

Abstract
Pathological hypertrophy of the heart is closely associated with endoplasmic reticulum stress (ERS), leading to maladaptations such as myocardial fibrosis, induction of apoptosis, and cardiac dysfunctions. Salubrinal is a known selective inhibitor of protein phosphatase 1 (PP1) complex involving dephosphorylation of phospho-eukaryotic translation initiation factor 2 subunit (p-eIF2)-α, the key signaling process in the ERS pathway. In this study, the effects of salubrinal were examined on cardiac hypertrophy using the mouse model of transverse aortic constriction (TAC) and cell model of neonatal rat ventricular myocytes (NRVMs). Treatment of TAC-induced mice with salubrinal (0.5 mg·kg−1·day−1) alleviated cardiac hypertrophy and tissue fibrosis. Salubrinal also alleviated hypertrophic growth in endothelin 1 (ET1)-treated NRVMs. Therefore, the present results suggest that salubrinal may be a potentially efficacious drug for treating pathological cardiac remodeling.
Keywords: eIF2α, ER stress, GRP, TGF-β, transverse aortic constriction
INTRODUCTION
Sustained pressure overload in the heart induces pathological hypertrophy with subsequent onset of myocardial fibrosis, induction of apoptosis, and cardiac dysfunctions (Berenji et al., 2005; Dickhout et al., 2011; Levy et al., 1990). At the cellular level, it is characterized by an increase in cell size, changes in gene expressions, and increase in protein synthesis (Huang et al., 2011). Therefore, the alleviation of hypertrophy, fibrosis, and apoptosis in the remodeled heart is considered a potential therapeutic strategy to prevent further progression to heart failure (Frey et al., 2004).
The endoplasmic reticulum (ER) as a major signal transduction organelle is very sensitive to alterations in cellular homeostasis. A variety of extracellular stimuli such as heat shock, ischemia, gene mutation, hypoxia, and increased protein synthesis can cause ER stress (ERS) leading to signal transduction to the cytoplasm and nucleus (Kaufman, 1999). ERS triggers a cytoprotective response called unfolded protein response (UPR) (Kaufman, 1999; Lee et al., 2010). As a primary step in UPR response, binding immunoglobulin protein (BiP)/glucose-regulated protein 78 (GRP78) binds to the unfolded/misfolded proteins, which in turn release the three sequestered key ER membrane signal transducers including activating transcription factor (ATF)-6, protein kinase-like ER kinase (PERK), and endoribonuclease inositol-requiring enzyme 1 (IRE-1) (Groenendyk et al., 2010; Ron and Walter, 2007). These transducers activate their downstream pathways, resulting in activation of various pro-survival mechanisms including up-regulation of ER chaperones such as GRP94, GRP78, and calreticulin (CRT), which enhances the ER-mediated maintenance of homeostasis (Ferri and Kroemer, 2001; Kaufman, 1999). However, prolonged stress switches the pro-survival activities of UPR to pro-apoptotic signaling mostly mediated by phospho-C-Jun-N-terminal kinase (p-JNK), CCAAT/-enhancer-binding protein homologous protein (CHOP), or caspase-3, or a combination of these molecules (Ron and Walter, 2007). Additionally, a cell-death response that involves cross-talk between the ER and mitochondria is activated, which involves the Bcl-2 family proteins (Kim et al., 2015a; Szegezdi et al., 2009).
A variety of accumulating evidence has shown that the pathogenesis of cardiac hypertrophy is associated with ERS (Okada et al., 2004; Park et al., 2012). Therefore, alleviation of ERS might have therapeutic benefit in treating several cardiac diseases (Lee et al., 2013; Park et al., 2012). The activation of the PERK pathway, which leads to phosphorylation of the eukaryotic translation initiation factor 2 subunit (eIF2)-α on Ser51 is cytoprotective during ERS (Boyce et al., 2005). Therefore, the selective chemical inhibition of eIF2α dephosphorylation is proposed as a strategy to alleviate ERS and has proven beneficial in the treatment of many diseases including cardiac (Li et al., 2015; Liu et al., 2014) and non-cardiac diseases (Boyce et al., 2005; Lee et al., 2010). Salubrinal, a cell permeable selective inhibitor of the cellular phosphatase complexes which dephosphorylates eIF2α, is reportedly cytoprotective against ERS-induced apoptosis (Boyce et al., 2005; Kim et al., 2008). In the present study, we evaluated the effects of salubrinal on pathological hypertrophy induced by transverse aortic constriction (TAC) in mice.
