

Abstract

Lomibuvir (1) is a non-nucleoside, allosteric inhibitor of the hepatitis C virus NS5B polymerase with demonstrated clinical efficacy. Further development efforts within this class of inhibitor focused on improving the antiviral activity and physicochemical and pharmacokinetic properties. Recently, we reported the development of this series, leading to compound 2, a molecule with comparable potency and an improved physicochemical profile relative to 1. Further exploration of the amino amide-derived side chain led to a series of lactam derivatives, inspired by the X-ray crystal structure of related thiophene carboxylate inhibitors. This series, exemplified by 12f, provided 3–5-fold improvement in potency against HCV replication, as measured by replicon assays. The synthesis, structure–activity relationships, in vitro ADME characterization, and in vivo evaluation of this novel series are discussed.
Keywords: Lomibuvir, non-nucleoside, hepatitis C, NS5B, polymerase, antiviral activity
Hepatitis C virus (HCV) infects
an estimated 80–115 million people worldwide.1 Chronic infection is associated with increased risk of
liver disease, fibrosis and cirrhosis, and hepatocellular carcinoma.2 In 2011, the HCV NS3 protease inhibitors boceprevir3 and telaprevir4 were
approved as the first direct-acting antiviral agents, administered
in combination with pegylated interferon α and ribavirin.5 In the following years, more efficacious agents
and combinations have been introduced, and these two protease inhibitor
drugs have been superseded.6,7 In our quest for new
chemical agents needed to decrease the duration of treatment and reduce
treatment side effects, we focused on developing inhibitors of the
NS5B polymerase, an approach anticipated to deliver an all-oral, interferon-free
treatment regimen when applied in combination with telaprevir. The
viral NS5B polymerase plays an essential role in the life cycle of
HCV, being responsible for viral genomic replication.8 Inhibitors of this enzyme in clinical development and practice
fall into two classes: nucleoside (or nucleotide) analogues and non-nucleoside,
allosteric inhibitors.9 Lomibuvir (1: also named VX-222 and VCH-222) is a selective, non-nucleoside
polymerase inhibitor that binds to thumb pocket 2 of the HCV NS5B
polymerase.10 A 3-day, monotherapy viral
kinetic study in which treatment-naïve patients with genotype
1 HCV infection were treated with 250, 500, or 750 mg lomibuvir twice
daily, or 1500 mg once daily, showed it to be well tolerated, and
patients achieved a mean HCV RNA reduction ranging from 3.1 to 3.4
log10.11 To continue to develop
this series, our program focused on improving physicochemical and
pharmacokinetic properties. As we recently reported, replacement of
the trans-4-hydroxycyclohexane ring in 1 with a glycine-derived amide (2) resulted in comparable
potency and improved physicochemical properties.12 During the course of this work, we established that although
compounds such as 3b showed higher activity than the
corresponding R-enantiomer 3a, the unsubstituted
compound 2 was sufficiently active, and we elected to
advance the unsubstituted (glycine) series. However, the question
remained whether the side chain was simply disadvantageous in the
wrong configuration, or could this region of the molecule be developed
to show a specific advantage, perhaps through engagement of target
interactions in the correct configuration. To further develop this
class of molecules, we applied a structure-guided design approach,
based on the X-ray crystal structures of the NS5B polymerase with
bound ligands of this class.
Based on the X-ray crystal structure of compound 4 bound to the thumb pocket 2 allosteric site of NS5B (PDB: 2GIR),13 compound 1 was docked, maintaining the same key contacts. The model illustrated in Figure 1 shows the trans-4-hydroxycyclohexyl group protruding from the binding site and in proximity to the surface residue Arg501 of the long helix forming the side of the binding pocket. Our objective was to capitalize on this proximity and derive additional binding interactions.
Figure 1.
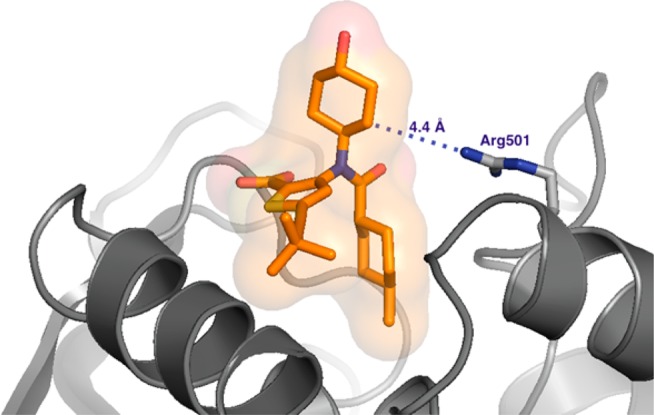
Model of 1 bound to HCV genotype 1b NS5B showing the proximity of the trans-4-hydroxycyclohexyl ring to Arg501.
