative stress biomarker [9]. However, the high cost limits the wide-range application of this method. Thus, clearly a simple and robust non-invasive method for non-invasive assessment of hepatic glutathione metabolism is needed.
Glutathione, γ-glutamylcysteinylglycine, is the major cellular anti-oxidant that efficiently quenches ROS, detoxifies reactive electrophile products of lipid peroxidation, and maintains the thiol status of proteins [10]. Glutathione also provides a reservoir to cysteine and regulates critical cellular processes as DNA synthesis and immune function via redox sensitive factors [11]. The spontaneous or glutathione peroxidase and/or glutathione S-transferase catalyzed reactions of glutathione with ROS, including hydroperoxides (ROOH), convert reduced glutathione (GSH) to its oxidized form (GSSG). To maintain the cellular redox status, in the physiological conditions GSSG is reduced back to GSH by glutathione reductase that utilizes NADPH as a reducing co-factor. However, in severe oxidative stress, when the ability of glutathione reductase is overwhelmed and/or NADPH is depleted, the GSSG accumulation leads to a reduced redox defense. The cellular oxidative stress can be further exacerbated via thiol-disulfide exchange reactions between GSSG and thiol-proteins that results in the formation of mixed protein-glutathione disulfides (GSSR, where R represents a thiol-protein). To support the thiol status of proteins, GSH is also consumed in thiol-disulfide exchange reactions catalyzed by thiol-transferase that produces additional GSSG. To prevent a shift in the redox equilibrium and to limit GSSR formation, GSSG is actively transported out of the cells [10,12].
With the unique ability of the hepatocytes to produce cysteine (the rate limiting substrate for GSH synthesis) through the transsulfuration pathway, the liver plays the central role in total body glutathione homeostasis [13]. In normal conditions, the plasma glutathione levels are essentially determined by the sinusoidal efflux of hepatic glutathione [12,14,15]. Indeed, in vivo measurements of glutathione efflux in fasted rats, based on hepatic arteriovenous concentration gradients, documented that the liver contributes to over 90% of the total glutathione secretion into circulation and therefore the rate of hepatic glutathione synthesis is balanced by its export into circulation [12]. This may have particular relevance to hepatic oxidative stress in NAFLD, because the alterations in plasma glutathione metabolism could be used as a surrogate of hepatic oxidative stress. However, the systematic oxidative stress results in immediate GSH trapping in the circulation [16,17] which precludes the utilization of plasma GSH/GSSG redox ratio as marker of hepatic oxidative stress.
Herein we have considered an alternative approach for the assessment of the hepatic glutathione metabolism and oxidative stress, namely, measurement of hepatic and/or plasma glutathione flux. Glutathione turnover has been successfully studied using radioactive and stable isotope labeled glutamate, cysteine or glycine [1,18,19]. These methods require a large amount of an expensive tracer, and in the case of stable isotopes, the infusion of labeled amino acids elevates amino acid levels and may disturb normal glutathione metabolism. For instance, cysteine availability regulates glutathione synthesis [10] and therefore the utilization of the cysteine tracer may artificially overestimate glutathione synthesis due to the tracer-induced increase in intracellular cysteine levels. We and others have developed the 2H2O-metabolic labeling approach to study lipid and protein kinetics in free living organisms [20–27]. 2H2O also has been used to assess the turnover rates of glutathione in animal models and humans [28,29]. Here we applied 2H2O-metabolic labeling approach along with the static measures of the glutathione redox ratio, to evaluate the effect of high fat diet containing cholesterol (also known as a Western type diet (WD)), on hepatic glutathione metabolism in low density lipoprotein receptor deficient (LDLR−/−) mice, a diet-induced mouse model of NAFLD. We aimed to determine whether the WD-induced alterations in hepatic glutathione metabolism in LDLR−/− mice would be associated with the changes in plasma glutathione flux.
2. Material and methods
2.1. Reagents
Standards of the tri-peptide Ala-Ala-Ala were purchased from Bachem Americas, Inc, (Torrance, CA). Common solvents and chemicals, including GSH, GSSG, dithiothreitol (DTT), l-cysteine, l-[2H3]cysteine, and iodoacetamide were purchased from Sigma-Aldrich (St. Louis, MO).
2.2. Animal studies
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the Cleveland Clinic and were performed in accordance to the NIH guidelines. Eight- to ten-week old male LDLR−/− mice were housed in pairs in our animal care facility with a 12:12 h light: dark cycle and had free access to food and water. Mice were randomized into two groups and fed an additional 12 weeks with a standard low fat diet (LFD, 20% kcal protein, 70% kcal carbohydrate and 10% kcal fat, Harlan Teklad), or a high saturated fat and cholesterol-containing diet (WD, TD88137, Harlan Teklad, Madison, WI, 45% kcal from fat, containing 21% of fat-saturated fat/total fat ratio = 0.64 and 0.2% cholesterol).
2.2.1. Glutathione turnover experiments
Glutathione turnover was measured using the 2H2O-metabolic labeling approach (Fig. 1A). The rational is that a bolus load of 2H2O rapidly equilibrates with total body water and amino acids. Thus, the time course 2H-labeling of glutathione represents its turnover rate (Fig. 1B). To obtain the baseline samples, three mice from each group were euthanized (pentabarbital, 50 mg/kg, intraperitoneal injection (i.p.)) and blood and liver samples were collected. The remaining mice (15 mice/group) received a loading dose of 2H2O saline solution (22 μL/g body weight by i.p.) and were given drinking water enriched with 2H2O (5%). Three mice from each group were euthanized (pentabarbital, 50 mg/kg, i.p.) after 4, 8, 24, and 72 h of 2H2O administration. Blood samples were collected through cardiac puncture. To avoid red blood cells hemolysis, blood was gently sampled using a syringe. Plasma was immediately isolated by centrifugation at 4 °C and was used for the analysis of 2H-labeling of the total body water and glutathione, as described below. Only plasma samples free of red blood cells hemolysis were used for the analyses. The heart was perfused with phosphate buffered saline (PBS) to wash the remaining blood. To ensure a complete washout of erythrocytes from hepatic vascular space, the PBS infusion (∼10–20 ml) continued until the liver tissue changes its color from red to light brown. At this point, the liver was removed, immediately frozen in liquid nitrogen and saved at −80 °C until further analyses. This approach eliminates the contamination of hepatic glutathione with the red blood cell-derived glutathione.
Fig. 1.