MATERIALS AND METHODS
Animal care and ethics statement
C57BL/6 mice were housed under controlled conditions at a temperature of 22°C with a 12 h light-dark cycle and 55–56% relative humidity. All investigations were performed in accordance with the protocols approved by the Gwangju Institute of Science and Technology Animal Care and Use Committee.
Transverse aortic constriction (TAC)
Nine-weeks-old C57BL/6 male mice (23–25 g) were used in this study. The operation procedure was followed as previously described (Park et al., 2012). Animals undergoing the sham surgery (same procedure without constriction) were considered as control group.
Administration of Salubrinal
Salubrinal (Calbiochem, USA) was reconstituted in phosphate buffered saline (PBS) containing 49.5% PEG 400 and 0.5% Tween 80, and the solution was filtered using a 0.2-μm sterile syringe filter. A dose of 0.5 mg·kg−1·day−1 salubrinal was administered by subcutaneous injection from the day of TAC surgery, once every day for 7 days for both Sal SHAM (n = 10) and Sal TAC (n = 10) animals. The dose was decided based on the previously reported pharmacokinetic results (Zhang et al., 2012). Vehicle (Veh) TAC (n = 10) and vehicle (Veh) SHAM (n = 9) received an equal volume of the vehicle (PBS containing 49.5% PEG 400 and 0.5% Tween 80). The animals were sacrificed after 7 days of salubrinal/vehicle administration.
Isolation of neonatal rat ventricular myocytes (NRVMs) and immunocytochemistry
Neonatal rat ventricular myocytes (NRVMs) were isolated using neonatal cardiomyocyte isolation system (Worthington Biochemical Corp., USA) as per manufacturer’s guidelines. The procedure was followed as described previously (Kim et al., 2015b). To induce cardiomyocyte hypertrophy, NRVMs were cultured in serum-free medium for 24 h and then 100 nM endothelin 1 (ET1, Sigma-Aldrich) was added for 24 h. For evaluating the cell surface area, the cardiomyocytes were cultured on 18 mm cover slips; fixed with 4% paraformaldehyde. The procedure for immunocytochemistry was followed as described previously (Park et al., 2012). The prepared cells were examined using an LSM 700 confocal microscope (Carl Zeiss Inc., Germany).
qRT-PCR
After extraction of total RNA from the mouse hearts using the TRIzol reagent (Invitrogen Life Technologies, USA), RNA was reverse-transcribed to produce cDNAs using the Prime Script RT reagent kit (TaKaRa, Japan). cDNA was used as a template for qRT-PCR using SYBR green dye (Kapa Biosystems, USA), and gene expression was normalized to β-actin. The sequences of the specific primers are described in Supplementary Table S1.
Western blotting
Whole heart homogenates and cell lysate were solubilized in RIPA buffer and protein concentrations were evaluated using BCA protein assay kit (Pierce, USA). Proteins (50 μg) were separated by SDS/PAGE (8–15% gel) and subsequently transferred to PVDF membranes. Western-blot analysis procedure was followed as previously described (Lee et al., 2013). The primary antibodies used in the study are KDEL (Abcam), p-IRE1α (Novus Biologicals), TGF-β (Cell Signaling), p-JNK (Santa Cruz), eIF2α (Santa Cruz), p-eIF2α (ser51) (Cell Signaling), ANP (Santa Cruz), BNP (Santa Cruz), and α-Tubulin (Santa Cruz) at 4°C overnight. Secondary antibodies (HRP-conjugated) were then applied at 25°C for 1 h, and ImageQuant LAS 4000 mini (GE Healthcare Bio-Sciences AB) was used to detect the western blot signals. The protein band intensities were analyzed by ImageJ software (NIH).
Histological analysis
The isolated mouse hearts were fixed with 4% paraformaldehyde, paraffin-embedded, and 6-μm thick sections were cut using a microtome (RM2135, Leica). Hematoxylin and eosin (H&E, Sigma-Aldrich) was used to stain the heart sections.
Statistical analysis
Results are shown as mean ± standard error of the mean (SEM) of three or more than three independent experiments. Statistical significance between multiple groups was determined using an analysis of variance (ANOVA) followed by Bonferroni test and t-test. Statistical significance was set at P < 0.05 or < 0.01.