Compounds of this series were prepared from commercially available (R)- and (S)-5-, 6-, and 7-membered lactams derived from α-amino acids as illustrated in Scheme 1. The lactams (5a–f) were phthaloyl-protected by treatment with ethyl 1,3-dioxoisoindoline-2-carboxylate to give products 6a–f. N-Methylation of the lactam was accomplished by treatment with NaH and MeI in DMF. Hydrazine deprotection of the phthaloyl group gave the desired lactams (8a–f), which were coupled to the thiophene 9(12) under Buchwald–Hartwig conditions to provide ester products 10a–f, which were converted to the corresponding amides 11a–f using trans-4-methylcyclohexylcarbonyl chloride. Hydrolysis of the methyl esters gave desired products 12a–12f. Experimental details are provided in the Supporting Information.
Scheme 1. General Synthesis of Compounds 12a–12f.

Reaction conditions: (a) ethyl 1,3-dioxoisoindoline-2-carboxylate, Na2CO3, H2O, RT; (b) NaH, MeI, DMF, 0 °C; (c) NH2NH2, MeOH, RT; (d) Pd(OAc)2, BINAP, Cs2CO3, toluene, 90 °C; (e) trans-4-methylcyclohexylcarbonyl chloride, pyridine, DCE, reflux; (f) LiOH, THF, H2O, RT
From modeling, we anticipated that the polymerase surface residue Arg501 might provide hydrogen bonding opportunities if a suitably oriented H-bond acceptor was engineered into the molecule. The side chain of this residue is located approximately 4 Å from the cyclohexyl group of 1 (Figure 1). Thus, introduction of strategically placed polar residues into the pendant ring might create this desired interaction. Consideration of the chiral derivatives 3a and 3b suggested that constraining the dimethylamide back onto the amino acid side chain would create lactams with potential to interact with NS5B surface residues. Thus, a series of lactam derivatives were synthesized that provided 3- to 5-fold improvements in potency against NS5B polymerase (genotype 1a and 1b), as measured by replicon assays,14 relative to 1 and 2. The genotype 1b data for these compounds is summarized in Table 1. Each of the lactams, whether 5-, 6-, or 7-membered, or of R- or S-configuration, demonstrate strong antiviral activity in the genotype 1b replicon assay, as measured by IC50 or IC90, and good inhibition in the presence of 40% human serum. In general, S-isomers were more potent than the corresponding R-isomers, but ring size was not a significant factor. The (S)-lactams were all comparable in activity to the acyclic S-Ala N,N-dimethylamide derivative 3b.
Table 1. Structure–Activity Relationships of the Lactam Series in the Anti-Viral Replicon Assay14.


All compounds were tested in duplicate. Values are means of at least two determinations ± standard deviation.
CC50 > 20 μM for all compounds tested.
IC50 in the presence of human serum at a final concentration of 40%.
Since each compound in the series 12a–12f was an effective antiviral agent, all were advanced to in vivo pharmacokinetic studies. Table 2 summarizes the IV and PO PK studies in rats. For the 5-membered lactams 12a and 12b, the R-enantiomer 12a shows a superior IV profile in terms of clearance (CL) and exposure (AUC); however, when accounting for protein binding, the unbound profiles are essentially the same. For the 6-membered lactams 12c and 12d, again the clearance and AUC of the R-enantiomer appears superior, but the unbound exposure of S-enantiomer 12d is notably better. Finally, for the 7-membered lactams 12e and 12f, the profiles are almost identical, though again the S-enantiomer 12f shows superior unbound exposure. Following oral dosing in rats, a similar outcome is observed: both S-enantiomers 12d and 12f show higher unbound exposure than their corresponding R-enantiomers 12c and 12e. For the 5-membered lactams, 12a and 12b show similar unbound exposure following PO dosing. Based on the strong antiviral activity and encouraging PK profiles, both 12d and 12f were further evaluated as potential development compounds.