Metabolic labeling of glutathione in mice with 2H2O. Panel A: Study design. Eight to ten week old LDLR−/− mice (18 mice/group) were fed either a standard low fat diet (LFD) or a high fat diet containing cholesterol (a Western type diet (WD)) for 12 weeks. After euthanizing 3 mice from each group, the remaining animals were loaded with bolus injection of 2H2O (22 μL/g body weight) and exposed to 5% 2H2O in their drinking water for different duration. Three mice from each group were euthanized at 4, 8, 24 and 72 h and blood and liver tissue were collected for future assays. Panel B: 2H2O rapidly equilibrates with total body water and 2H incorporates into amino acids including glutamic acid, cysteine, and glycine. The first step in glutathione synthesis involves glutamic acid and cysteine with the production of γ-glutamylcysteine dipeptide via γ-glutamylcysteine synthetase (GCS). In the second step glutathione synthase (GS) catalyzes coupling of γ-glutamylcysteine with glycine. 2H-glutamic acid, 2H-cysteine and 2H-glycine along with their unlabeled analogs are incorporated into glutathione.
2.3. Analytical methods
2.3.1. Total body water enrichment
The [2H]-enrichment of total body water was measured using a modification [30] of the acetone exchange method [31]. Briefly, 5 μL of plasma parallel with the calibration curve samples was incubated with 2 μL of 10 N KOH, and 5 μL of pure acetone in a 2 ml glass screw-cap GC vial at room temperature for 4 h. The acetone from headspace was directly injected (1 μL) for gas chromatography–mass spectrometry GC–MS analysis in electron impact mode. Acetone was monitored at ions m/z 58 (M0), 59 (M1) and 60 (M2). The regression equation from the calibration curve was used to calculate the 2H-enrichment of total body water.
2.3.2. Sample preparation for hepatic glutathione analysis
2.3.2.1. Total glutathione (GSH+GSSR)
Liver samples (50 mg) were homogenized in 500 μL deionized water on ice using a Tissue Tearor (model 985370, Biospec Products Inc. Bartlesville, OK). To isolate insoluble particles, the sample was centrifuged at 14,000g for 15 min at 4 °C. The supernatant (100 μL) corresponding to 10 mg liver tissue was spiked with 10 nmol (10 μL of 1 mM) Ala-Ala-Ala, an internal standard. The sample was reduced with 10 μL of 0.2 M DTT solution at room temperature for 30 min. The reduced thiols were subsequently alkylated with 10 μL of 0.4 M iodoacetamide solution at room temperature in the dark for 30 min. To precipitate proteins, the sample was treated with cold acetone (750 μL, −20 °C) and further incubated at −20 °C for 2 h. After centrifugation, the supernatant was removed and evaporated under nitrogen gas to dryness and the residue was reconstituted into 30 μL of 0.1% formic acid. Hepatic glutathione content was quantified using regression equation derived from glutathione calibration curve which was constructed using 10 μL of glutathione solutions with the concentrations of 0.5 μM, 5 μM, 10 μM, 20 μM and a constant amount (10 pmol) of the internal standard Ala-Ala-Ala. The standard solution samples were reduced and alkylated using the same procedure as described above for the biological samples.
2.3.2.2. Reduced glutathione (GSH)
Sample preparation for the analysis of reduced glutathione was similar to those described for the total glutathione assay, except that samples were immediately alkylated with iodoacetamide without reduction with DTT.
2.3.3. Sample preparation for plasma glutathione analysis
Since the reduced glutathione was plasma was rapidly oxidized, the kinetic analyses were performed based on total glutathione assay. Two nmole Ala-Ala-Ala (an internal standard) was added into 50 μL plasma sample. The sample was reduced with DTT (50 μL of 0.2 M) and alkylated with iodoacetamide (50 μL of 0.4 M) as described above. After the precipitation of plasma proteins, the supernatant containing glutathione was concentrated using ion-exchange column chromatography (AG 50W-X8 resin, hydrogen form). The column was first washed with water (5 ml), and then glutathione was eluted with the 4 N ammonium hydroxide solution in water (3 ml). The eluent was dried and the residue was reconstituted in 20 μL 0.1% formic acid prior to analysis.
2.3.4. Glutathione assay by mass spectrometry
Glutathione was analyzed by a nano-flow HPLC coupled with electrospray ionization ion trap tandem mass spectrometry (LC–MS/MS). Ten μL of glutathione sample solutions were loaded onto a reversed phase nano-column (C18, 75 μm × 100 mm, 10 μm, Newobjective). Chromatographic separation was performed by an Eksigent NanoLC-1D Plus HPLC (Eksigent Technologies Inc.) using a mobile phase A (0.1% formic acid in water) and B (0.1% formic acid in 95% acetonitrile). A 15-min gradient started with 100% of phase A. After 2 min, phase B was increased to 50% in 3 min and then kept at 50% for 2 min. Subsequently, phase B was decreased to 0% in 1 min and the column was equilibrated for 7 min. Tandem mass spectra were recorded on a Finnigan LTQ (Thermo Electron Corp., Bremen, Germany) operated in a positive ion mode. The peptides were introduced to ion source at a flow rate of 350 nL/min via a non-coated PicoFrit emitter (FS360-75-15-N, New Objective Inc., Woburn, MA) at 2.5 kV. The inlet capillary temperature was maintained at 175 °C. Each full MS scan spectrum between 150 and 750 (m/z) was followed by a 2 zoom scans of 2 single reaction monitoring (SRM) experiments. The first SRM scan targeted on the alkylated glutathione precursor 365.2 (m/z) with the transition 365.2→236.2, and the zoom scan ranged from 234 to 244 (m/z). To include all isotopomers of the intact glutathione molecule, the precursor ion 365.2 (m/z) was selected with a 5 Da isolation window (364.5–369.5 m/z). This covers all isotopomers, including M0, M1, M2, and M3 (> 99.9% of overall isotopic distribution). To improve the statistics of spectral accuracy, average of ∼200 spectra were acquired along the glutathione elution peak. The second SRM scan targeted the internal standard Ala-Ala-Ala precursor 232.2 (m/z) with the transition 232.2→143.2 using a 5 Da precursor isolation window (231.5–236.5 m/z) and the zoom scan of product ion ranged from 140 to 150 (m/z).
