

ABSTRACT
The development of induced pluripotent stem cells (iPSCs) like never before has opened novel opportunity to study diseases in relevant cell types. In our recent study, Williams syndrome (WS), a rare genetic neurodevelopmental disorder, that is caused by hemizygous deletion of 25–28 genes on chromosome 7, is of interest because of its unique cognitive and social profiles. Little is known about haploinsufficiency effect of those deleted genes on molecular and cellular phenotypes at the neural level due to the lack of relevant human cellular model. Using the cellular reprogramming approach, we reported that WS iPSC-derived neural progenitor cells (NPCs) has increased apoptosis and therefore increased doubling time, which could be rescued by complementation of frizzled 9, one of the genes typically deleted in WS. Moreover, WS iPSC-derived CTIP2-positive pyramidal neurons exhibit morphologic alterations including longer total dendrites and increasing dendritic spine number. In addition, WS iPSC-derived neurons show an increase in calcium transient frequency and synchronized activity likely due to increased number of dendritic spines and synapses. Our work integrated cross-level data from genetics to behavior of WS individuals and revealed altered cellular phenotypes in WS human NPCs and neurons that could be validated in other model systems such as magnetic resonance imaging (MRI) in live subjects and postmortem brain tissues.
KEYWORDS: disease modeling, iPSC, Williams syndrome
Williams Syndrome (WS) is a genetic neurodevelopmental disorder where 25–28 genes on chromosome 7 (7q11.23) are hemizygously deleted (∼1.5 megabases) as a result of unequal crossing over of homologous chromosome during meiosis.1 Haploinsufficiency of these genes causes multiple symptoms including supravalvular aortic stenosis (SVAS), visuospatial deficits, mild to moderate mental retardation, infantile hypercalcemia, craniofacial features (elfin-like faces), hypersensitivity to sound, strong language skills and a very unique social behavior.2,3 Most of WS patients do not live to normal life expectancy. The death is solely the result of SVAS as elastin which is encoded by ELN, is insufficiently produced to form elastic fibers lining the aorta. Thinner elastic fibers then are compensated by increase in number of smooth muscle cells, resulting in less flexible thickened aorta.4 Unlike patients with most neurodevelopmental disorders including autism spectrum disorders (ASD), Rett syndrome, and down syndrome, who have poor social skills,5-7 WS patients, on the other hand, are hypersocial and attracted to strangers with the over use of expressive language.8 Although varied in intensity, the hypersociability is still observed across cultures where children with WS always score relatively higher than typically developing (TD) ones.9
WS cases were first reported in 1961 by Dr. John Cyprian Phipps Williams, a New Zealander cardiologist and his colleagues.10 He noticed that 4 patients with SVAS also shared other common phenotypes including facial features and intellectual disabilities. Nowadays, patients are routinely diagnosed by detection of absence of one copy of ELN using fluorescent in situ hybridization (FISH) test.11 The majority of diagnosed WS patients (98%) have classical/typical deletion whereas the others have atypical deletion which could be either as large as ∼3.5 Mb12 or as small as 83.6 kb (ELN and LIMK1 deletion),13 resulting in variation in phenotypes. Interestingly, WS individuals with smaller deletion exhibit fewer traits compared with classical/typical WS individuals, suggesting a potential link between the missing typical WS phenotypes and genes spared in the genome of those atypical WS individuals.
In the past decades, to reveal mechanism underlying WS characteristics and ultimately establish causal links between genes and WS phenotypes, different models have been used. Human peripheral tissue such as lymphoblastoid cells and fibroblasts provide gene expression profiles that suggest possible alteration in molecular mechanism in different target cell types, including neurons and smooth muscle cells.14 Verification of such data was not possible at that time as no live source of those human target cell types were available. Single-gene knockout mouse models were also generated.15-18 While most of heterozygous mice for each gene exhibited some relevant phenotypes, one of them (Fkbp6) showed no phenotype at all unless both alleles were knocked out,19 suggesting that symptomatic traits could be the result of either interaction between more than one gene or the physiologic differences between mouse and human. Recently, a mouse model for WS with full hemizygous deletion has been successfully created and recapitulated most of phenotypes in human including total brain volume reduction, increased social interaction, hypersensitivity to sound and mild motor deficits.20 Unfortunately, visuospatial learning and memory of WS mice were not tested in this study. While promising, one caveat of this mouse model is that expressive language observed in human could not be understandably assessed in mouse, if any. Functional studies of each gene attributing to WS phenotype were summarized in Table 1.
Table 1.
