

Abstract
Purpose:
To assess the effects of Aleglitazar on hyperglycaemia-induced apoptosis.
Methods:
We incubated human cardiomyocytes, cardiomyocytes from cardiac-specific peroxisome proliferator-activated receptor-γ knockout or wild-type mice in normoglycaemic or hyperglycaemic conditions (glucose 25 mM). Cells were treated with different concentrations of Aleglitazar for 48 h. We measured viability, apoptosis, caspase-3 activity, cytochrome-C release, total antioxidant capacity and reactive oxygen species formation in the treated cardiomyocytes. Human cardiomyocytes were transfected with short interfering RNA against peroxisome proliferator-activated receptor-α or peroxisome proliferator-activated receptor-γ.
Results:
Aleglitazar attenuated hyperglycaemia-induced apoptosis, caspase-3 activity and cytochrome-C release and increased viability in human cardiomyocyte, cardiomyocytes from cardiac-specific peroxisome proliferator-activated receptor-γ knockout and wild-type mice. Hyperglycaemia reduced the antioxidant capacity and Aleglitazar significantly blunted this effect. Hyperglycaemia-induced reactive oxygen species production was attenuated by Aleglitazar in both human cardiomyocyte and wild-type mice cardiomyocytes. Aleglitazar improved cell viability in cells exposed to hyperglycaemia. The protective effect was partially blocked by short interfering RNA against peroxisome proliferator-activated receptor-α alone and short interfering RNA against peroxisome proliferator-activated receptor-γ alone and completely blocked by short interfering RNA to both peroxisome proliferator-activated receptor-α and peroxisome proliferator-activated receptor-γ.
Conclusion:
Aleglitazar protects cardiomyocytes against hyperglycaemia-induced apoptosis by combined activation of both peroxisome proliferator-activated receptor-α and peroxisome proliferator-activated receptor-γ in a short-term vitro model.
Keywords: Aleglitazar, peroxisome proliferator-activated receptor-α and peroxisome proliferator-activated receptor-γ agonist, hyperglycaemia, cardiomyocytes, apoptosis
Introduction
Type 2 diabetes mellitus (T2DM) is associated with an increased incidence of myocardial infarction and diabetic cardiomyopathy.1–4 Clinical guidelines state that the increased cardiovascular risk in patients with T2DM can be reduced by controlling dyslipidemia, blood pressure, body weight and hyperglycaemia.5 However, most patients still do not achieve recommended goals for these risk factors.6 This highlights the need for further decreasing the cardiovascular risk in individuals with T2DM. Diabetic cardiomyopathy, characterized by left ventricular hypertrophy, myocardial apoptosis, fibrosis and dysfunction, is a common complication of T2DM.3,4 T2DM is associated with poorer outcomes following myocardial infarction.7 Cardiac hypertrophy is associated with deactivation of peroxisome proliferator-activated receptor-α (PPARα).8 We and others have shown that peroxisome proliferator-activated receptor-γ (PPARγ) agonists, especially of the thiazolidinedione family, attenuate myocardial infarct size and post-ischaemic dysfunction in normal and diabetic animals.9–12 However, the protective effects of pioglitazone were reported to be independent of cardiomyocyte PPARγ activation.9 There are data showing that PPARα agonists also protect against ischaemia-reperfusion injury.13–17 Aleglitazar (Ale) is a non-thiazolidinedione dual PPARα and PPARγ agonist that has glucose-lowering and lipid-modifying effects.18,19 Ale was developed for the potential treatment of hyperglycaemia and dyslipidemia in patients with T2DM. It was shown that Ale protects the pancreas, kidneys and eyes of male Zucker diabetic fatty (ZDF) rats against diabetic complications.20 Studies suggested that Ale could minimize PPAR-related adverse effects and weight gain in the treatment of patients with T2DM.18 Preclinical and phase I studies have shown favourable effects of Ale on glycaemic control, insulin sensitivity and dyslipidemia.19 The aim of this study was to assess whether dual activation of PPARα and PPARγ by Ale would have beneficial anti-apoptosis effects in human and murine cardiomyocytes exposed to hyperglycaemia. The data generated from these in vitro experiments might have potential therapeutic implications for the treatment of diabetic cardiomyopathy. During the conduction of the study, however, Roche announced that following the results of a regular safety review of the Ale AleCardio phase III trial,21,22 the independent Data and Safety Monitoring Board (DSMB) has recommended halting the trial due to safety signals and lack of efficacy with respect to reduction in cardiovascular events and mortality. Based on this recommendation, the manufacturer (F. Hoffmann-La Roche Ltd, Basel, Switzerland) has decided to terminate the AleCardio trial and all other trials involving Ale (http://www.roche.com/media/store/releases/med-cor-2013-07-10.htm). However, some aspects of the drug have not been adequately evaluated. Evidence from several recent large, randomized, controlled trials suggests that improved glycaemic control is associated with a reduced risk of cardiovascular disease (CVD), but that this effect may be greater in individuals with a shorter duration of diabetes and without CVD, and that it may take many years of good glycaemic control to translate into cardiovascular (CV) risk reduction.23–25 So, Ale may reduce cardiomyocyte apoptosis induced by high glucose of the newly diagnosed patients with diabetes and dyslipidemia and attenuate the progression of atherosclerosis.