2.3.5. Cysteine assay by mass spectrometry
Briefly, 100 mg frozen liver sample was spiked with 10 nmol [2H3]cysteine and homogenized in 200 μL of water solution of 75 mM DTT. After centrifugation, the supernatant was isolated and proteins were precipitated with 700 μL of cold acetone (−20°C, 1 h). The supernatant was isolated and dried in the speedvac. The sample was reconstituted in 50 μL of 50% acetonitrile water solution with 0.1% trifluoroacetic acid. The sample was separated on a HILIC Silica column (2.1 × 150 mm, 5 μm, Waters Atlantis) using mobile phase A (99.8% water, 0.2% acetic acid) and B (99.8% acetonitrile, 0.2% acetic acid) with a 0.3 ml/min flow rate and a 30-min gradient. After 2 min conditioning of the column with 95% mobile phase B, B was reduced to 50% in 8 min and kept at 50% for 4 min. Subsequently solvent B was reduced to 5% in 4 min and kept at 5% for 5 min. Finally, solvent B rose from 5% to 95% in 0.5 min and the column was equilibrated at 95% B for 6.5 min. The cysteine was analyzed with the precursor and product ions transition 122→76 m/z (collision energy 15.0) for endogenous cysteine, and 125→79 m/z (collision energy 15.0) for [2H3]cysteine, respectively. In addition, the transitions 122→59 m/z (collision energy 20.0) and 125→62 m/z (collision energy 20.0) with further loss of NH3 were monitored for cysteine and [2H3]cysteine. To confirm that all cysteine molecules were in the reduced state, another pair of transitions for the oxidized (dimerized) cysteine 241→152 m/z (collision energy 15.0) and [2H3]cysteine 247→158 m/z (collision energy 15.0) were also monitored. Other mass spectrometer instrument parameters were as follows: Capillary (kV) 3.00, Cone (V) 30, Source Temperature (°C) 150, Desolvation Temperature (°C) 300, Cone Gas Flow (L/h) 150, Desolvation Gas Flow (L/h) 757, Multiplier (V) 500, and Collision Cell Pressure (mbar) 2.79e−3.
2.3.6. Glutathione–4-hydroxynonenal (GSH–4-HNE) assay by mass spectrometry
GSH–4-HNE was analyzed as described [32]. Briefly, powder-frozen liver samples (∼100mg) spiked with 0.1 nmol of GSH-[2H11]–4-HNE conjugate as an internal standard, were homogenized in 2 ml of 100 mM iodoacetamide and 2 mM butylated hydroxytoluene (BHT) in 10 mM ammonium bicarbonate buffer, pH 9.8, together with 4 ml of acetonitrile and 2 ml of chloroform. Then the supernatant was completely dried after centrifugation, and the residue was reconstituted in 100 μL of Milli-Q water prior to LC–MS/MS analysis. The precursor and product ions for GSH–4-HNE and GSH-[2H11]–4-HNE with transitions m/z 464→308, and 475→308, respectively, were monitored.
2.3.7. 4-HNE assay by mass spectrometry
4-HNE was analyzed as previously described [33]. Briefly, after spiking samples with custom made [2H11]–4-HNE (internal standard), the aldehyde group was reduced to the hydroxyl group with sodium borodeuteride. Samples were derivatized with trimethylsilyl reagent and analyzed using GC–MS in electron impact mode.
2.3.8. Total lipid peroxidation assay
Lipid peroxidation was determined by measuring thiobarbituric acid reactive substances (TBARS) including malonyl dialdehyde (MDA) using the commercial kit provided by Cayman Chemical Co. In brief, 100 μL of homogenate (25 mg liver in 250 μL of RIPA buffer) was mixed with 100 μL of 10% tricholoroacetic acid, and mixed with 1.33 mL of a color reagent which was composed of 37 mM barbituric acid solubilized in 1.75 M acetic acid and 0.35 M sodium hydroxide. The mixture was incubated at 100 °C for 1 h and measured spectrophotometrically at 535 nm.
2.4. Calculations
The time course 2H enrichment of glutathione was used to calculate its turnover rate constant. The fractional synthesis rate (FSR) of glutathione was determined based on a single compartmental model by fitting a time course of total labeling of a peptide (Epeptide (t))to an exponential rise curve equation:
| (1) |
where Ess and k represent the plateau labeling and the FSR of glutathione, respectively.
Total labeling of glutathione was calculated as the weighted sum of the molar percent enrichment (MPE) of individual mass isotopomers (Mi), with i = 0,…, n, as previously described [22]:
| (2) |
The molar percent enrichment (MPE) of individual mass isotopomers was calculated based on the areas of unlabeled (monoisotopic) and labeled isotopomers using the following formula:
| (3) |
where AMi represents the area under the curve for ith isotopomer.
Although our approach does not require the background correction for natural enrichment, the isotopic excess (net labeling) due to 2H incorporation was calculated as the difference of total MPE and the baseline MPE calculated for the control mice.
| (4) |
The production rates (PR) of glutathione was calculated as the product of FSR and glutathione levels:
| (5) |
where the hepatic PR is expressed in μmolgww−1 h−1 and the hepatic glutathione levels represents μmol of total glutathione per g wet liver weight (gww). The PR and plasma glutathione levels are expressed in μmol L−1 h−1 and μmolL−1 (μM), respectively. Calculation of the FSR values based on multiple animals precluded us normalization of the data based on the total pool size. Therefore, this expression of PR does not account for the differences in body weights of mice in LFD and WD groups.
2.5. Data Presentation and Statistics
The statistical significances of the differences of reduced, oxidized and total glutathione levels and production rates between the LFD and a WD experiments were tested using two-tailed t-test with equal variance. Data shown in time course figures are the excess labeling of glutathione. To account for the variabilities in the total body water labeling, the data was normalized for the body water 2H-enrichment in each mouse and the asymptotic labeling of each curve. Each symbol in the figures corresponds to three mice experiments. An exponential transformation was used to model 2H-enrichment of glutathione as the outcome with time, diet and the time-diet interaction as the independent variables. For this purpose, normalized GSH turnover data was fit to an exponential 3P growth model. Differences in parameters were assessed by evaluating their ratio and 95% confidence interval. All analyses were performed using statistic software JMP (version 10.0.0, SAS Institute, Cary, NC).
3. Results
Recently, we have shown that LDLR−/− mice on a WD develop obesity, dyslipidemia (increased plasma triglycerides and LDL-cholesterol, and reduced HDL-cholesterol), insulin resistance, fatty liver, hepatic inflammation and mild fibrosis-all markers of NAFLD [34]. In this study, we have explored the utility of plasma glutathione turnover measurements as a proxy of hepatic glutathione metabolism related to oxidative stress. Hepatic and plasma glutathione turnovers were estimated using the 2H2O-metabolic labeling approach. Prior to administering 2H2O, mice consumed ∼2.5–3 g food per day and exhibited normal growth. No adverse effect(s) were observed during 2H2O treatment (mice consumed the same amount of food and no weight loss was observed during the 3 day labeling experiment).