Mouse and human disease model for genes hemizygously deleted in Williams syndrome.
| Phenotypes observed in single-gene mouse model |
|||
|---|---|---|---|
| Gene | Heterozygotes | Homozygotes | Phenotypes observed in rare atypical WS |
| NOL1R | N/A | N/A | N/A |
| TRIM50 | N/A | N/A | N/A |
| FKBP6 | No phenotype observed19 | Infertility in male19 | N/A |
| FZD9 | Moderate increase in apoptosis in developing dentate gyrus and visuospatial learning/memory deficits49 | High increase in apoptosis in developing dentate gyrus and visuospatial learning/memory deficits49 | N/A |
| Depletion of developing B cell50 | |||
| BAZ1B | Moderate craniofacial abnormalities 51 | High craniofacial abnormalities and lethality after birth /xref> | N/A |
| BCL7B | N/A | N/A | N/A |
| TBL2 | N/A | N/A | N/A |
| MLXIPL | N/A | Lipogenesis reduction52 | N/A |
| VPS37D | N/A | N/A | N/A |
| DNAJC30 | N/A | N/A | N/A |
| WBSCR22 | N/A | N/A | N/A |
| STX1A | No learning and memory deficits53,54 | Impaired long-term potentiation and memory consolidation53 | N/A |
| High embryonic lethality54 | |||
| ABHD11 | N/A | N/A | N/A |
| CLDN3 | N/A | N/A | N/A |
| CLDN4 | N/A | N/A | N/A |
| WBSCR27 | N/A | N/A | N/A |
| WBSCR28 | N/A | N/A | N/A |
| ELN | Increase in elastic lamellae and smooth muscle in arteries4 | Embryonic lethality due to arterial obstruction17 | Supravalvular aortic stenosis55 |
| LIMK1 | Generated but never characterized56 | Altered spine morphology, long-term potentiation, and fear responses56 | Impaired visuospatial constructive cognition13 |
| ELF4H | N/A | N/A | N/A |
| LAT2 | N/A | T-cell activation abnormalities57 | N/A |
| RFC2 | N/A | N/A | N/A |
| CLIP2 | Moderate growth deficiency and hippocampal dysfunction18 | Moderate growth deficiency and hippocampal dysfunction18 | Motor and cognitive deficits58 |
| GTF2IRD1 | No phenotype observed59 | Craniofacial abnormalities59 | Visuospatial construction41,61 |
| Mild growth retardation and decrease in fear and aggression60 | Mild growth retardation and decrease in fear and aggression60 | ||
| WBSCR23 | N/A | N/A | N/A |
| GTF2i | Increase in social interaction with unfamiliar mice40 | Embryonic lethality40 | Hypersociability41 |
| Visuospatial construction61 | |||
N/A, neither mouse model nor atypical WS patients for particular gene was available
The use of patient-derived iPSCs to study molecular mechanism underlying pathogenesis has overcome those major obstacles the other disease models faced in the past. Upon reprogramming, these iPSCs possess the abilities closely resembling embryonic stem cells (ESCs) i.e. to self-renew and to differentiate into diverse range of cell types of 3 germ layers (endoderm, mesoderm and endoderm). By ectopically expressing 4 transcription factors (Oct4, Sox2, Klf4 and c-Myc) in adult human fibroblasts, the Yamanaka group was able to induce differentiated cells back to their pluripotent state and demonstrated that these human iPSCs were similar to human ESCs in terms of morphology, proliferation, gene expression and ability to differentiate into several cell types of 3 germ layers.21 Such advancement has opened up new possibilities that could greatly benefit the medical research, including therapeutic applications and disease modeling.22-26 First, unlike primary patient cells, which are limited in quantity, iPSCs provide infinite storable source for studies requiring large amount of cells such as high-throughput drug screening. Second, human iPSC-derived cells are more physiologically relevant than animal models and available transformed cell lines. Ultimately, iPSCs allows parallel interdisciplinary studies such as behavioral profiling and organ function and activity monitoring, to be performed in the same individual, facilitating the better interpretation of results integrated across all levels.
Because of unique well-defined genetic background of WS and our interdisciplinary collaboration, we reprogrammed the somatic cells collected from participants in cross-level studies and generated different neural cells to explore the phenotypes in NPCs and neurons (Fig. 1). We have described neuronal phenotypes of NPCs and neurons derived from WS patient together with parallel validation in postmortem neurons.27
Figure 1.