Material and methods
Isolation of cardiomyocytes from cardiac-specific PPARγ knockout mice
Cardiac-specific inducible PPARγ KO (PPARγCKO) mice were generated by crossing B6.Cg-Tg(Myh6-cre/Esr1)1Jmk/J mice with B6.129-Ppargtm2Rev/J mice in our laboratory, as described before.9 Mice carrying both loxp and Cre transgene were selected to perform the studies. PPARγ deletion in cardiomyocytes was achieved after 7 days of tamoxifen administration (TAM; 20 mg/kg/day intraperitoneal injections). Cardiomyocytes from 3-month-old PPARγCKO or wild-type (WT) mice were isolated using methods previously described with minor modification.9 Mice were heparinized (5 IU/g) and anaesthetized with ketamine (60 mg/kg) and xylazine (6 mg/kg). Hearts were excised and cannulated via the aorta and connected to the perfusion apparatus. Hearts were perfused for 3 min at a rate of 3 mL/min with calcium-free media. Hearts were perfused with the modified Eagle’s medium (MEM) containing Type-II collagenase (1 mg/mL) and 20 µM CaCl2 at a rate of 3 mL/min for 15 min. After perfusion, atria were removed and ventricles were cut into small pieces and minced in collagenase solution for 6 min. Myocytes were washed and plated on laminin-coated plate in media 199 with 4% fetal bovine serum (FBS) and 100 U penicillin and returned to the incubator for attachment (1 h). After attachment, non-attached, rounded cells were washed away and the media was replaced with fresh serum-free medium to remove all non-myocytes. After additional 16 h, the medium was replaced with normal culture medium (MEM) with Hanks’ Balanced Salt Solution, supplemented with 0.1 mg/mL bovine serum albumin and penicillin 100 U/mL at 37°C in a 2% CO2 incubator. After 24 h, when the 88% of the cultured myocytes showed rod-shaped, and viability was 90%, we started the experiments. Knocking out of PPARγ in the cardiomyocytes was confirmed by immunoblotting and reverse transcription polymerase chain reaction (RT-PCR).
Human cardiomyocytes
Primary human cardiomyocytes (HCM) are cultured cardiac myocytes. These cells have been used as a model for drug development and predictive toxicity testing. HCMs have been used in studies of human gene expression,26 oxidative stress,27 diabetes and apoptosis.28 HCMs are isolated from human adult heart tissue29 and were purchased from ScienCell Research Laboratories (USA).
Determine the dosage range of Ale in cardiomyocytes culture
At first, we determined the concentration range of Ale that will not cause toxicity in vitro. Lactate dehydrogenase (LDH) was measured as an indicator of cell viability [LDH release detection kit (Roche)]. HCMs and mCM-WT cardiomyocytes were seeded in 200 µL medium for 96-well plate with a density of 8 × 102 cells/mm2 and then treated with or without Ale at different concentrations (0–40 nM) for different time points (12, 24 or 48 h).30 Ale was dissolved in 0.1% dimethyl sulfoxide (DMSO). LDH activity was measured with two replicates for each condition at an absorbance of 490 nm. WT cardiomyocytes not exposed to Ale served as a control. Myocytes from one mouse were used for each experiment. In total, six PPARγCKO mice and six WT mice were used. Cytoplasmic enzyme release was shown as a ratio of the released LDH into the media to the total LDH (release plus cellular content) at the end of treatment.
To determine the effect of Ale on apoptosis, caspase-3 activity and mitochondrial cytochrome-C release
To explore the effect of Ale on cardiomyocytes apoptosis and leakage of cytochrome-C (Cyt-c) induced by hyperglycaemia (HG, glucose 25 mM), we incubated cardiomyocytes from human (HCMs) or mice (PPARγCKO or WT) in normoglycaemic condition (NG, glucose 5.5 mM) or hyperglycaemic conditions (HG, glucose 25 mM). Cells were treated with different concentrations of Ale for 48 h. The degree of intracellular DNA fragmentation (apoptosis) was quantified using the Cell Death Detection ELISAPLUS kit (Roche, Hertfordshire, UK) per manufacturer instruction.30 Caspase-3 activity was performed using a colorimetric activity assay kit according to the manufacturer’s instructions. The cells were harvested and then suspended in the cell lysis buffer to obtain cell lysate. Protein concentration was determined using Lowry Protein Assay, and 200 mg protein of cell lysate was incubated in 100 mL of reaction buffer containing 5 mL of caspase-3 substrate (4 mM DEVD-pNA) in 96-well plates. The reaction buffer contained 1% NP-40, 20 mM Tris–HCl (pH 7.5), 137 mM NaCl and 10% glycerol. The samples were incubated in the dark and caspase-3 activity was evaluated using a spectrophotometer at 405 nm.9 We also measured translocation of cytochrome-C from mitochondria into cytosol during apoptosis. Cytosolic fractions were obtained at the end of the treatments and cell lysate was prepared. The release of mitochondrial cytochrome-C into the cytosol was determined by ELISA (MBL International, Woburn, MA), per the manufacturer instructions. The change in colour was monitored at a wavelength of 450 nm using a plate reader (Molecular Devices, Sunnyvale, CA). Measurements were performed in duplicate, and the Cyt-c content was expressed as OD450 per mg protein.9 A total of 6 PPARγCKO mice and 6 WT were used.
Viability assays
Cells were pretreated with different concentrations of Ale, and cell death was induced by exposure of cells to HG. Cells incubated in NG condition served as a control. The viability of cells (with and without exposure to HG) was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay kit [R&D systems (Minneapolis, MN, USA)]. The HG-induced cell death was examined by counting trypan-blue stained cells, and the results were expressed as a percentage of the total cells counted, based on the ability of live cells to exclude trypan blue. For each well, we counted 250–300 cells. 3-[4,5-yl]-2,5-diphenyltetrazolium bromide (MTT, 0.5 mg/mL) was used to treat the cells for 4 h at 37°C. The attached cells were lysed in 2-isopropanol containing 0.04 M HCl, and the amount of metabolized MTT was determined using a microplate reader.9 A total of 4 PPARγCKO mice and 4 WT mice were used.
Total antioxidant capacity assay
The total antioxidant capacity of Ale in cells was evaluated by Trolox equivalent antioxidant capacity assay using a standard antioxidant assay kit (Cayman, Ann Arbor, MI, USA). Briefly, the cells were homogenized on ice in a buffer containing 5 mM potassium phosphate; 0.9% NaCl; and 0.1% glucose, pH 7.4. The samples were centrifuged, and the supernatant were collected for the assay according to manufactory instruction, and the absorbance was read at 405 nm using a microplate reader. A total of 6 PPARγCKO mice and 6 WT mice were used.