RESULTS
Salubrinal alleviated ERS responses in pressure overload cardiac hypertrophy
To examine the effect of salubrinal on ERS responses, we used 1 μM thapsigargin for 12 h to induce ERS in NRVMs with or without 75 μM salubrinal. The results showed that salubrinal significantly reduced ERS response in NRVMs (Supplementary Fig. S1). As previously reported (Okada et al., 2004; Park et al., 2012), the expression levels of ERS markers such as p-IRE, GRP78, GRP94, and p-eIF2α were significantly upregulated in mice subjected to TAC after 1 week. To examine whether salubrinal alleviated the upregulated ERS markers, the expression levels of the ERS markers were examined. Administration of salubrinal for 1 week (0.5 mg/kg/day) reduced the expression levels of p-IRE, GPR94, and GRP78 compared with vehicle administration (Fig. 1), suggesting that salubrinal alleviates the pressure-overload-associated ERS responses in the TAC heart. As expected, salubrinal increased the ratio of p-eIF2α/eIF2α in the salubrinal-treated TAC group compared to the vehicle-treated TAC group, by inhibiting the PP1-induced dephosphorylation of p-eIF2α (Fig. 1). Phosphorylation of eIF2α has been shown to be involved in translational inhibition (Boyce et al., 2005), which may suggest an additional mechanism involved in reducing the UPR by attenuation of protein synthesis.
Fig. 1. Salubrinal (Sal) attenuated endoplasmic reticulum stress (ERS) responses in transverse aortic constriction (TAC)-induced hypertrophic hearts.
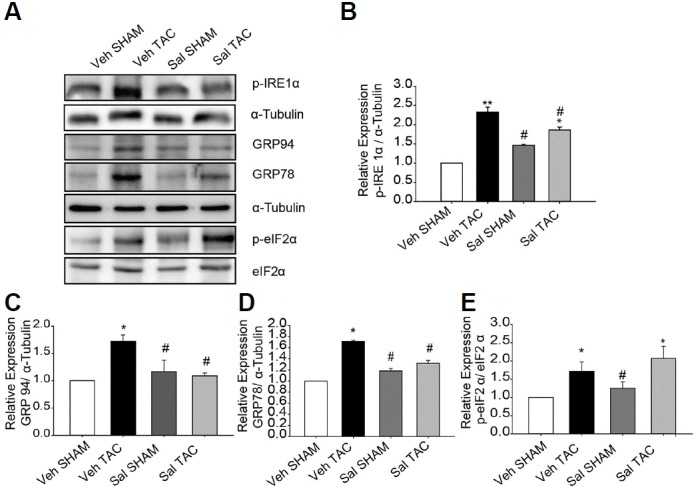
(A–E) Protein expression levels of p-IRE1-α, GRP78, GRP94, and p-eIF2-α. All data represent mean ± SEM; *P < 0.05, **P < 0.01 vs. Veh SHAM, and #P < 0.05 vs. Veh TAC. (n = 3)
Salubrinal reduced pressure overload cardiac hypertrophy
To examine the effects of salubrinal on cardiomyocyte hypertrophy, immunocytochemistry was performed on NRVMs treated with 100 nM ET1 for 24 h with or without 75 μM salubrinal. The results showed that 100 nM ET1 increased the cell surface area approximately 2-fold (Figs. 2A and 2B); however, the addition of salubrinal abolished the increase in cell size. We then investigated whether salubrinal also affected pressure overload-induced cardiac hypertrophy using the mouse model of TAC. Subcutaneous administration of salubrinal reduced the increment in heart weight (HW)/body weight (BW) induced by TAC (Figs. 3A–3C). Results of the qRT-PCR and western blot analysis also showed that the enhanced expression levels of the hypertrophic markers natriuretic peptide A/ANP (NPPA) and natriuretic peptide B/BNP (NPPB) were significantly decreased in salubrinal-treated TAC mice in comparison to vehicle-treated TAC mice (Fig. 3). Collectively, our results suggest that salubrinal attenuated pressure-overload cardiac hypertrophy.
Fig. 2. Salubrinal (Sal) reduced cardiomyocyte hypertrophy.