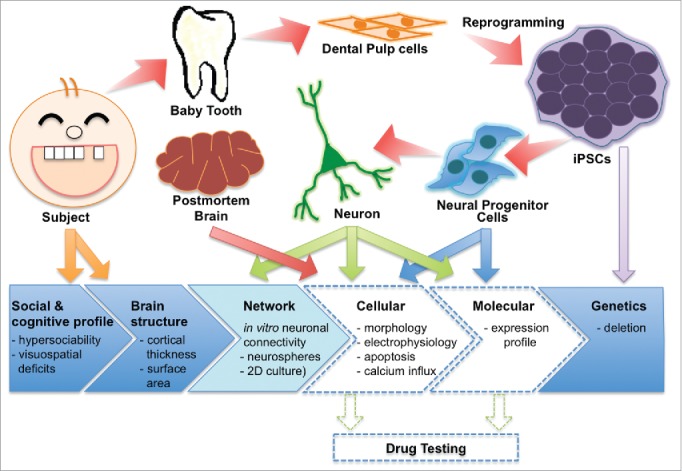
Modeling WS using human iPSCs: Overview. A model to study the neurodevelopmental aspect of WS subjects. We derived iPSCs from dental pulp cells extracted from deciduous tooth of WS individuals whose neurocognitive and social profile was confirmed. Then, WS patient-derived iPSCs were differentiated into neural progenitor cells (NPCs) and neurons for the investigation of specific cellular phenotypes, such as morphology, electrophysiology, apoptosis, calcium influx and expression profile. Concurrently, we analyzed the morphology of postmortem cortical neurons on same subtype we analyzed in the iPSC-derived neuronal culture. Dashed arrows represent future/possible studies.
Despite the availability of current protocols for neuronal differentiation, a major concern in disease modeling using iPSC-derived neural cells that could lead to misinterpretation of the findings is the variation in the neuronal populations generated in vitro from each batch of differentiation. We have characterized as much as possible our neuronal culture derived in the study by performing 3 independent analyses. First, a C1 Fluidigm single cell analyses for 96 different genes across several cortical iPSC-derived NPCs and neurons in the different typical WS, atypical WS and TD lines revealed that while NPC populations were very homogeneous among the 3 genotypes, there were consistent differences in sub-populations of post-mitotic neurons. The data clearly showed that the majority of neurons derived from our protocol had a cortical excitatory biased identity (60–80%), some inhibitory neurons (20%) and some glia cells (∼30%). Our results also revealed non-significant differences in the number of neurons expressing different cortical layers and neurotransmitters between WS and TD. Second, target gene expression analyses showed the difference in expression level of several genes between WS and TD neurons. Finally, unbiased RNAseq analyses in iPSC-derived sorted neurons confirmed the cortical excitatory nature of our differentiation and pointed out the specific metabolic pathways that are uniquely affected in typical WS compared with TD and atypical WS neurons.
WS cellular phenotypes could be observed early on in precursor cells. WS NPCs have increased doubling time due to an increase in apoptosis, possibly contributing to the decrease in cortical surface area that we observed in WS brains and to the previously reported overall reduction in WS brain volume.28 Moreover, because of the lack of such NPC phenotype in one atypical WS subject, we were able to restrict this phenomenon to the frizzled 9 (FZD9) gene, one of 25–28 genes hemizygously deleted in typical WS, which encodes transmembrane G protein-coupled receptor for Wnt signaling pathway, Using gain and loss of function of FZD9 itself as well as targeted gene expression analysis of Wnt signaling downstream of FZD9 and treatment of GSK3 inhibitor, we confirmed the contribution of FZD9 to NPC survival through canonical Wnt pathway. While we could not exclude the contribution of other genes to other equally relevant phenotypes, our results support that FZD9 is directly responsible for the increased cell death in WS NPCs. As we discussed above, the data open the possibility that FZD9 is responsible for a cortex with less surface area in WS subjects and likely contributes to the disproportional subtypes of neurons in WS.
WS-derived cortical neurons layer V/VI showed several morphometric alterations, including higher total dendritic length and higher number of dendritic protrusions/spines. Rather than the compensatory effect of the surviving NPCs and neurons growing more dendrites and being more active due to the death of WS NPCs, we proved that these phenotypes are intrinsic to WS neurons. We performed a time-lapse of neuronal differentiation among the 3 genotypes by seeding iPSC-derived neurons at the same density and measuring the total dendritic length over time. The result clearly showed a fast growth rate in typical WS neurons compared with TDs and atypical WS. Moreover, we traced TD neurons differentiated from TD NPCs plated at different cellular densities. Within the range of 300–1200 cells/mm2, we detected no significant differences in the total dendritic length, segment number and spine density. Thus, we conclude that the observed neuronal phenotypes are intrinsic to WS neurons and not due to a compensatory effect.