Table 2. Rat IV and PO Pharmacokinetic Profiles of Lactams 12a–12f.
| 12a | 12b | 12c | 12d | 12e | 12f | |
|---|---|---|---|---|---|---|
| Rat ppba | 99.8 | 98.1 | 99.96 | 98.9 | 99.8 | 99.2 |
| Rat IV PK Parameters | ||||||
| nb | 3 | 2 | 3 | 3 | 3 | 3 |
| dosec | 1 | 1 | 0.5 | 0.5 | 1 | 1 |
| AUC0-INFd | 1.74 ± 1.86 | 0.229 ± 0.038 | 25.4 ± 20.7 | 8.77 ± 7.31 | 1.13 ± 0.12 | 1.48 ± 0.82 |
| Unbound AUC0-INFe | 3.48 ± 3.72 | 4.35 ± 0.71 | 10.2 ± 8.3 | 96.5 ± 80 | 2.26 ± 0.23 | 11.8 ± 6.6 |
| CLf | 22.5 ± 24.1 | 73.7 ± 12.1 | 1.19 ± 1.67 | 8.30 ± 13.3 | 15.9 ± 1.59 | 16.1 ± 6.8 |
| Unbound CLf | 11,300 ± 12000 | 3880 ± 640 | 2980 ± 4180 | 755 ± 1210 | 7950 ± 800 | 2010 ± 850 |
| T1/2g | 2.73 ± 0.56 | 3.07 ± 2.54 | 5.57 ± 3.08 | 6.68 ± 3.75 | 2.79 ± 0.82 | 2.12 |
| Vssh | 1.93 ± 1.97 | 6.04 ± 3.52 | 0.221 ± 0.167 | 1.17 ± 1.20 | 1.05 | 1.95 ± 0.81 |
| Rat PO PK Parameters | ||||||
| nb | 2 | 2 | 3 | 3 | 3 | 3 |
| dosec | 3 | 3 | 1.5 | 1.5 | 3 | 3 |
| AUC0-INFd | 3.32 ± 3.98 | 0.176 ± 0.060 | 24.4 ± 29.1 | 6.39 ± 9.45 | 3.53 ± 2.55 | 3.82 ± 2.56 |
| Unbound AUC0-INFe | 6.64 ± 7.96 | 3.34 ± 1.14 | 9.76 ± 11.6 | 70.3 ± 104 | 7.06 ± 5.10 | 30.6 ± 20.5 |
| Cmaxi | 1.31 ± 1.5 | 0.119 ± 0.006 | 2.54 ± 2.15 | 0.751 ± 0.728 | 0.589 ± 0.21 | 1.04 ± 0.452 |
| %F | 64 | 26 | 32 | 24 | 122 | 97 |
| T1/2g | 6.49 ± 4.77 | 4.66 ± 1.89 | 6.13 ± 3.85 | 4.80 ± 4.36 | 4.31 ± 2.68 | 2.91 ± 1.94 |
Plasma protein binding, %.
Number of animals.
mg/kg.
μg·h/mL.
ng·h/mL.
mL/min/kg.
hours.
L/kg.
μg/mL.
A comparison of the in vitro ADME profiles of 12d and 12f revealed little distinction. Both compounds show high plasma protein binding (∼99%) and acceptable stability in rat and human liver microsomes (Supporting Information Table S2). While both 12d and 12f show no propensity for CYP inhibition (3A4, 2D6, 2C9 all >30 μM), 12d did exhibit CYP induction potential, as evaluated in human PXR-gene mediated activation, whereas 12f showed no induction potential (Table 3). A similar propensity for PXR gene activation has been reported with related thiophene carboxylate NS5B inhibitors.15 Based on this difference, 12f was selected for further profiling.
Table 3. PXR Data.
| Compound | PXR EC50(μM) | PXR max. activation vs controla | PXR max. activationb |
|---|---|---|---|
| 12c | 20.4 | 7.3-fold | 28% |
| 12d | 17.3 | 15.1-fold | 63% |
| 12e | >30 | NDc | 16% |
| 12f | >30 | NDc | 10% |
Control contains 0.1% DMSO.
Relative to 20 μM rifampicin.
Not detectable.
Further in vivo profiling is summarized in Table 4. IV pharmacokinetic profiles in dog and monkey showed good exposure and moderate clearance.
Table 4. Pharmacokinetic Parameters for Compound 12f Following a Single IV Bolus Dose of 1 mg/kg in Dogs and Monkeys.
| Species | Dog | Monkey |
|---|---|---|
| AUC0-INFa | 1.45 ± 0.815 | 0.443 ± 0.094 |
| Unbound AUC0-INFb | 10.1 ± 5.7 | 4.43 ± 0.94 |
| CLc | 15.0 ± 6.4 | 33.5 ± 7.17 |
| Unbound CLc | 2140 ± 910 | 3350 ± 717 |
| T1/2d | 1.39 ± 0.21 | 5.44 ± 0.55 |
| VSSe | 1.59 ± 0.58 | 5.27 ± 3.39 |
μg·h/mL.
ng·h/mL.
mL/min/kg.
hours.