To characterize the mass spectrometric signature of glutathione, the baseline samples (t = 0, no 2H2O treated) were analyzed by an ion-trap LTQ system using MS/MS scans. The alkylated glutathione is eluted at 4.2 min with good chromatographic properties free of interfering peaks (Fig. 2A). The molecular formula of alkylated (with iodoacetamide) glutathione (GluCys(carbamidomethyl)Gly) was confirmed by the presence of an ion with m/z 365.17 [M+H]+ corresponding to protonated molecular ion (Fig. 2B). Collision-indiced dissociation of molecular ion in MS/MS scan yields dipeptide fragment ions with m/z of 236.03 and 290.18 corresponding to Cys(carbamidomethyl)Gly and GluCys(carbamidomethyl), respectively (Fig. 2D). In addition, an ion with m/z of 347.06 corresponding to neutral loss of water molecule from molecular ion also was present in the MS/MS spectrum. We tuned and validated the utility of the LTQ mass spectrometer for the optimal measurement of isotopic ratios of glutathione. The isotopic distribution analysis of glutathione's molecular ion in the zoom scan revealed that the interfering ions from other species contaminate the isotopic envelope (Fig. 2C). This precluded us from using the molecular ion when quantifying the 2H-enrichment. In contrast, the baseline isotope distribution of the alkylated glutathione fragment (Cys(carbamidomethyl)Gly) ion (Fig. 2E, first panel), was similar to the theoretical values calculated using web-based Protein Prospector application (http://prospector.ucsf.edu/prospector/mshome.htm). To evaluate the spectral accuracy and reproducibility of isotopic ratio measurements in LTQ mass spectrometer, the isotopic distribution of glutathione standard was analyzed in replicates (n = 5) and compared with the theoretical values. The isotope distribution of this fragment ion was very consistent with the coefficient variation of < 1% (Supplementary Table 1A). There were slightly higher errors in the isotope distribution measurements, when we analyzed the baseline glutathione samples (t = 0 h) from mice liver and plasma (Supplementary Table 1B). Still, the biological variability between samples was < 1%. Furthermore, when we analyzed the time course labeling of reduced (GSH) and total glutathione (GSH+GSSR) from the same liver samples, we observed very small variability between different samples at the same time points (less than 2% error) and the error did not vary according to either enrichment levels or time (Supplementary Table 2). Therefore, we used this fragment ion to quantify the 2H enrichment of glutathione in biological samples. Although the Cys(carbamidomethyl)Gly fragment incorporates less 2H than the molecular ion of glutathione and lower 2H enrichment may potentially affect the accuracy of measurements, in this study we found that Cys(carbamidomethyl)Gly already has ∼6% 2H-enrichment after 4 h of 2H2O administration. Fig. 2E shows the expected (albeit gradual) increase in normalized 2H enrichment of the fragment ion Cys (carbamidomethyl)Gly during 8 h of 2H2O treatment as reflected in the relative increase of M1 and M2 isotopomers compared to monoisotopic M0 isotopomer.
Fig. 2.
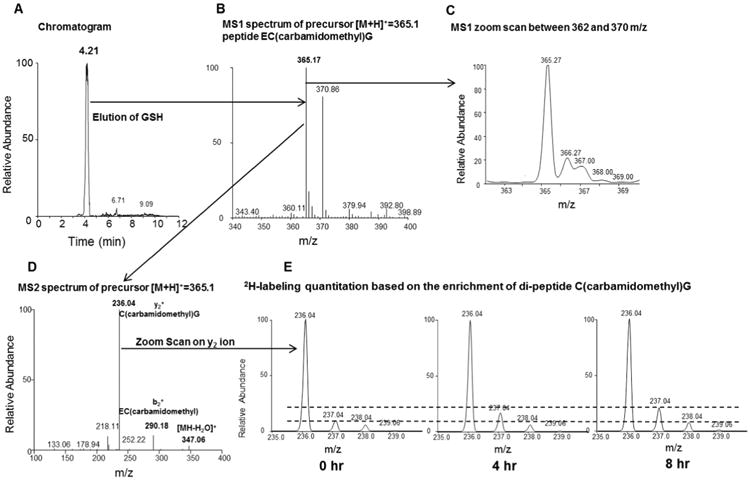
Glutathione analysis by LC–MS/MS. Panel A: Carbamidomethylated glutathione derivative is eluted at 4.2 min. Panel B: MS1 spectrum of precursor ion of carbamidomethylated glutathione with m/z 365.15 in positive ion mode. Panel C: MS1 zoom scan of precursor ion monitored between 362 m/z and 370 m/z. Panel D: Collision-indiced dissociation of derivatized glutathione yields abundant cysteine(carbamidomethyl)glycine ion at m/z 236.04 along with other fragment ions. Panel E: The zoom scanned spectra of cysteine(cabamidomethyl)glycine fragment ion before and after (4 h and 8 h) of 2H2O treatment. The dashed lines are added to aid in visualizing differences in the isotopic abundance.
To assess the effect of WD on hepatic oxidative stress based on the glutathione redox ratio, we quantified the hepatic levels of reduced and total glutathione. The calibration curve of glutathione concentration (Supplementary Fig. 1) is linear (r2 = 0.996) over a large range of glutathione abundance with the detection limit of 5 pmol. We used the regression equation of this curve for the quantification of reduced and total glutathione. AWD significantly increased the total glutathione levels in both plasma (16.98 ± 6.77 μmol/gww in LFD group vs. 40.41 ± 9.07 in WD group, P < 0.05, Table 1) and the liver (2.95 ± 0.35 μmol/gww in LFD group vs. 3.62 μ 0.67 in WD group, P < 0.05, Table 1). Although a WD led to a slight decrease in hepatic reduced glutathione the changes were not significant (Fig. 3A). Thus, the WD-induced changes in total hepatic glutathione were related to the increased content of oxidized and DTT-reducible forms of glutathione (GSSG+GSSR) (Fig. 3B), estimated as the difference of total and reduced glutathione (0.44 ± 0.13 μmol/gww in LFD vs. 1.12 ± 0.44 μmol/gww in WD, P < 0.05). This was also reflected in the glutathione redox ratio, i.e. a WD led to more than a 2-fold decrease in GSH/GSSR ratio (P < 0.05). Since a WD did not lead to any significant changes in hepatic reduced glutathione levels, to assess the hepatic oxidative stress we quantified lipid peroxidation by measuring thiobarbituric acid reactive substances (TBARS) including malonyl dialdehyde (MDA) by calorimetric assay and specifically 4-HNE by mass spectrometry as other markers of oxidative stress. We did not observe any differences in neither TBARS nor 4-HNE between LFD and WD groups (Supplementary Fig. 2A and B), indicating that our mouse model did not exhibit any significant changes in lipid peroxidation that is characteristic to severe oxidative stress.