Detection of reactive oxygen species generation
To determine the effect of Ale on reactive oxygen species (ROS) formation, cardiomyocytes were treated as above. 50 µM 2′7′-dichlorofluorescin (DCF)-diacetate was added for 30 min at 37°C. The intracellular ROS concentration was quantified by the measurement of DCF fluorescence intensity using an excitation wavelength of 485 nm and emission wavelength of 524 nm. A total of 5 PPARγCKO mice and 5 WT mice were used.
Statistical analysis
Results are reported as mean ± standard error (SE). Data were compared using analysis of variance (ANOVA) with Sidak corrections for multiple comparisons. Values of p < 0.05 were considered statistically significant.
Results
Effects of Ale on cytotoxicity
At first we looked for the effects of escalating concentrations of Ale on LDH release to assess for potential toxicity in HCM and wild-type mouse cardiomyocytes (mCM-WT). Cells were incubated for 12, 24 or 48 h with escalating concentrations of Ale, and the percent difference in LDH release was calculated (Figure 1). Ale at concentrations of 0.1 to 20 µM did not significantly increase LDH release compared to vehicle alone. However, at concentrations of 30 and 40 µM, there was a significant increase in LDH release. Therefore, in the next experiments, we used Ale at concentration up to 20 µM. The reported through blood levels of Ale in humans are 0.0017–0.01 µM.31
Figure 1.

Effects of Aleglitazar on cytotoxicity in human cardiomyocytes (HCM) and wild-type mice cardiomyocytes (mCM-WT).
The cells were incubated with different concentrations of Aleglitazar (0–40 µM) for 12, 24 or 48 h. *p < 0.05 versus 0.0 µM Ale group (n = 4 in each group).
Effects of Ale on HG-induced apoptosis, caspase-3 activity and cytochrome-C release
Cells were incubated at normoglycaemic condition (NG, glucose 5.5 mM) or hyperglycaemic conditions (HG, glucose 25 mM) with escalating concentration of Ale (0.01–20 µM) for 48 h (Figure 2). To evaluate the effect of Ale on HG-induced apoptosis, we measured DNA fragmentation (a late marker of apoptosis) and caspase-3 activity (an early marker of apoptosis).32 HG increased apoptosis, caspase-3 activity and cytochrome-C release in HCM, mCM-WT and mCM-PPARγ KO cardiomyocytes. Ale, dose dependently, decreased apoptosis, caspase-3 activity and cytochrome-C release. The effects were significantly different from HG without Ale at 1.0–20 µM for HCM and mCM-WT and 10–20 µM for the mCM-PPARγ KO cardiomyocytes, suggesting that at lower concentrations most of the protective effects of Ale are PPARγ dependent (Figure 2), whereas at higher concentration, PPARα activation partially mediates the protective effects.
Figure 2.

Effects of Aleglitazar on apoptosis, caspase-3 activity and cytochrome-C induced by HG in human cardiomyocytes (HCM), wild-type mouse cardiomyocytes (mCM-WT) and cardiac-specific inducible PPARγ KO mice cardiomyocytes (mCM-PPARγ KO). Caspase-3 activity is expressed as a ratio of the activity in each group/the activity in the normoglycaemic (NG) group. *p < 0.05 versus 0.0 µM Ale group (n = 6 in each group).
NG: normoglycaemia; HG: hyperglycaemia.
Cell viability (MTT)
HCM, mCM-WT and mCM-PPARγ-KO cardiomyocytes were incubated in NG or HG without or with escalating concentrations of Ale (Figure 3). There was a significant decrease in cell viability in the cultures exposed to HG without Ale. Ale at concentrations of 0.5–20, 1.0–20 and 10–20 µM increased cell viability in the HCM, mCM-WT and mCM-PPARγ-KO cardiomyocytes, respectively. Yet, at 20 µM, the protective effect of Ale was smaller in the mCM-PPARγ-KO cardiomyocytes than in the HCM and mCM-WT cardiomyocytes. These findings suggest that the protection effect exerted by Ale at lower concentrations (1.0 µM) is PPARγ dependent, whereas at higher concentrations (10 and 20 µM) it is mediated by both PPARα and PPARγ activation (Figure 3).
Figure 3.

Cell viability (MTT) in cells exposed to hyperglycaemia (HG) without and with escalating concentrations of Ale. Results are expressed as percentage of the normoglycaemia (NG) group. *p < 0.001 versus 0.0 µM Ale group (n = 6 in each group).
Antioxidant capacity
Exposure to HG for 48 h reduced the antioxidant capacity of the cardiomyocytes. Ale, dose dependently, blunted the effects of HG. Again in mCM-PPARγ-KO cardiomyocytes, Ale had a significant effect only at concentrations of 10 and 20 µM. In contrast, Ale had a significant effect at 1.0 µM in HCM and mCM-WT, suggesting that at lower concentrations the effect of Ale is mainly PPARγ dependent (Figure 4).
Figure 4.