The morphometric alterations reported for WS cortical neurons go in the opposite direction of neurons derived from autism spectrum disorders, where the social cognition is impaired.23,25,29,30 Although the findings of the differences in the number of dendritic segments, branching points and trees did not agree between the iPSC model and postmortem tissue, we did observed a similar trend for WS iPSC-derived neurons to have a higher number of dendritic segments and branching points, as observed in WS postmortem brains. It is possible that iPSC-derived neurons lack the dynamics from environmental inputs, and deficiencies in dendritic segments and branching points may occur later in development, resulting in the only partial dendritic changes observed in postmortem specimens. Nonetheless, the fact that we did found similar morphological alternations in both the 2-month-old iPSC-derived neurons in the dish, and in postmortem cortical neurons of WS subjects, not only supports the hypothesis that the iPSC model is capable of recapitulating early neuronal pathology in WS individuals but also suggests that these changes occur early in development and produce life-long effects.
Increases in calcium signaling frequency exhibited in typical WS neurons can be caused by several possibilities including defects in electrical activity generated by changes in intrinsic excitability, in synaptic connectivity, in calcium channels or in calcium pumps. We showed that typical WS neurons could actually form more excitatory synapses compared with TDs. The hyper-connectivity in WS neuronal networks was further validated by electrophysiological recordings using multi-electrode arrays at different time points. Thus, changes in calcium signaling are likely a reflection of the alteration in synaptic activity in WS neurons.
We have shown that, similar to other neurodevelopmental disorders, WS is characterized by pathologies at both macroscopic and neuronal levels. As pathologies in the organization of dendritic trees and dendritic spines have been long known to represent a feature of mental retardation and developmental disorders,31-35 functional interpretations of the differences with TD subjects was hindered by the lack of methods to test neuronal activity and to connect the genetics and molecular aspects of diseases with the function and morphology of the neurons. Thus, we have demonstrated that iPSC-derived neurons with morphological changes largely comparable to the ones in the postmortem cortex, displayed differences in doubling time of NPCs and calcium transients in response to activity, both of which may underlie morphological changes observed in individual neurons.27
Although the generation of heterogeneous populations of neuron subtypes and astrocytes from iPSCs in vitro was achieved in our study, they were 2-dimensional and, therefore, less complex, and much less mature compared with our actual sophisticated brains. Thus, 2D neuronal cultures could possibly recapitulate only cellular defects occurring during early stages of brain development and the findings obtained from such models must be cautiously interpreted. Recently, the development of 3D culture system resulting in brain organoids has been reported,36-38 offering more closely relevant in vitro models for mature brain organization and cellular network. Different differentiation protocols have been developed and fine-tuned to generate organoids with different particular brain region identity,37,39 potentially allowing study of pathological effects focusing on specific yet complex 3D brain-like structures. When fully established, 3D models would greatly advance disease modeling research especially for complex multigenic neurodevelopmental disorders affecting multiple brain regions like WS.
Future goals
GTF2i and its role in neurons
One of the most interesting genes hemizygously deleted in WS is GTF2I (general transcription factor 2i). Because there were several previous studies on this gene compared with others in the WS deleted regions, GTF2I is a good candidate for further scientific explorations using reprogrammed cells. Heterozygous knockout GTF2I mice exhibited increased social interaction with ‘stranger’ mice but no alteration in learning and memory.40 In atypical WS patient, the subject with hemizygous deletion of all genes except GTF2I showed less social interaction with strangers.41 According to the expression profile of WS fibroblast and lymphoblastoid cells, GTF2I was downregulated in WS compared with non-affected individuals.14 It is ubiquitously expressed in the mouse brain during early development and become restrict to cerebellar Purkinje cells and hippocampal interneurons in adult.42 Upon ligand stimulation (such as epidermal growth factor), GTF2I is tyrosine phosphorylated, resulting in enhancement of transcriptional activity.43 Besides the role as transcription factor, it has been recently shown that GTF2I might contribute to calcium channel regulation. GTF2I, when activated, competes with TRPC3 (canonical transient receptor potential cation channel 3) for binding to phospholipase C (PLC) via split PH domain, which results in decrease in accumulation of TRPC3 at cell membrane and consequently an inhibition of calcium influx.44 In WS neurons, haploinsufficiency of GTF2I could potentially lead to an increase in TRPC3 accumulation at cell membrane and therefore an increase in calcium influx. Thus, it is interesting to determine the roles of GTF2I as transcription factor and calcium channel regulator in WS and TD iPSC-derived neurons. The findings will shed light on the cellular basis of differential response to stimulation in WS neurons mediated in part by the activity of GTF2I.