Table 1.
Effect of a Western diet (WD) on hepatic and plasma glutathione turnover in LDLR−/− mice. The production rates (PR) of total glutathione in the liver and plasma were calculated based on respective total glutathione levels and turnover rate constants. Data are presented as means ± SD. The content of total glutathione (GSH+DTT-reducible forms of GSH) are expressed in μmol equivalents of GSH.
| Glutathione | LFD | WD | P | |
|---|---|---|---|---|
| Levels | Liver (μmol/gww) | 2.95 ± 0.35 | 3.62 ± 0.67* | <0.05 |
| Plasma (μM) | 16.98 ± 6.77 | 40.41 ± 9.07* | <0.05 | |
| FSR | Liver (pool/h) | 0.25 ± 0.04 | 0.18 ± .03* | <0.05 |
| Plasma (pool/h) | 0.43 ± 0.08 | 0.21 ± 0.02* | <0.05 | |
| PR | Liver (μmol gww−1 h−1) | 0.73 ± 0.14 | 0.68 ± 0.13 | NS |
| Plasma (μmol L−1 h−1) | 6.95 ± 2.13 | 8.38 ± 1.75 | NS |
NS: not significant. *P < 0.05.
Fig. 3.

Effect of a Western diet on hepatic glutathione redox ratio in LDLR−/− mice. Hepatic content of reduced glutathione (GSH) (Panel A), total glutathione (GSH+GSSGR) (presented in Table 1) was analyzed by mass spectrometry as described in Section 2. Hepatic oxidized glutathione (GSSR) content (Panel B) was calculated as the difference of total and reduced glutathione. Glutathione redox ratio (GSH/GSSR) in the liver (Panel C) was calculated based on hepatic content of reduced and oxidized glutathione. Data are presented as mean ± SD (n = 6/group). *P < 0.05.
Next, we applied the 2H2O-metabolic labeling approach to assess the hepatic glutathione turnover in LDLR−/− mice. Bolus loading of pure 2H2O followed by the administration of 5% 2H2O in the drinking water results in a steady-state body water labeling ∼3.0–3.5% (Supplementary Fig. 3). In our orientation experiment, we compared the hepatic turnover rates of reduced and total (reduced+oxidized) glutathione in mice fed a LFD. A single compartmental model of the time course labeling of both the reduced and the total glutathione revealed that the turnover rate constants were almost identical (Supplementary Fig. 4). Consistent with a previous report [29], these results suggest that reduced and oxidized glutathione pools equilibrate rapidly and have similar turnover rates. Therefore, in the following experiments the effect of a WD on hepatic glutathione metabolism was assessed based on the total glutathione turnover measured using 3 mice at each time point. The increased pool size of total glutathione in a WD group was associated with a significant reduction of glutathione FSR in this group (0.25 ± 0.04 in LFD vs. 0.18 ± 0.03, P < 0.05) (Fig. 4A, Table 1). Similar, but more dramatic changes (2 fold decrease, P < 0.05), were observed in total plasma glutathione turnover (Fig. 4B). This was related to > 2 fold increase in the plasma glutathione pool size (Table 1). The absolute quantification of glutathione flux revealed that these changes were not translated into the production rates of either hepatic or plasma glutathione in a WD (Table 1). Taken together, these results suggest that the changes in both hepatic and plasma glutathione levels are attributed to their FSRs but not the PRs.
Fig. 4.

Glutathione turnover assessed using 2H2O-metabolic labeling approach. After intraperitoneal bolus load, 2H2O was administered in drinking water (5%) for up to 72 h. Effect of a Western diet on the turnover rate constant of hepatic (Panel A) and plasma (Panel B) glutathione LDLR−/− mice fed with a standard low fat diet (black) and a Western diet (red). *P < 0.05.
As a major cellular anti-oxidant, glutathione scavenges free radicals and reactive electrophiles, including lipid peroxidation product 4-HNE [35]. To assess whether the reduced glutathione redox ratio was due to HNE trapping, we measured the hepatic levels of GSH–4-HNE adducts. Although, GSH–4-HNE levels slightly increased in the WD group, it was not significantly different compared to LFD-fed animals (0.14 ± 0.06 nmol/gww in LFD vs. 0.16 ± 0.05 in WD).
Finally, to assess whether the WD-induced reduction in hepatic glutathione production was due to the depletion of cysteine, the rate limiting substrate for glutathione synthesis, we measured the hepatic cysteine content using LC–MS/MS. No differences were observed in the hepatic cysteine levels between LFD (0.50 ± 0.06 μmol/gww) and WD (0.49 ± 0.09 μmol/gww) groups, indicating that reduced glutathione synthesis was not due to sequestration of hepatic cysteine.
4. Discussion
In this study, for the first time, we simultaneously characterized the hepatic glutathione redox status and kinetics in a diet-induced mouse model of NAFLD. We demonstrated that a WD results in the altered hepatic glutathione metabolism as reflected by the decreased glutathione redox ratio and a decrease in both hepatic and plasma glutathione turnovers in LDLR−/− mice. However, these changes in glutathione metabolism were not associated with the hepatic oxidative stress measured based on markers of lipid per-oxidation. These findings suggest that plasma glutathione turnover could be used as a proxy of hepatic glutathione metabolism that reflects early defense response to oxidative insult.
Although both human [7,36] and experimental NAFLD [37–39] are associated with chronic oxidative stress, there is an inconsistency in the literature in relation to changes in the glutathione status in NAFLD. While serum levels of glutathione increased in patients with NASH [40], hepatic glutathione content seems to be lower in patients with both steatosis and NASH [7]. On the other hand, an initial increase (3 day) then a decrease of total hepatic glutathione has been reported in animal models of NAFLD [38]. Similar results, i.e. an initial increase and then a reduced level of cellular glutathione, were observed in HepG2/C3A cells after 48 h of palmitate treatment, an in vitro model of fatty liver [37]. Since there is no data on oxidized and reduced glutathione levels in these studies, it is impossible to estimate the oxidative stress based on the glutathione redox ratio. In contrast, simultaneous unbiased measurements of both reduced and total glutathione in the liver by mass spectrometry in our study allows one to assess a WD-induced changes in glutathione metabolism.