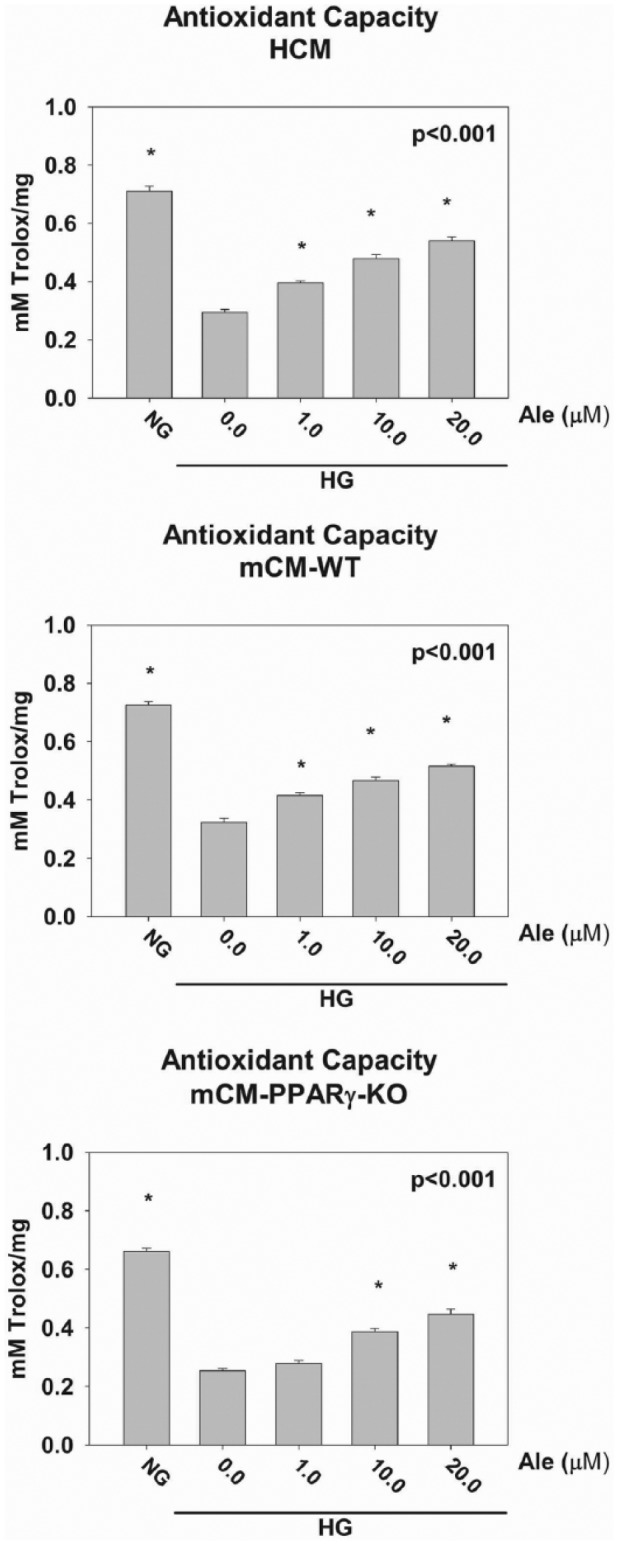
Exposure to HG for 48 h reduced the antioxidant capacity of the cardiomyocytes. Ale dose dependently blunted the effects of HG. *p < 0.05 versus 0.0 M Ale group (n = 6 per group).
NG: normoglycaemia; HG: hyperglycaemia.
ROS generation
Treatment of cardiomyocytes with HG for 48 h significantly increased ROS levels. This effect of HG on ROS production was significantly attenuated by 1.0, 10 and 20 µM of Ale in both HCM and mCM-WT cardiomyocytes, whereas only at 10 and 20 µM of Ale in mCM-PPARγ KO cardiomyocytes, suggesting again that at lower concentrations, the effect of Ale is PPARγ dependent. Moreover, at 10 and 20 µm, more attenuation was seen in the HCM and mCM-WT cardiomyocytes than in the mCM-PPARγ KO cardiomyocytes, suggesting involvement of both the PPARα and PPARγ receptors (Figure 5).
Figure 5.
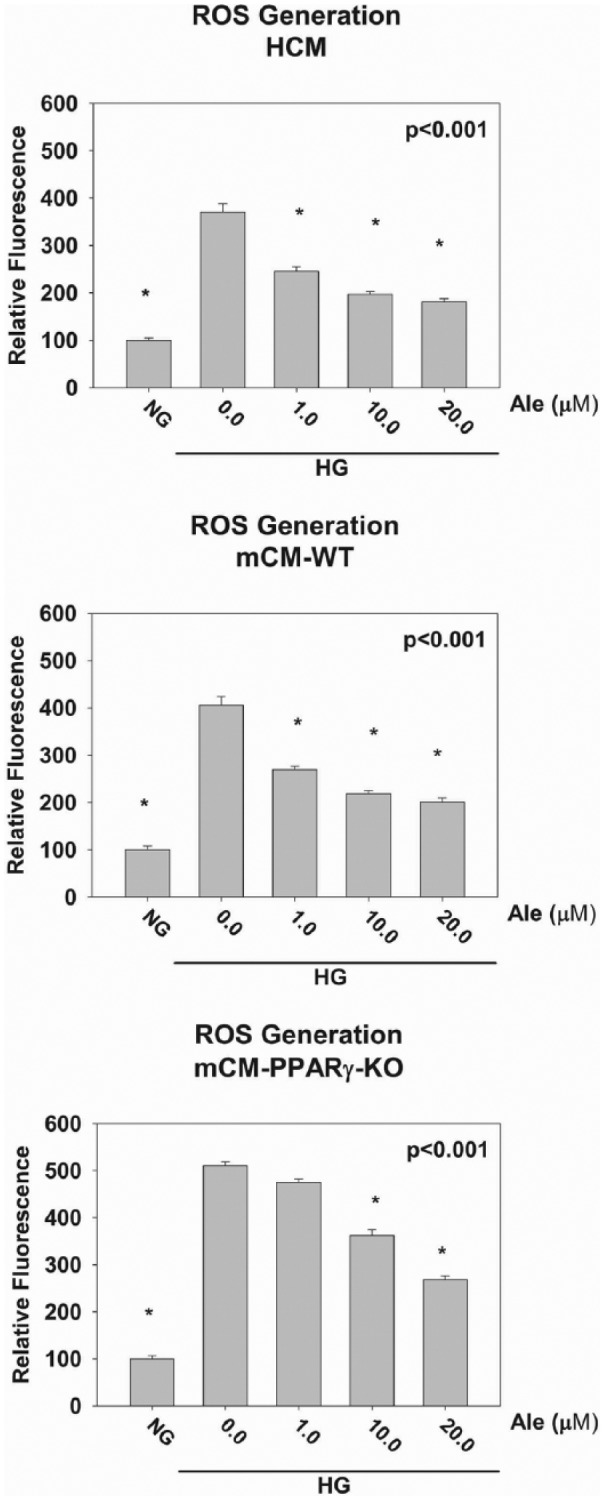
Effects of Aleglitazar on ROS generation caused by HG. *p < 0.05 versus 0.0 µM Ale group (n = 6 per group).