L/kg.
An in vitro hepatocyte uptake study showed that 12f was actively transported, with 5-fold higher concentration in hepatocytes at 37 °C than at 4 °C after 15 min. Correspondingly, a rat bile duct cannulation study showed elimination primarily in bile; 35% of the dose was excreted intact in bile, suggesting both active uptake and efflux mechanisms are involved. Finally, tissue distribution studies of 12f in rats showed that exposure was highest in the liver and the second highest in plasma; it was significantly lower, by a factor of at least 10, in other major organs (Figure 2). This profile was considered ideal for an orally delivered drug targeting a disease of the liver.
Figure 2.

Mean plasma and tissue concentration–time profile of compound 12f in male Sprague–Dawley rats.
To develop the thiophene carboxylate class of allosteric HCV NS5B inhibitors, we applied both structure-guided design and conformational constraint to identify compounds with enhanced antiviral activity, possibly resulting from additional binding interactions between protein and ligand. We successfully developed a new series of compounds containing a lactam motif that provided further potency, possibly as a result of the desired designed interactions between the compounds and NS5B residue Arg501. Compound 12f is 3–5-fold more potent against NS5B polymerase relative to 1, as measured by replicon assays, and it possesses pharmaceutical properties and an in vivo profile consistent with parameters desirable for further development.
Acknowledgments
The authors wish to thank Lucille L’Heureux for replicon assay work, Olga Futer for project management, Scott Carrier and Melina Panitsidis for bioanalytical work, Zhengqi Yi for protein binding, Leena Laitinen for hepatocyte uptake data, Greg May for chiral separations, and Barry Davis for high resolution mass spectra.
Glossary
Abbreviations
- HCV
hepatitis C virus
- NS5B
nonstructural protein 5B
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00479.
General experimental details and synthetic procedures, analytical data, and microsomal stability data (PDF)
Author Present Address
§ (S.K.D.) Research Institute of the McGill University Health Center, 1001 Decarie Boulevard, Montréal, QC H4A 3J1, Canada.
Author Present Address
∥ (N.M.) Arbutus Biopharma Inc., 3805 Old Easton Road, Doylestown, PA 18902, USA.
Author Present Address
⊥ (B.G.R.) Biogen, 225 Binney Street, Cambridge, MA 02142, USA.
The authors declare the following competing financial interest(s): All of the authors of this manuscript are current or former employees of Vertex Pharmaceuticals.
Supplementary Material
References
- Gower E.; Estes C.; Blach S.; Razavi-Shearer K.; Razavi H. Global Epidemiology and Genotype Distribution of the Hepatitis C Virus Infection. J. Hepatol. 2014, 61 (1, Supplement), S45–S57. 10.1016/j.jhep.2014.07.027. [DOI] [PubMed] [Google Scholar]
- Shepard C. W.; Finelli L.; Alter M. J. Global Epidemiology of Hepatitis C Virus Infection. Lancet Infect. Dis. 2005, 5 (9), 558–567. 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- Njoroge F. G.; Chen K. X.; Shih N.-Y.; Piwinski J. J. Challenges in Modern Drug Discovery: A Case Study of Boceprevir, an HCV Protease Inhibitor for the Treatment of Hepatitis C Virus Infection. Acc. Chem. Res. 2008, 41 (1), 50–59. 10.1021/ar700109k. [DOI] [PubMed] [Google Scholar]
- Perni R. B.; Almquist S. J.; Byrn R. A.; Chandorkar G.; Chaturvedi P. R.; Courtney L. F.; Decker C. J.; Dinehart K.; Gates C. A.; Harbeson S. L.; Heiser A.; Kalkeri G.; Kolaczkowski E.; Lin K.; Luong Y. P. C.; Rao B. G.; Taylor W. P.; Thomson J. A.; Tung R. D.; Wei Y.; Kwong A. D.; Lin C. Preclinical Profile of VX-950, a Potent, Selective, and Orally Bioavailable Inhibitor of Hepatitis C Virus NS3–4A Serine Protease. Antimicrob. Agents Chemother. 2006, 50 (3), 899–909. 10.1128/AAC.50.3.899-909.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlütter J. Therapeutics: New Drugs Hit the Target. Nature 2011, 474 (7350), S5–S7. 10.1038/474S5a. [DOI] [PubMed] [Google Scholar]