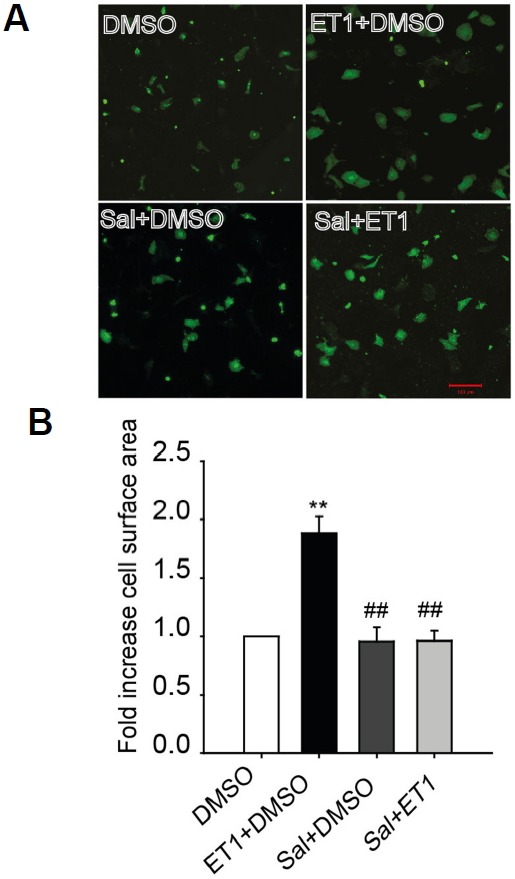
(A) Immunofluorescence imaging showing cell sizes of NRVMs treated with 100 nM endothelin1 (ET1) in the presence or absence of 75 μM salubrinal (scale bar, 100 μm). (B) Fold increase in cell surface area compared to dimethyl sulfoxide (DMSO) control. All data represent mean ± SEM; *P < 0.05, **P < 0.01 vs. DMSO control and #P < 0.05, ##P < 0.01 vs. ET1. (n = 100 cells for each group)
Fig. 3. Salubrinal (Sal) reduced cardiac hypertrophy.
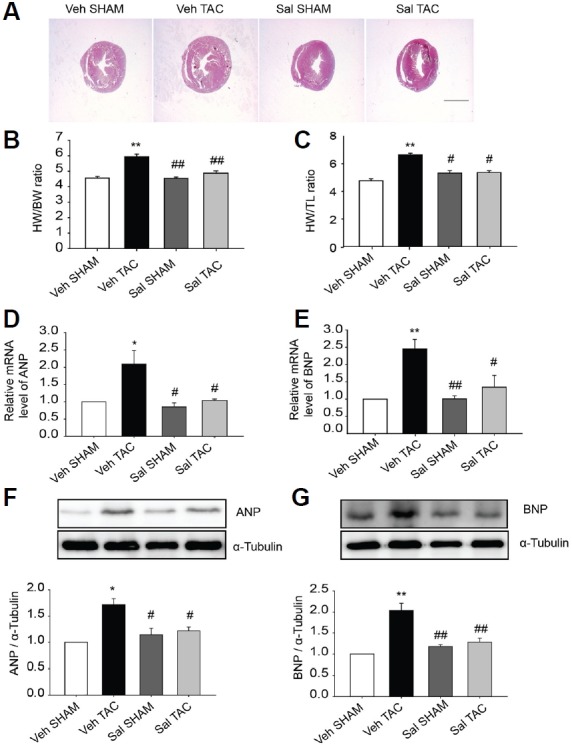
(A) Cross sections of hearts under different experimental conditions (scale bar, 2 mm). (B) HW/BW and (C) HW/TL ratios for the various experimental groups (n = 6–8). Salubrinal (Sal) reduced fetal gene expression in TAC induced cardiac hypertrophy. (D and E) Relative mRNA levels of ANP (NPPA), and BNP (NPPB) were evaluated using qRT-PCR (n = 5–6). (F, G) Protein expression levels of ANP and BNP (n = 3–5). All data represent mean ± SEM; *P < 0.05, **P <0.01 vs. Veh SHAM and #P < 0.05, ##P <0.01 vs. Veh TAC.
Salubrinal attenuated cardiac fibrosis signaling in pressure overload cardiac hypertrophy
The effects of salubrinal on fibrosis of the heart during pressure overload cardiac hypertrophy were examined using TGF-β signaling genes. Significantly decreased expression of TGF-β was noticed in the salubrinal-treated TAC mice compared to vehicle-treated TAC mice. Our results further showed that the transcription of fibrosis marker genes including pro-collagen isoforms collagen1α1 and collagen3α1 were greatly enhanced in vehicle-treated TAC mice compared to the other groups (Figs. 4B and 4C). These results suggest that salubrinal attenuated TGF-β mediated fibrosis pathways in TAC-induced mouse hypertrophied hearts.
Fig. 4. Salubrinal (Sal) attenuated TGF-β pathway signaling molecules.