The effects of PPARγ and PPARα silencing on the protective effect of Ale on cell viability in cardiomyocytes exposed to HG
HG reduced cell viability. Ale (20 µM) improved viability. The protective effect was partially blocked by siRNA to PPARα (siPPARα) or PPARγ (siPPARγ). When both PPARα and PPARγ were silenced, the protective effect was completely lost. These experiments show that the protective effect in both HCM and mCM-WT is mediated via combined activation of PPARα and PPARγ (Figure 6).
Figure 6.

The effects of PPARγ and PPARα silencing on the protective effect of Ale on cell viability in cardiomyocytes exposed to HG. *p < 0.05 versus HG; #p < 0.05 versus HG + Ale (n = 6 per group).
Discussion
In this study, we found that activation of both PPARα and PPARγ by Ale increased cell viability and decreased HG-induced increased apoptosis, caspase-3 activity and Cyt-c release in a dose-dependent manner. HG reduced the antioxidant capacity of the cardiomyocytes and increased ROS concentrations, whereas Ale attenuated these adverse effects of HG. It seems that at low concentration, the protective effects of Ale are mediated mainly by PPARγ activation, whereas at higher concentration both PPARα and PPARγ are involved. An alternative explanation is that PPARα activation facilitates or has synergistic effects with the effects of PPARγ activation. siRNA against PPARα or PPARγ partially blocked the protective effect. However, silencing both PPARα and PPARγ completely blocked the effect of Ale.
HG induces myocardial apoptosis, leading to diabetic cardiomyopathy.33 HG induces Cyt-c release.33 The leakage of Cyt-c from mitochondria into the cytoplasm is known to activate caspases and initiate apoptosis.34 Previous studies revealed that ischaemia/reperfusion promotes myocardial apoptosis via mitochondrial Cyt-c-mediated caspase-3 activation and is dependent on ROS production.33 Werner et al.35 reported that Ale reduces oxidative stress-induced apoptosis in circulating angiogenic cells. In this study, HG-induced cardiomyocyte apoptosis, caspase-3 activation and Cyt-c release were dose dependently attenuated by Ale. Furthermore, Ale improved viability of the cells exposed to HG.
The protective effects of metformin against ischaemia-reperfusion injury are PPARα-dependent.36 Metformin attenuated the decrease in state 3 respiration and complexes I and II and decreased mitochondrial permeability transition pore (PTP) opening. It was suggested that PPARα activation by metformin abolishes oxidative stress-induced physical interactions between PPARα and cyclophilin D and thus with inhibition of PTP formation.37 On the other hand, some of the cardioprotective effects of PPARα activation are related to activation of adenosine monophosphate-activated protein (AMP) kinase,37 stimulation of fatty acid β-oxidation with improved adenosine triphosphate (ATP) production,38,39 and to its anti-inflammatory properties.40 Therefore, by activating PPARα, Ale can attenuate mitochondria-mediated cell death (as shown in this study as attenuation of Cyt-c release). Others identified insulin-like growth factor-1 as the downstream target of PPARα activation that mediates the protective effect.15 Moreover, it has been shown that PPARα agonists protect the heart against ischaemia-reperfusion injury by upregulating the prosurvival mediators PI3-Kinase/Akt, endothelial nitric oxide synthase and endothelin-1.11,13,14
The protective effects of PPARγ agonists are also ascribed to activation of the prosurvival mediators PI3-Kinase/Akt and cyclooxygenase 2 (COX2).9–11 Thus, by activation of both the PPARα and PPARγ receptors, additive effects on the activation of the prosurvival pathways are expected.
A direct association between hyperglycaemia and oxidative stress in diabetic injuries has been shown.41,42 Ho et al.43 demonstrated that exposure of human umbilical vein endothelial cells in vitro to HG caused a significant increase in ROS formation in association with caspase-3 activation and apoptosis. Addition of ascorbic acid as an antioxidant agent to the cell culture suppressed the activation of caspase-3 and the induction of apoptotic cell death.43 Mitochondria from diabetic hearts have morphological and functional damage coupled with lipid oxidation and ROS generation.44,45 Furthermore, ROS formation induced by hyperglycaemia can ultimately cause cardiomyocyte apoptosis.28,46,47 In this study, the effects of HG on ROS production and antioxidant capacity were significantly attenuated by 1.0, 10 and 20 µM of Ale in both the HCM and mCM-WT, whereas only at 10 and 20 µM of Ale in the mCM-PPARγ KO cardiomyocytes. Ale was significantly effective in improving cell viability at concentrations higher than 0.5, 1.0 and 10.0 µM in HCM, mCM-WT and mCM-PPARγ-KO, respectively. The findings revealed that the protection effects exerted by Ale at lower concentrations are probably PPARγ dependent, whereas at higher concentration, PPARα activation partially mediates the effect. Our experiments using siRNAs to PPARα and PPARγ further support this observation. In a previous study, we showed that Ale increased the DNA binding capacity of both PPARα and PPARγ (relatively more increase in PPARα than PPARγ in HCM, whereas more increase in PPARγ than PPARα in mCM-WT).30 Silencing PPARα alone or PPARγ alone partially blocked the effect of Ale on cell viability in both HCM and mCM-WT, whereas silencing both PPARα and PPARγ completely blocked the protective effect. This suggests that in contrast to pioglitazone,9 Ale does not have PPAR-independent effects.