Though the redox ratio is a valuable marker of cellular oxidative insult, it does not indicate whether the change in the ratio is due to an increased GSH utilization or a reduced production. Since the cellular GSH level is regulated by its synthesis and utilization we aimed to assess total glutathione flux using 2H2O-based metabolic labeling approach. In contrast to labeled amino acids, 2H2O rapidly equilibrates with total body water (including intracellular fluids) and it readily labels amino acids within 20 min without perturbing the precursor pool size [25,41]. Based on an abundant literature which suggests that plasma glutathione originates from the liver, we assessed glutathione flux in the circulation and directly in the liver. Our results show that a WD leads to a reduction in hepatic glutathione synthesis that is associated with hepatic oxidative stress. The concurrent measurements of hepatic and plasma glutathione flux in our study enabled us to validate plasma glutathione turnover as a proxy of hepatic glutathione metabolism.
The mechanism through which a WD-induces an alteration in glutathione turnover is not completely clear. The glutathione synthesis is regulated by γ-glutamylcysteine synthetase (GCS) activity, cysteine availability and GSH feedback inhibition [10]. Since intracellular cysteine levels are close to the Km value of GCS, the cysteine availability is one of the key factors that regulates glutathione synthesis. However, our results show that the WD-induced alterations in hepatic glutathione metabolism were not associated with intracellular cysteine depletion, suggesting that the impaired GSH synthesis was not due to limited hepatic cysteine availability. Furthermore, it has been shown that plasma concentrations of total cysteine are increased but that total plasma glutathione levels are reduced in obese subjects with NAFLD [42], suggesting a possible GCS-mediated impairment of cysteine utilization for glutathione synthesis. Mechanistically, when GSH levels are high, the glutathione biosynthesis is down-regulated through the non-allosteric inhibition of GCS by feed-back inhibition with GSH, while oxidative stress-induced increased utilization of GSH initiates a feed-forward loop that activates GCS. Our results show that a WD-mediated reduction in GSH synthesis was not associated with the altered GSH levels (Fig. 3A) which rules out the regulation of GCS by the altered levels of GSH. However, it is plausible that in our diet-induced mouse model of NAFLD, the insufficient activity of GCS cannot sustain an increased demand for GSH synthesis. This is also consistent with the finding that the 129 C/T polymorphism in the promoter region of the GCLC gene encoding the catalytic subunit of GCS in humans is associated with NASH [43]. Therefore, defective GCS activity could be the cause of reduced hepatic glutathione synthesis, however the mechanism of reduced glutathione synthesis observed in this study warrants future investigations.
NAFLD is characterized with hyperlipidemia-induced oxidative stress [4]. As a major cellular anti-oxidant, glutathione scavenges free radicals, detoxifies electrophiles and maintains thiol status of proteins. Glutathione trapping of aldehydes with the formation of GSH–aldehyde adducts detoxifies reactive lipid peroxidation products [32]. In this study, we measured GSH–4-HNE adduct levels to determine whether GSH utilization in this pathway is involved in the reduced redox ratio. Our results suggest that a WD does not increase GSH–4-HNE levels therein suggesting that glutathione oxidation, but not its trapping with lipid peroxidation product, is the major pathway of glutathione utilization. This is also consistent with our finding that a WD-induced decrease in the glutathione redox ratio was not associated with increased production of lipid peroxidation products. Rather we observed a trend forward to the WD-induced decrease in both hepatic TBARS and 4-HNE levels (Supplementary Fig. 2A and 2B), suggesting activation of other compensatory pathways of oxidative defense. This may also explain our observations that despite decreased FSR of glutathione, the hepatic levels of reduced glutathione did not change significantly. Thus, the resilience of the liver to oxidative insult in our mouse model conserved GSH pool that was not affected by a WD. Based on available gene expression data, it is expected that a WD-induced up-regulation of glutathione peroxidase and down-regulation of glutathione reductase result in impaired glutathione recycling [44] (Fig. 5). This would increase the total glutathione pool that leads to decreased FSR of glutathione without consequential significant changes in GSH levels. Impaired GSSG reduction to GSH would accumulate NADPH that could be channeled to fatty acid synthesis. Consistent with this notion, recently we have demonstrated that a WD-induced insulin resistance leads to increased de novo lipogenesis in this model [34].
Fig. 5.
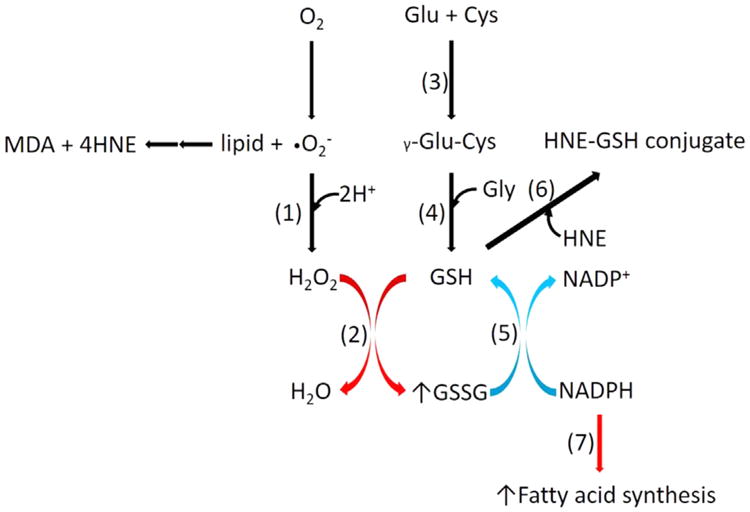
A Western diet-induced alterations in hepatic glutathione metabolism. A Western diet activates glutathione peroxidase but deactivates glutathione reductase that result in accumulation of oxidized glutathione without any significant changes in lipid peroxidation products, including GSH–4-HNE. Impaired glutathione recycling increases the total glutathione pool that leads to reduced FSR of glutathione without significant changes in GSH levels. Impaired GSSG reduction to GSH would accumulate NADPH that could be channeled to fatty acid synthesis. (1) superoxide dismutase, (2) glutathione peroxidase, (3) glutamate cysteine ligase, (4) glutathione synthetase, (5) glutathione reductase, (6) glutathione-S transferase, (7) fatty acid synthethase. Red and blue colored arrows show up-regulated and down-regulated reactions or pathways, respectively.