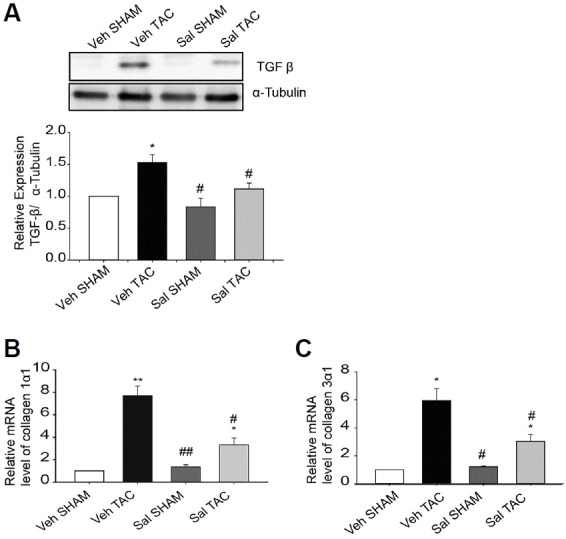
(A) TGF-β expression in whole heart homogenates from SHAM and TAC-induced mice treated with vehicle or salubrinal (0.5 mg·kg−1·day−1 ) for 1 week (n = 3). (B, C) Transcripts for collagen isoforms (n = 3–6) were evaluated using qRT-PCR in hearts of SHAM and TAC-induced mice after treatment with vehicle or salubrinal. All data are mean ± SEM; *P < 0.05, **P <0.01 vs. Veh SHAM, #P < 0.05, ##P <0.01 vs. Veh TAC.
Salubrinal attenuated elevated expression levels of apoptotic marker genes in TAC mouse heart
To study the effect of salubrinal on ERS-induced apoptosis in NRVMs, 1 μM thapsigargin (TG) was used as an ERS inducer for 12 h, in presence or absence of salubrinal. The results show that salubrinal led to decreased expression levels of the pro-apoptotic markers, cleaved caspase-3, and CHOP, and decreased the ratio of Bax/Bcl-2 (Supplementary Fig. S2), indicative of reduced apoptosis. These results were supported by terminal deoxy-nucleotidyl-transferase-mediated dUTP nick-end labeling (TUNEL) assay, which demonstrated a reduced number of TUNEL-positive cells when treated with salubrinal. (Supplementary Figs. S2C and S2D). Previously, we provided evidence treatment with chemical chaperon 4-phenylbutyric acid (PBA) attenuated the TAC-induced apoptosis (Park et al., 2012). In the present study, we evaluated the expression levels of ER initiated apoptotic marker p-JNK in vehicle-treated SHAM and TAC, as well as salubrinal-treated SHAM and TAC mice to determine whether salubrinal had similar effects in the heart. As shown in (Supplementary Fig. S3), administration of salubrinal significantly decreased the TAC-induced upregulation of p-JNK expression, suggesting that TAC-induced increase in apoptosis can be prevented by salubrinal.
DISCUSSION
Pathological cardiac hypertrophy induced by pressure overload is closely associated with ERS due to Ca2+ dysregulation and increased protein synthesis (Li et al., 2015; Okada et al., 2004). Microscopic examination has also shown that the ER develops markedly in the hypertrophic hearts, as a compensatory response to the elevated protein synthesis in the ER (Okada et al., 2004). Since salubrinal was identified as a specific inhibitor of protein phosphatase 1 (PP1), which involves dephosphorylation of p-eIF2α and inhibition of the downstream PERK-eIf2α pathway (Boyce et al., 2005), we investigated whether salubrinal has inhibitory effects on the ERS-mediated cardiac hypertrophy in an animal model of TAC. The present findings showed that salubrinal (1) attenuated ERS, as indicated by downregulation of ERS markers such as GRP94 and GRP78 in TAC mice (Fig. 1); (2) effectively alleviated cardiac hypertrophy in TAC mice, as indicated by the reduced HW/BW ratio, decreased ANP and BNP expression levels (Fig. 3), and (3) attenuated tissue fibrosis and decreased the expression of the fibrosis markers in TAC mice (Fig. 4), In addition, salubrinal also attenuated elevated apoptosis in TAC mice (Supplementary Fig. S3).