In contrast to our encouraging results with short-term in vitro exposure, a recent clinical study found that long-term Ale did not reduce the risk of cardiovascular events and mortality in patients with T2DM and recent acute coronary syndrome.22 Benardeau et al.20 also showed that Ale improved glycaemic control and protected the pancreatic beta-cells and the kidneys of ZDF rats. Many agents that have shown promising results in in vitro and in vivo animal models failed to show beneficial effects in the clinical setting. The potential factors that could explain these discrepancies have been discussed.48,49 These factors include the animal models, length of therapy, dose and concentrations of drugs and interactions with other agents commonly administrated in the clinical setting. Specifically, here we assessed the short-term protective effects of Ale against high concentrations of glucose (25 mM = 450 mg/dL) in vitro. These short-term protective effects may not be translated to long-term protective effects in the clinical setting, especially as most of the diabetic patients are not exposed to such high glucose concentrations with the concomitant use of anti-diabetic medications. Another important issue is the drug concentration. Wright et al.50 have shown that the effects of both Ale and muraglitazar on the expression of various genes in primary HCM change at different drug concentrations. Similar findings have been described in primary human hepatocytes.51 Ale had a more potent effect on PPARα than on PPARγ activation in HCM, whereas in the mCM-WT, PPARγ activation was more prominent, it might be that in the concentrations achieved in the clinical setting, these favourable effects cannot be reached. We used drug concentrations slightly higher than the reported through levels in patients (0.0017–0.01 µM).31 Thus, the long-term effects of Ale in the clinical setting could be different from those seen in our short-term experiments. And finally, we cannot exclude that long-term exposure to the drug may induce adverse effects or drug–drug interactions that could potentially offset the protective effects against high glucose levels. An additional explanation could be the duration of diabetes before exposure to the drug. Evidence from several recent large, randomized, controlled trials suggests that improved glycaemic control is associated with a reduced risk for CVD, but that this effect may be greater in individuals with a shorter duration of diabetes and without CVD, and that it may take many years of good glycaemic control to translate into CV risk reduction.52,53 It remains unknown whether or not Ale can improve cardiovascular outcomes in patients with short-term T2DM. Unfortunately, further clinical studies and the use of Ale in the clinical setting were terminated (http://www.roche.com/media/media_releases/med-cor-2013-07-10.htm).
The investigational agent, muraglitazar, was associated with an apparent excess of adverse cardiac ischaemic events in an aggregate analysis of small phase-2 and phase-3 trials,54 whereas development of another dual agonist, tesaglitazar, was also terminated due to renal toxicity.55 Yet, newer dual PPARα/γ agonists are being developed.56–58 Thus, overcoming drug interactions and adverse effects, dual PPARα/γ might still show efficacy in reducing cardiovascular morbidity and mortality in patients with T2DM. However, the exact spectrum and magnitude of gene activation by each of the specific dual PPARα/γ agonists differ and is also affected by the drug concentration.50,51 Thus, results, obtained with one dual PPARα/γ agonist at a specific concentration, cannot be inferred to the other PPARα/γ agonist agents.
The reported serum trough levels of Ale in humans are 0.0017–0.01 µM.31 The concentrations that were associated with a significant effect in our in vitro experiments are much higher. Therefore, it is unclear whether similar protective effects can be seen in in vivo animal models or in the clinical setting. Yet, we found that at 5 mg/kg/day, Ale reduced experimental infarct size in db/db mice exposed to 30 min coronary artery occlusion followed by 24 h reperfusion.30
The limitation of the study is that no in vivo experiments were done to clarify whether the favourable antioxidant, anti-hypertrophic and cardioprotective effects of Ale, observed in short-term in vitro exposure, can be seen in diabetic animals. Future studies are warranted to address these issues. Furthermore, our findings cannot be extrapolated to the clinical practice.
Conclusion
Ale protects cardiomyocytes against hyperglycaemia-induced apoptosis by combined activation of both PPARα and PPARγ in an in vitro short-term model.
Acknowledgments
Yan Chen and Hongmei Chen contributed equally to this work.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding: This work was supported by the grants from F. Hoffmann-La Roche Ltd, National Natural Science Foundation of China (81160035 and 81460044), Yunnan Provincial Science and Technology Department (2014HB031 and 2011FZ119) and Yunnan Provincial Bureau of Health (D201211). M.B. has received research grants from AstraZeneca, Boehringer Ingelheim, Eli Lilly and Novo Nordisk. He has received lecture fees from Takeda Pharmaceuticals and is a consultant to Merck and Genentech. Y.Y. and Y.B. received research grants from AstraZeneca and Boehringer Ingelheim. Y.B. received lecture fees from AstraZeneca.
References
- 1. Haffner SM, Lehto S, Ronnemaa T, et al. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339: 229–234. [DOI] [PubMed] [Google Scholar]
- 2. Harjai KJ, Stone GW, Boura J, et al. Comparison of outcomes of diabetic and nondiabetic patients undergoing primary angioplasty for acute myocardial infarction. Am J Cardiol 2003; 91: 1041–1045. [DOI] [PubMed] [Google Scholar]
- 3. Rubler S, Dlugash J, Yuceoglu YZ, et al. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 1972; 30: 595–602. [DOI] [PubMed] [Google Scholar]
- 4. Hayat SA, Patel B, Khattar RS, et al. Diabetic cardiomyopathy: mechanisms, diagnosis and treatment. Clin Sci 2004; 107: 539–557. [DOI] [PubMed] [Google Scholar]