One of the primary objectives of our study was to identify a sensitive non-invasive surrogate marker of hepatic oxidative stress applicable to pre-clinical human studies. Although recently, the NMR technique coupled with [2-13C]glycine was introduced to measure hepatic glutathione synthesis, the low sensitivity of this method requires high levels of expensive tracer administration [9]. Furthermore, ∼50–65% [2-13C]glycine enrichment in plasma resulted in a 3–5 fold increase in intracellular and plasma glycine levels. Since glycine supplementation increases glutathione production [18,45], it is likely that this approach overestimates the synthesis rate of glutathione. Here, as an alternative approach, we propose to use 2H2O for non-invasive investigation of hepatic glutathione metabolism. 2H2O is widely used in human studies for glucose, lipid, protein and DNA turnover studies. At lower levels, e.g. ∼0.5–2% of total body water [46, 47], 2H2O appears to be well tolerated in humans and therefore the application of 2H2O in human studies for measurement of glutathione turnover in circulation may allow non-invasively measuring the hepatic glutathione metabolism and oxidative stress in NAFLD and related diseases [29].
In summary, this study using the heavy water based metabolic labeling approach provides strong evidence for alterations in hepatic and plasma glutathione metabolism in a diet-induced mouse model of NAFLD in vivo. Suppressed hepatic glutathione synthesis in combination with the increased demand on glutathione utilization and decreased conversion of GSSR to GSH leads reduced to the GSH/GSSR ratio, an early maker of oxidative insult. It remains to be determined whether stimulated glutathione synthesis and the reversal of reduced glutathione redox ratio can be achieved by an anti-oxidant based treatment of patients with NAFLD.
Supplementary Material
Acknowledgments
This work was supported in parts by the National Institutes of Health Grant 5R21RR025346-03 (TK) and the American Heart Association Grants 13IRG14700011 and 15GRNT25500004 (TK), 12GRNT12050453 (GFZ) and RO1GM112044 (TK, BW), and by the Clinical and Translational Science Collaborative of Cleveland (TK).
Abbreviations
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- ROS
reactive oxygen species
- FSR
fractional synthesis rate
- GS–MS
gas chromatography mass spectrometry
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- LFD
low fat diet
- WD
Western diet
- DTT
dithiothreitol
- TBARS
thiobarbituric acid reactive substances
- 4-HNE
4-hydroxynonenal
- GCS
γ-glutamylcysteine synthetase
- GS
glutathione synthetase
- Mass isotopomers are designated as Mn
where n is the number of atomic mass units above the molecular weight of the unlabeled isotopomer M
Footnotes
Appendix A. Supplementary material: Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed. 2016.03.032.
References
- 1.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346(16):1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 2.Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006;290(5):G852–G858. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- 3.Begriche K, et al. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6(1):1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52(1):59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Bedogni G, et al. Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology. 2005;42(1):44–52. doi: 10.1002/hep.20734. [DOI] [PubMed] [Google Scholar]
- 6.Salomone F, Godos J, Zelber-Sagi S. Natural antioxidants for non-alcoholic fatty liver disease: molecular targets and clinical perspectives. Liver Int. 2015 doi: 10.1111/liv.12975. [DOI] [PubMed] [Google Scholar]
- 7.Videla LA, et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci. 2004;106(3):261–268. doi: 10.1042/CS20030285. [DOI] [PubMed] [Google Scholar]
- 8.Koek GH, Liedorp PR, Bast A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin Chim Acta. 2011;412(15–16):1297–1305. doi: 10.1016/j.cca.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 9.Skamarauskas JT, et al. Noninvasive in vivo magnetic resonance measures of glutathione synthesis in human and rat liver as an oxidative stress biomarker. Hepatology. 2014;59(6):2321–2330. doi: 10.1002/hep.26925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 1999;13(10):1169–1183. [PubMed] [Google Scholar]
- 11.Wouters MA, et al. Thiol-based redox signalling: rust never sleeps. Int J Biochem Cell Biol. 2011;43(8):1079–1085. doi: 10.1016/j.biocel.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Lauterburg BH, Adams JD, Mitchell JR. Hepatic glutathione homeostasis in the rat: efflux accounts for glutathione turnover. Hepatology. 1984;4(4):586–590. doi: 10.1002/hep.1840040402. [DOI] [PubMed] [Google Scholar]
- 13.Beatty PW, Reed DJ. Involvement of the cystathionine pathway in the biosynthesis of glutathione by isolated rat hepatocytes. Arch Biochem Biophys. 1980;204(1):80–87. doi: 10.1016/0003-9861(80)90009-0. [DOI] [PubMed] [Google Scholar]
- 14.Burk RF, Hill KE. Reduced glutathione release into rat plasma by extrahepatic tissues. Am J Physiol. 1995;269(3 Pt 1):G396–G399. doi: 10.1152/ajpgi.1995.269.3.G396. [DOI] [PubMed] [Google Scholar]
- 15.Ookhtens M, Mittur AV, Erhart NA. Changes in plasma glutathione concentrations, turnover, and disposal in developing rats. Am J Physiol. 1994;266(3 Pt 2):R979–R988. doi: 10.1152/ajpregu.1994.266.3.R979. [DOI] [PubMed] [Google Scholar]
- 16.Griffith OW, Tate SS. The apparent glutathione oxidase activity of gamma-glutamyl transpeptidase. Chemical mechanism. J Biol Chem. 1980;255(11):5011–5014. [PubMed] [Google Scholar]
- 17.Anderson ME, Meister A. Dynamic state of glutathione in blood plasma. J Biol Chem. 1980;255(20):9530–9533. [PubMed] [Google Scholar]
- 18.Sekhar RV, et al. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care. 2011;34(1):162–167. doi: 10.2337/dc10-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Capitan P, et al. Gas chromatographic-mass spectrometric analysis of stable isotopes of cysteine and glutathione in biological samples. J Chromatogr B Biomed Sci Appl. 1999;732(1):127–135. doi: 10.1016/s0378-4347(99)00273-x. [DOI] [PubMed] [Google Scholar]
- 20.Brunengraber DZ, et al. Influence of diet on the modeling of adipose tissue triglycerides during growth. Am J Physiol Endocrinol Metab. 2003;285(4):E917–E925. doi: 10.1152/ajpendo.00128.2003. [DOI] [PubMed] [Google Scholar]