The role of salubrinal in alleviating cardiac hypertrophy is attributed to its inhibitory action on protein synthesis (Boyce et al., 2005). This effect is mediated via maintenance of p-eIF2α by the direct inhibition of PP1, and may also involve decreased growth arrest and DNA damage-inducible protein 34 (GADD34), which is negatively regulated by CHOP expression (Fu et al., 2010). In this study, salubrinal treatment decreased the cell size (Fig. 2) and protein synthesis (data not shown) in cardiomyocytes, which was increased after ET1 treatment. Therefore, enhanced inhibition of protein synthesis may enable cardiomyocyte recovery from ERS by balancing the cellular protein folding capacity (Fig. 5). The salubrinal treatment reduced the ERS marker chaperone proteins GRP78 and GRP94 that were upregulated by TAC (Fig. 1). This is consistent with our hypothesis that the translational inhibition by salubrinal leads to decreased protein synthesis in the ER (Boyce et al., 2005) and, therefore, alleviates UPR in the ER (Fig. 5).
Fig. 5. Schematic representation of the events initiated in TAC-operated mouse heart and salubrinal treated TAC-operated mouse heart.
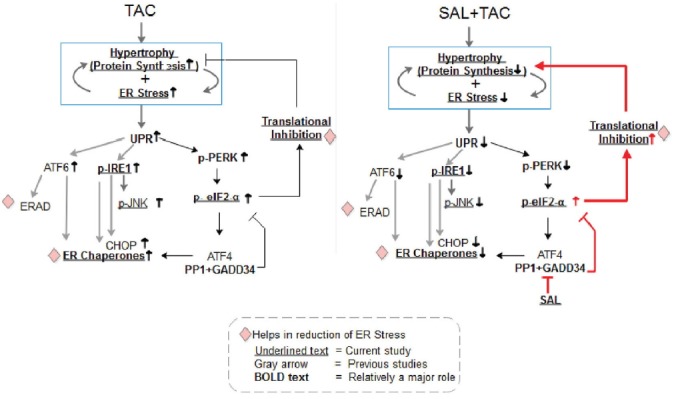
The visual model shows that salubrinal treatment increases the abundance of p-eIF2α leading to attenuation of protein synthesis and reduction of cardiac hypertrophy and ER stress/UPR. The Sal TAC hearts showed a decreased p-IRE, GRP78, GRP94, and p-JNK levels.
TGF-β signaling is associated with various cardiac pathologies such as ERS-induced hypertrophy (Park et al., 2012), induction of EMT and heart failure (Kuwahara et al., 2002; Rosenkranz et al., 2002), myofibroblast transdifferentiation, and enhanced extracellular matrix protein synthesis (Annes et al., 2003; Desmouliere et al., 1993). In the present study we noticed salubrinal administration remarkably decreased the expression of TGF-β (Fig. 4). Consistently, the targets of the Smad cascade, pro-collagen isoforms collagen1α1 and collagen3α1 transcripts, which were greatly enhanced in TAC mice were alleviated by treatment with salubrinal. These results are in agreement with an earlier study in which salubrinal has shown a reduction in myocardial fibrosis in myocardial infarction (MI) model of heart failure (Liu et al., 2014).
It is known that ERS-induced myocardial apoptosis is one of the mechanisms involved in cardiac remodeling (Dickhout et al., 2011; Kim et al., 2008; Nickson et al., 2007; Park et al., 2012; Younce and Kolattukudy, 2010). The involvement of JNK, c-Jun, NF-KB and other pathways has been reported in ERS-induced hypertrophy and apoptosis (Hamid et al., 2011). In this study, we showed that salubrinal attenuated the expression of p-JNK in TAC mice (Supplementary Fig. S3), indicating that salubrinal suppressed the ERS-induced cardiac cell death. Salubrinal abrogating activation of the c-Jun pathway that arises from ERS has been observed to prevent progression to cardiac failure in an earlier study (Liu et al., 2014). Also, it is interesting to note that salubrinal induces translational repression, which in turn decreases the total UPR response leading to cardioprotection (Fig. 5). Owing to extensive cross-talk between signaling pathways (Park et al., 2012), the underlying mechanisms have not been fully elucidated yet.