- 5. Graham I, Atar D, Borch-Johnsen K, et al. European guidelines on cardiovascular disease prevention in clinical practice: executive summary. Atherosclerosis 2007; 194: 1–45. [DOI] [PubMed] [Google Scholar]
- 6. Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA 2004; 291: 335–342. [DOI] [PubMed] [Google Scholar]
- 7. Lehto S, Pyorala K, Miettinen H, et al. Myocardial infarct size and mortality in patients with non-insulin-dependent diabetes mellitus. J Intern Med 1994; 236: 291–297. [DOI] [PubMed] [Google Scholar]
- 8. Barger PM, Brandt JM, Leone TC, et al. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest 2000; 105: 1723–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Birnbaum Y, Long B, Qian J, et al. Pioglitazone limits myocardial infarct size, activates Akt, and upregulates cPLA2 and COX-2 in a PPAR-gamma-independent manner. Basic Res Cardiol 2011; 106: 431–446. [DOI] [PubMed] [Google Scholar]
- 10. Ye Y, Hu Z, Lin Y, et al. Downregulation of microRNA-29 by antisense inhibitors and a PPAR-gamma agonist protects against myocardial ischaemia-reperfusion injury. Cardiovasc Res 2010; 87: 535–544. [DOI] [PubMed] [Google Scholar]
- 11. Ye Y, Lin Y, Manickavasagam S, et al. Pioglitazone protects the myocardium against ischemia-reperfusion injury in eNOS and iNOS knockout mice. Am J Physiol Heart Circ Physiol 2008; 295: H2436–H2446. [DOI] [PubMed] [Google Scholar]
- 12. Makino N, Maeda T, Oyama J, et al. Improving insulin sensitivity via activation of PPAR-gamma increases telomerase activity in the heart of OLETF rats. Am J Physiol Heart Circ Physiol 2009; 297: H2188–H2195. [DOI] [PubMed] [Google Scholar]
- 13. Bulhak AA, Jung C, Ostenson CG, et al. PPAR-alpha activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: involvement of the PI3-Kinase/Akt and NO pathway. Am J Physiol Heart Circ Physiol 2009; 296: H719–H727. [DOI] [PubMed] [Google Scholar]
- 14. Bulhak AA, Sjoquist PO, Xu CB, et al. Protection against myocardial ischaemia/reperfusion injury by PPAR-alpha activation is related to production of nitric oxide and endothelin-1. Basic Res Cardiol 2006; 101: 244–252. [DOI] [PubMed] [Google Scholar]
- 15. El Azzouzi H, Leptidis S, Bourajjaj M, et al. Peroxisome proliferator-activated receptor (PPAR) gene profiling uncovers insulin-like growth factor-1 as a PPARalpha target gene in cardioprotection. J Biol Chem 2011; 286: 14598–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yue TL, Bao W, Jucker BM, et al. Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation 2003; 108: 2393–2399. [DOI] [PubMed] [Google Scholar]
- 17. Yue TL, Nerurkar SS, Bao W, et al. In vivo activation of peroxisome proliferator-activated receptor-delta protects the heart from ischemia/reperfusion injury in Zucker fatty rats. J Pharmacol Exp Ther 2008; 325: 466–474. [DOI] [PubMed] [Google Scholar]
- 18. Hansen BC, Tigno XT, Benardeau A, et al. Effects of aleglitazar, a balanced dual peroxisome proliferator-activated receptor alpha/gamma agonist on glycemic and lipid parameters in a primate model of the metabolic syndrome. Cardiovasc Diabetol 2011; 10: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benardeau A, Benz J, Binggeli A, et al. Aleglitazar, a new, potent, and balanced dual PPARalpha/gamma agonist for the treatment of type II diabetes. Bioorg Med Chem Lett 2009; 19: 2468–2473. [DOI] [PubMed] [Google Scholar]
- 20. Benardeau A, Verry P, Atzpodien EA, et al. Effects of the dual PPAR-alpha/gamma agonist aleglitazar on glycaemic control and organ protection in the Zucker diabetic fatty rat. Diabetes Obes Metab 2013; 15: 164–174. [DOI] [PubMed] [Google Scholar]
- 21. Erdmann E, Califf R, Gerstein HC, et al. Effects of the dual peroxisome proliferator-activated receptor activator aleglitazar in patients with Type 2 Diabetes mellitus or prediabetes. Am Heart J 2015; 170: 117–122. [DOI] [PubMed] [Google Scholar]
- 22. Lincoff AM, Tardif JC, Schwartz GG, et al. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: the AleCardio randomized clinical trial. JAMA 2014; 311: 1515–1525. [DOI] [PubMed] [Google Scholar]
- 23. Leiter LA. Outcome trials on the effects of glycemic control on cardiovascular risk in type 2 diabetes mellitus – what do we know and what will we learn from the ongoing trials? Medicographia 2013; 35: 40–47. [Google Scholar]
- 24. Group UPDSU. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352: 837–853. [PubMed] [Google Scholar]
- 25. Zoungas S, de Galan BE, Ninomiya T, et al. Combined effects of routine blood pressure lowering and intensive glucose control on macrovascular and microvascular outcomes in patients with type 2 diabetes: new results from the ADVANCE trial. Diabetes Care 2009; 32: 2068–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hussein S, Michael P, Brabant D, et al. Characterization of human septic sera induced gene expression modulation in human myocytes. Int J Clin Exp Med 2009; 2: 131–148. [PMC free article] [PubMed] [Google Scholar]
- 27. Li SY, Li Q, Shen JJ, et al. Attenuation of acetaldehyde-induced cell injury by overexpression of aldehyde dehydrogenase-2 (ALDH2) transgene in human cardiac myocytes: role of MAP kinase signaling. J Mol Cell Cardiol 2006; 40: 283–294. [DOI] [PubMed] [Google Scholar]
- 28. Li SY, Sigmon VK, Babcock SA, et al. Advanced glycation endproduct induces ROS accumulation, apoptosis, MAP kinase activation and nuclear O-GlcNAcylation in human cardiac myocytes. Life Sci 2007; 80: 1051–1056. [DOI] [PubMed] [Google Scholar]
- 29. Wei L, Lu J, Feng L, et al. HIF-1alpha accumulation upregulates MICA and MICB expression on human cardiomyocytes and enhances NK cell cytotoxicity during hypoxia-reoxygenation. Life Sci 2010; 87: 111–119. [DOI] [PubMed] [Google Scholar]
- 30. Qian J, Chen H, Birnbaum Y, et al. Aleglitazar, a balanced dual PPARalpha and -gamma agonist, protects the heart against ischemia-reperfusion injury. Cardiovasc Drugs Ther 2016; 30: 129–141. [DOI] [PubMed] [Google Scholar]