- 21.Turner SM, et al. Measurement of TG synthesis and turnover in vivo by 2H2O incorporation into the glycerol moiety and application of MIDA. Am J Physiol Endocrinol Metab. 2003;285(4):E790–E803. doi: 10.1152/ajpendo.00402.2002. [DOI] [PubMed] [Google Scholar]
- 22.Kasumov T, et al. Measuring protein synthesis using metabolic (2)H labeling, high-resolution mass spectrometry, and an algorithm. Anal Biochem. 2011;412(1):47–55. doi: 10.1016/j.ab.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kasumov T, et al. 2H2O-based high-density lipoprotein turnover method for the assessment of dynamic high-density lipoprotein function in mice, Arterioscler. Thromb Vasc Biol. 2013;33(8):1994–2003. doi: 10.1161/ATVBAHA.113.301700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kasumov T, et al. Assessment of cardiac proteome dynamics with heavy water: slower protein synthesis rates in interfibrillar than subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol. 2013;304(9):H1201–H1214. doi: 10.1152/ajpheart.00933.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L, et al. Plasma proteome dynamics: analysis of lipoproteins and acute phase response proteins with 2H2O metabolic labeling. Mol Cell Proteomics. 2012;11(7) doi: 10.1074/mcp.M111.014209. M111 (014209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boros LG, et al. Ethanol diversely alters palmitate, stearate, and oleate metabolism in the liver and pancreas of rats using the deuterium oxide single tracer. Pancreas. 2009;38(2):e47–e52. doi: 10.1097/mpa.0b013e318199fea4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao GG, et al. Determination of protein synthesis in vivo using labeling from deuterated water and analysis of MALDI-TOF spectrum. J Appl Physiol. 1985;2008(3):828–836. doi: 10.1152/japplphysiol.00976.2007. [DOI] [PubMed] [Google Scholar]
- 28.Cabral CB, et al. Estimating glutathione synthesis with deuterated water: a model for peptide biosynthesis. Anal Biochem. 2008;379(1):40–44. doi: 10.1016/j.ab.2008.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kombu RS, et al. Dynamics of glutathione and ophthalmate traced with 2H-enriched body water in rats and humans. Am J Physiol Endocrinol Metab. 2009;297(1):E260–E269. doi: 10.1152/ajpendo.00080.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah V, et al. Headspace analyses of acetone: a rapid method for measuring the 2H-labeling of water. Anal Biochem. 2010;404(2):235–237. doi: 10.1016/j.ab.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 31.Yang D, et al. Assay of low deuterium enrichment of water by isotopic exchange with [U-13C3]acetone and gas chromatography-mass spectrometry. Anal Biochem. 1998;258(2):315–321. doi: 10.1006/abio.1998.2632. [DOI] [PubMed] [Google Scholar]
- 32.Sadhukhan S, et al. Glutathionylated 4-hydroxy-2-(E)-alkenal enantiomers in rat organs and their contributions toward the disposal of 4-hydroxy-2-(E)-nonenal in rat liver. Free Radic Biol Med. 2014;70:78–85. doi: 10.1016/j.freeradbiomed.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Q, et al. Dietary regulation of catabolic disposal of 4-hydroxynonenal analogs in rat liver. Free Radic Biol Med. 2012;52(6):1043–1053. doi: 10.1016/j.freeradbiomed.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kasumov T, et al. Ceramide as a mediator of non-alcoholic fatty liver disease and associated atherosclerosis. PLoS One. 2015;10(5):e0126910. doi: 10.1371/journal.pone.0126910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volkel W, et al. Glutathione conjugates of 4-hydroxy-2(E)-nonenal as biomarkers of hepatic oxidative stress-induced lipid peroxidation in rats. Free Radic Biol Med. 2005;38(11):1526–1536. doi: 10.1016/j.freeradbiomed.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 36.Nobili V, et al. Glutathione metabolism and antioxidant enzymes in patients affected by nonalcoholic steatohepatitis. Clin Chim Acta. 2005;355(1–2):105–111. doi: 10.1016/j.cccn.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 37.Garcia MC, Amankwa-Sakyi M, Flynn TJ. Cellular glutathione in fatty liver in vitro models. Toxicol In Vitro. 2011;25(7):1501–1506. doi: 10.1016/j.tiv.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 38.Grattagliano I, et al. Severe liver steatosis correlates with nitrosative and oxidative stress in rats. Eur J Clin Invest. 2008;38(7):523–530. doi: 10.1111/j.1365-2362.2008.01963.x. [DOI] [PubMed] [Google Scholar]
- 39.Pacana T, et al. Dysregulated hepatic methionine metabolism drives homocysteine elevation in diet-induced nonalcoholic fatty liver disease. PLoS One. 2015;10(8):e0136822. doi: 10.1371/journal.pone.0136822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koruk M, et al. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann Clin Lab Sci. 2004;34(1):57–62. [PubMed] [Google Scholar]
- 41.Belloto E, et al. Determination of protein replacement rates by deuterated water: validation of underlying assumptions. Am J Physiol Endocrinol Metab. 2007;292(5):E1340–E1347. doi: 10.1152/ajpendo.00488.2006. [DOI] [PubMed] [Google Scholar]
- 42.Pastore A, et al. Plasma levels of homocysteine and cysteine increased in pediatric NAFLD and strongly correlated with severity of liver damage. Int J Mol Sci. 2014;15(11):21202–21214. doi: 10.3390/ijms151121202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oliveira CP, et al. Association of polymorphisms of glutamate–cystein ligase and microsomal triglyceride transfer protein genes in non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2010;25(2):357–361. doi: 10.1111/j.1440-1746.2009.06001.x. [DOI] [PubMed] [Google Scholar]
- 44.Renaud HJ, et al. Effect of diet on expression of genes involved in lipid metabolism, oxidative stress, and inflammation in mouse liver-insights into mechanisms of hepatic steatosis. PLoS One. 2014;9(2):e88584. doi: 10.1371/journal.pone.0088584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruiz-Ramirez A, et al. Glycine restores glutathione and protects against oxidative stress in vascular tissue from sucrose-fed rats. Clin Sci. 2014;126(1):19–29. doi: 10.1042/CS20130164. [DOI] [PubMed] [Google Scholar]
- 46.Previs SF, et al. Quantifying rates of protein synthesis in humans by use of 2H2O: application to patients with end-stage renal disease. Am J Physiol Endocrinol Metab. 2004;286(4):E665–E672. doi: 10.1152/ajpendo.00271.2003. [DOI] [PubMed] [Google Scholar]
- 47.Wang D, et al. Characterization of human plasma proteome dynamics using deuterium oxide. Proteomics Clin Appl. 2014;8(7–8):610–619. doi: 10.1002/prca.201400038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.