The data obtained in the present study suggests that salubrinal is cardioprotective and, therefore, may be a potentially efficacious therapeutic agent for treating ERS-mediated cardiovascular diseases. However, excessive phosphorylation of eIF2α by high doses of salubrinal could cause problems in other types of cells and tissues (Cnop et al., 2007; Gao et al., 2013). It was previously shown that pretreatment with salubrinal abolished the neuroprotective effects of ischemic preconditioning, in permanent focal ischemia (Gao et al., 2013). Studies in pancreatic beta cells showed that salubrinal-induced eIF2-α phosphorylation potentiated the pro-apoptotic effects of free fatty acids (Cnop et al., 2007). But in the heart salubrinal has shown, anti-apoptotic (Liu et al., 2012) and cardioprotective effects (Liu et al., 2014). However, these effects of salubrinal could be due to changes in levels of several molecules involved in a broad spectrum of cellular pathways that interact with ERS pathways. Therefore, future studies such as systems biological approaches involving integrative global cellular pathway analysis will be necessary to identify adverse effects of salubrinal. In the current study, there was no evidence of nonspecific toxicity by administration of salubrinal because we observed no effects on weight gain, visceral organ weights (Supplementary Table S2) or physical activity in the study groups.
The effects of salubrinal on ERS-associated diseases have been evaluated in several tissues (Boyce et al., 2005; Lee et al., 2010; Teng et al., 2014); however, to date there is no report related to cardiac hypertrophy. Therefore, in this study, we report that subcutaneous injection of salubrinal (0.5 mg·kg−1·day−1) reduced pressure overload-induced cardiac hypertrophy, fibrosis, and apoptosis with associated improvement in heart function. Furthermore, these results suggest that treatment with salubrinal may be a promising therapeutic strategy to suppress maladaptive alterations in the ERS signaling pathway in cardiac pathologies.
Supplementary Information
ACKNOWLEDGMENTS
This work was supported by the 2016 GIST Research Institute (GRI) Grant, and the NRF grant funded by the Korean Ministry of Science, ICT & Future Planning (NRF-2013M3A9A7046297).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Annes J.P., Munger J.S., Rifkin D.B. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Berenji K., Drazner M.H., Rothermel B.A., Hill J.A. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol. 2005;289:H8–H16. doi: 10.1152/ajpheart.01303.2004. [DOI] [PubMed] [Google Scholar]
- Boyce M., Bryant K.F., Jousse C., Long K., Harding H.P., Scheuner D., Kaufman R.J., Ma D., Coen D.M., Ron D., et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Cnop M., Ladriere L., Hekerman P., Ortis F., Cardozo A.K., Dogusan Z., Flamez D., Boyce M., Yuan J., Eizirik D.L. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J Biol Chem. 2007;282:3989–3997. doi: 10.1074/jbc.M607627200. [DOI] [PubMed] [Google Scholar]
- Desmouliere A., Geinoz A., Gabbiani F., Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–111. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickhout J.G., Carlisle R.E., Austin R.C. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res. 2011;108:629–642. doi: 10.1161/CIRCRESAHA.110.226803. [DOI] [PubMed] [Google Scholar]
- Ferri K.F., Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3:E255–263. doi: 10.1038/ncb1101-e255. [DOI] [PubMed] [Google Scholar]
- Frey N., Katus H.A., Olson E.N., Hill J.A. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- Fu H.Y., Okada K., Liao Y., Tsukamoto O., Isomura T., Asai M., Sawada T., Okuda K., Asano Y., Sanada S., et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122:361–369. doi: 10.1161/CIRCULATIONAHA.109.917914. [DOI] [PubMed] [Google Scholar]
- Gao B., Zhang X.Y., Han R., Zhang T.T., Chen C., Qin Z.H., Sheng R. The endoplasmic reticulum stress inhibitor salubrinal inhibits the activation of autophagy and neuroprotection induced by brain ischemic preconditioning. Acta Pharmacol Sin. 2013;34:657–666. doi: 10.1038/aps.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenendyk J., Sreenivasaiah P.K., Kim D.H., Agellon L.B., Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res. 2010;107:1185–1197. doi: 10.1161/CIRCRESAHA.110.227033. [DOI] [PubMed] [Google Scholar]
- Hamid T., Guo S.Z., Kingery J.R., Xiang X., Dawn B., Prabhu S.D. Cardiomyocyte NF-κB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–138. doi: 10.1093/cvr/cvq274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Zhang H., Shao Z., O’Hara K.A., Kopilas M.A., Yu L., Netticadan T., Anderson H.D. Suppression of endothelin-1-induced cardiac myocyte hypertrophy by PPAR agonists: role of diacylglycerol kinase zeta. Cardiovasc Res. 2011;90:267–275. doi: 10.1093/cvr/cvq401. [DOI] [PubMed] [Google Scholar]