- Bidell M. R.; McLaughlin M.; Faragon J.; Morse C.; Patel N. Desirable Characteristics of Hepatitis C Treatment Regimens: A Review of What We Have and What We Need. Infect. Dis. Ther. 2016, 5 (3), 299–312. 10.1007/s40121-016-0118-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Development of Antiviral Drugs for the Treatment of Hepatitis C at an Accelerating Pace. Rev. Med. Virol. 2015, 25 (4), 254–267. 10.1002/rmv.1842. [DOI] [PubMed] [Google Scholar]
- Ivashkina N.; Wölk B.; Lohmann V.; Bartenschlager R.; Blum H. E.; Penin F.; Moradpour D. The Hepatitis C Virus RNA-Dependent RNA Polymerase Membrane Insertion Sequence Is a Transmembrane Segment. J. Virol. 2002, 76 (24), 13088–13093. 10.1128/JVI.76.24.13088-13093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofia M. J.; Chang W.; Furman P. A.; Mosley R. T.; Ross B. S. Nucleoside, Nucleotide, and Non-Nucleoside Inhibitors of Hepatitis C Virus NS5B RNA-Dependent RNA-Polymerase. J. Med. Chem. 2012, 55 (6), 2481–2531. 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- Biswal B. K.; Cherney M. M.; Wang M.; Chan L.; Yannopoulos C. G.; Bilimoria D.; Nicolas O.; Bedard J.; James M. N. G. Crystal Structures of the RNA-Dependent RNA Polymerase Genotype 2a of Hepatitis C Virus Reveal Two Conformations and Suggest Mechanisms of Inhibition by Non-Nucleoside Inhibitors. J. Biol. Chem. 2005, 280 (18), 18202–18210. 10.1074/jbc.M413410200. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Torres M.; Lawitz E.; Conway B.; Kaita K.; Sheikh A. M.; Ghalib R.; Adrover R.; Cooper C.; Silva M.; Rosario M.; Bourgault B.; Proulx L.; McHutchison J. G. 31. Safety And Antiviral Activity Of The HCV Non-Nucleoside Polymerase Inhibitor VX-222 In Treatment-Naive Genotype 1 HCV-Infected Patients. J. Hepatol. 2010, 52 (Supplement 1), S14. 10.1016/S0168-8278(10)60033-5. [DOI] [Google Scholar]
- Court J. J.; Poisson C.; Ardzinski A.; Bilimoria D.; Chan L.; Chandupatla K.; Chauret N.; Collier P. N.; Das S. K.; Denis F.; Dorsch W.; Iyer G.; Lauffer D.; L’Heureux L.; Li P.; Luisi B. S.; Mani N.; Nanthakumar S.; Nicolas O.; Rao B. G.; Ronkin S.; Selliah S.; Shawgo R. S.; Tang Q.; Waal N. D.; Yannopoulos C. G.; Green J. Discovery of Novel Thiophene-Based, Thumb Pocket 2 Allosteric Inhibitors of the Hepatitis C NS5B Polymerase with Improved Potency and Physicochemical Profiles. J. Med. Chem. 2016, 59 (13), 6293–6302. 10.1021/acs.jmedchem.6b00541. [DOI] [PubMed] [Google Scholar]
- Le Pogam S.; Kang H.; Harris S. F.; Leveque V.; Giannetti A. M.; Ali S.; Jiang W.-R.; Rajyaguru S.; Tavares G.; Oshiro C.; Hendricks T.; Klumpp K.; Symons J.; Browner M. F.; Cammack N.; Nájera I. Selection and Characterization of Replicon Variants Dually Resistant to Thumb- and Palm-Binding Nonnucleoside Polymerase Inhibitors of the Hepatitis C Virus. J. Virol. 2006, 80 (12), 6146–6154. 10.1128/JVI.02628-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger N.; Lohmann V.; Bartenschlager R. Enhancement of Hepatitis C Virus RNA Replication by Cell Culture-Adaptive Mutations. J. Virol. 2001, 75 (10), 4614–4624. 10.1128/JVI.75.10.4614-4624.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes-Seeman D.; Boiselle C.; Capacci-Daniel C.; Chopra R.; Hoffmaster K.; Jones C. T.; Kato M.; Lin K.; Ma S.; Pan G.; Shu L.; Wang J.; Whiteman L.; Xu M.; Zheng R.; Fu J. Design and Synthesis of Lactam–thiophene Carboxylic Acids as Potent Hepatitis C Virus Polymerase Inhibitors. Bioorg. Med. Chem. Lett. 2014, 24 (16), 3979–3985. 10.1016/j.bmcl.2014.06.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.