- 31. Younk LM, Uhl L, Davis SN. Pharmacokinetics, efficacy and safety of aleglitazar for the treatment of type 2 diabetes with high cardiovascular risk. Expert Opin Drug Metab Toxicol 2011; 7: 753–763. [DOI] [PubMed] [Google Scholar]
- 32. Lin H, Qian J, Castillo AC, et al. Effect of miR-23 on oxidant-induced injury in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 2011; 52: 6308–6314. [DOI] [PubMed] [Google Scholar]
- 33. Cai L, Li W, Wang G, et al. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 2002; 51: 1938–1948. [DOI] [PubMed] [Google Scholar]
- 34. De Moissac D, Gurevich RM, Zheng H, et al. Caspase activation and mitochondrial cytochrome C release during hypoxia-mediated apoptosis of adult ventricular myocytes. J Mol Cell Cardiol 2000; 32: 53–63. [DOI] [PubMed] [Google Scholar]
- 35. Werner CM, Schirmer SH, Gensch C, et al. The dual PPARalpha/gamma agonist aleglitazar increases the number and function of endothelial progenitor cells: implications for vascular function and atherogenesis. Br J Pharmacol 2014; 171: 2685–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barreto-Torres G, Parodi-Rullan R, Javadov S. The role of PPARalpha in metformin-induced attenuation of mitochondrial dysfunction in acute cardiac ischemia/reperfusion in rats. Int J Mol Sci 2012; 13: 7694–7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barreto-Torres G, Hernandez JS, Jang S, et al. The beneficial effects of AMP kinase activation against oxidative stress are associated with prevention of PPARalpha-cyclophilin D interaction in cardiomyocytes. Am J Physiol Heart Circ Physiol 2015; 308: H749–H758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lam VH, Zhang L, Huqi A, et al. Activating PPARalpha prevents post-ischemic contractile dysfunction in hypertrophied neonatal hearts. Circ Res 2015; 117: 41–51. [DOI] [PubMed] [Google Scholar]
- 39. Ravingerova T, Carnicka S, Ledvenyiova V, et al. Upregulation of genes involved in cardiac metabolism enhances myocardial resistance to ischemia/reperfusion in the rat heart. Physiol Res 2013; 62(suppl. 1): S151–S163. [DOI] [PubMed] [Google Scholar]
- 40. Ibarra-Lara ML, Sanchez-Aguilar M, Soria E, et al. Peroxisome proliferator-activated receptors (PPAR) downregulate the expression of pro-inflammatory molecules in an experimental model of myocardial infarction. Can J Physiol Pharmacol 2016; 94: 634–642. [DOI] [PubMed] [Google Scholar]
- 41. Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404: 787–790. [DOI] [PubMed] [Google Scholar]
- 42. Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 1999; 48: 1–9. [DOI] [PubMed] [Google Scholar]
- 43. Ho FM, Liu SH, Liau CS, et al. High glucose – induced apoptosis in human endothelial cells is mediated by sequential activations of c-Jun NH2-terminal kinase and caspase-3. Circulation 2000; 101: 2618–2624. [DOI] [PubMed] [Google Scholar]
- 44. Shen X, Zheng S, Thongboonkerd V, et al. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am J Physiol Endocrinol Metab 2004; 287: E896–E905. [DOI] [PubMed] [Google Scholar]
- 45. Ye G, Metreveli NS, Donthi RV, et al. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 2004; 53: 1336–1343. [DOI] [PubMed] [Google Scholar]
- 46. Kuo WW, Wang WJ, Lin CW, et al. NADPH oxidase-derived superoxide anion-induced apoptosis is mediated via the JNK-dependent activation of NF-kappaB in cardiomyocytes exposed to high glucose. J Cell Physiol 2012; 227: 1347–1357. [DOI] [PubMed] [Google Scholar]
- 47. Crespo FL, Sobrado VR, Gomez L, et al. Mitochondrial reactive oxygen species mediate cardiomyocyte formation from embryonic stem cells in high glucose. Stem Cells 2010; 28: 1132–1142. [DOI] [PubMed] [Google Scholar]
- 48. Hausenloy DJ, Baxter G, Bell R, et al. Translating novel strategies for cardioprotection: the Hatter Workshop Recommendations. Basic Res Cardiol 2010; 105: 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Birnbaum GD, Birnbaum I, Ye Y, et al. Statin-induced cardioprotection against ischemia-reperfusion injury: potential drug-drug interactions. Lesson to be learnt by translating results from animal models to the clinical settings. Cardiovasc Drugs Ther 2015; 29: 461–467. [DOI] [PubMed] [Google Scholar]
- 50. Wright MB, Ebeling M, Steiner G, et al. Abstract 510: gene set enrichment analysis of the dual PPAR-α/agonists aleglitazar and muraglitazar reveals distinct metabolic signatures in human cardiomyocytes. Arterioscler Thromb Vasc Biol 2012; 32: A510. [Google Scholar]
- 51. Deehan R, Maerz-Weiss P, Catlett NL, et al. Comparative transcriptional network modeling of three PPAR-alpha/gamma co-agonists reveals distinct metabolic gene signatures in primary human hepatocytes. PLoS ONE 2012; 7: e35012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Holman RR, Paul SK, Bethel MA, et al. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359: 1577–1589. [DOI] [PubMed] [Google Scholar]
- 53. Nathan DM, Cleary PA, Backlund JY, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353: 2643–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA 2005; 294: 2581–2586. [DOI] [PubMed] [Google Scholar]
- 55. AstraZeneca. AstraZeneca discontinues development of Galida (tesaglitazar), http://www.sec.gov/Archives/edgar/containers/fix047/901832/000095010306001449/dp02741_6k.htm (accessed 21 March 2014).
- 56. De Filippis B, Linciano P, Ammazzalorso A, et al. Structural development studies of PPARs ligands based on tyrosine scaffold. Eur J Med Chem 2015; 89: 817–825. [DOI] [PubMed] [Google Scholar]
- 57. Lee HS, Chang M, Lee JE, et al. Carcinogenicity study of CKD-501, a novel dual peroxisome proliferator-activated receptors alpha and gamma agonist, following oral administration to Sprague Dawley rats for 94–101 weeks. Regul Toxicol Pharmacol 2014; 69: 207–216. [DOI] [PubMed] [Google Scholar]
- 58. Piemontese L, Fracchiolla G, Carrieri A, et al. Design, synthesis and biological evaluation of a class of bioisosteric oximes of the novel dual peroxisome proliferator-activated receptor alpha/gamma ligand LT175. Eur J Med Chem 2014; 90: 583–594. [DOI] [PubMed] [Google Scholar]