- Kaufman R.J. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Kim I., Xu W., Reed J.C. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discovery. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Kim D.Y., Kim H.R., Kim K.K., Park J.W., Lee B.J. NELL2 function in protection of cells against endoplasmic reticulum stress. Mol Cells. 2015a;38:145–150. doi: 10.14348/molcells.2015.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.O., Song D.W., Kwon E.J., Hong S.E., Song H.K., Min C.K., Kim D.H. miR-185 Plays an anti-hypertrophic role in the heart via multiple targets in the calcium-signaling pathways. PLoS One. 2015b;10:e0122509. doi: 10.1371/journal.pone.0122509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara F., Kai H., Tokuda K., Kai M., Takeshita A., Egashira K., Imaizumi T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–135. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- Lee D.Y., Lee K.S., Lee H.J., Kim D.H., Noh Y.H., Yu K., Jung H.Y., Lee S.H., Lee J.Y., Youn Y.C., et al. Activation of PERK signaling attenuates Abeta-mediated ER stress. PLoS One. 2010;5:e10489. doi: 10.1371/journal.pone.0010489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.H., Kwon E.J., Kim D.H. Calumenin has a role in the alleviation of ER stress in neonatal rat cardiomyocytes. Biochem Biophys Res Commun. 2013;439:327–332. doi: 10.1016/j.bbrc.2013.08.087. [DOI] [PubMed] [Google Scholar]
- Levy D., Garrison R.J., Savage D.D., Kannel W.B., Castelli W.P. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- Li R.J., He K.L., Li X., Wang L.L., Liu C.L., He Y.Y. Salubrinal protects cardiomyocytes against apoptosis in a rat myocardial infarction model via suppressing the dephosphorylation of eukaryotic translation initiation factor 2α. Mol Med Rep. 2015;12:1043–1049. doi: 10.3892/mmr.2015.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.L., Li X., Hu G.L., Li R.J., He Y.Y., Zhong W., Li S., He K.L., Wang L.L. Salubrinal protects against tunicamycin and hypoxia induced cardiomyocyte apoptosis via the PERK-eIF2alpha signaling pathway. Journal of geriatric cardiology: JGC. 2012;9:258–268. doi: 10.3724/SP.J.1263.2012.02292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Wang J., Qi S.Y., Ru L.S., Ding C., Wang H.J., Zhao J.S., Li J.J., Li A.Y., Wang D.M. Reduced endoplasmic reticulum stress might alter the course of heart failure via caspase-12 and JNK pathways. Can J Cardiol. 2014;30:368–375. doi: 10.1016/j.cjca.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Nickson P., Toth A., Erhardt P. PUMA is critical for neonatal cardiomyocyte apoptosis induced by endoplasmic reticulum stress. Cardiovasc Res. 2007;73:48–56. doi: 10.1016/j.cardiores.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K., Minamino T., Tsukamoto Y., Liao Y., Tsukamoto O., Takashima S., Hirata A., Fujita M., Nagamachi Y., Nakatani T., et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- Park C.S., Cha H., Kwon E.J., Sreenivasaiah P.K., Kim D.H. The chemical chaperone 4-phenylbutyric acid attenuates pressure-overload cardiac hypertrophy by alleviating endoplasmic reticulum stress. Biochem Biophys Res Commun. 2012;421:578–584. doi: 10.1016/j.bbrc.2012.04.048. [DOI] [PubMed] [Google Scholar]
- Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rosenkranz S., Flesch M., Amann K., Haeuseler C., Kilter H., Seeland U., Schluter K.D., Bohm M. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1) Am J Physiol Heart Circ Physiol. 2002;283:H1253–1262. doi: 10.1152/ajpheart.00578.2001. [DOI] [PubMed] [Google Scholar]
- Szegezdi E., Macdonald D.C., Ni Chonghaile T., Gupta S., Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296:C941–953. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- Teng Y., Gao M., Wang J., Kong Q., Hua H., Luo T., Jiang Y. Inhibition of eIF2alpha dephosphorylation enhances TRAIL-induced apoptosis in hepatoma cells. Cell Death Dis. 2014;5:e1060. doi: 10.1038/cddis.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younce C.W., Kolattukudy P.E. MCP-1 causes cardiomyoblast death via autophagy resulting from ER stress caused by oxidative stress generated by inducing a novel zinc-finger protein, MCPIP. Biochem J. 2010;426:43–53. doi: 10.1042/BJ20090976. [DOI] [PubMed] [Google Scholar]
- Zhang P., Hamamura K., Jiang C., Zhao L., Yokota H. Salubrinal promotes healing of surgical wounds in rat femurs. J Bone Miner Metab. 2012;30:568–579. doi: 10.1007/s00774-012-0359-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.