Molecular pathway underlying inverse social behaviors in ASD and WS
ASD, which are mainly characterized by deficits in verbal communication, impaired social interaction, and limited and repetitive interests and behavior, affect about 1 in 68 children in the United States.45 Although some cases belong to familial forms, majority of them are sporadic.46,47 Studies of WS could possibly contribute to the understanding in molecular mechanism underlying poor social skills of ASD. Unequal crossing over during meiosis of WS deletion region results in not only hemizygous deletion but also its duplication. The studies reported the cases where the children with this duplication have ASD-like phenotypes including poor social skill, language impairment and developmental delay,48 suggesting dosage effect of these 25–28 genes on opposite social behaviors. The discovery of molecular pathway alteration in opposite directions in WS and patients with duplication will presumably shed light on hidden players controlling social behavior.
As demonstrated in our previously published studies,23,25 iPSCs could be used to investigate the functional consequences of MECP2 mutation (Rett syndrome) and TRPC6 disruption (non-syndromic autism) on neurons derived from those patients. iPSC-derived neurons from both diseases shared common phenotypes including altered calcium influx, morphological alterations and significant reduction in glutamatergic synapses. These lines of evidence suggest that common pathways, leading to neuronal alterations, may be involved in the etiology of ASDs. In our WS study, same parameters were measured in WS iPSC-derived neurons. Interestingly, while ASD iPSC-derived neurons had decreased total length and fewer branching points compared with control neurons, WS iPSC-derived neurons, on the other hand, had increased total dendritic length and more branching trees. In other words, ASD-derived neurons were less elaborated on their dendritic ramification than WS neurons. We also found that, contrasting to ASD iPSC-derived neurons, calcium transient frequency in WS iPSC-derived neurons was higher compared with control neurons. Using this iPSC system and genome editing technology, we would be able to investigate the impact of haploinsufficiency of each of WS genes in human neurons and explore the hypothesis that different copy number variations can lead to different/opposite biologic effects among WS, idiopathic ASD, ASD-like WS duplication and TD iPSC-derived neurons.
Summary
We revealed WS neuronal phenotypes at cellular levels using iPSCs and differentiation protocols (see summary in Fig. 2). Simple observation of slow growth in typical WS NPCs prompted us to investigate further in NPC proliferation and apoptosis. While proliferation was not altered in all WS NPCs, we found significant increase in apoptosis in only typical WS, not atypical NPCs, suggesting that gene regulating apoptosis in NPCs was spared in atypical WS genome. We showed that the FZD9 is specifically responsible for an increase in apoptosis in typical WS NPCs as the treatment of shFZD9 in TD NPCs and overexpression of FZD9 in typical WS NPCs mimicked the phenotypes of typical WS and TD NPCs, respectively. We also demonstrated that typical WS iPSC-derived CTIP2+ (layer V/VI) neurons had longer total dendritic length, increased number of dendritic spines and more number of dendritic trees, indicating that they were more elaborate. Most of phenotypes were similarly observed in postmortem cortical layerV/VI neurons except number of dendritic trees. Instead, postmortem neurons had higher number of segments and branching points, which, nevertheless, suggest that they were more elaborate. Moreover, typical WS iPSC-derived neurons also exhibited calcium transient frequency alteration. Thus, the iPSC system is a powerful model for studying complex disease like WS. Further investigation of particular genes could provide more insight into molecular mechanism underlying etiology of WS and the basis of the human social behavior.
Figure 2.
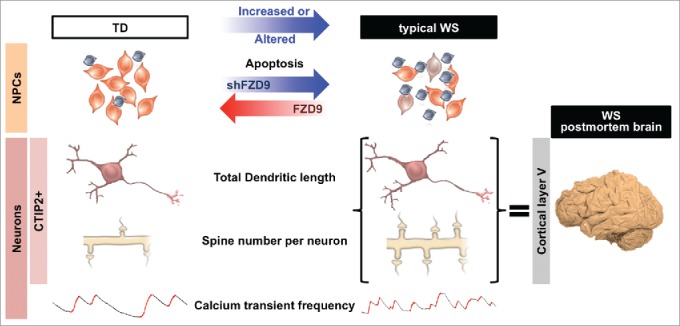
Phenotypic alterations of typical WS iPSC-derived NPCs and neurons. Typical WS NPCs had increased apoptosis compared with TD NPCs. Phenocopy was achieved in TD NPCs treated with shRNA against FZD9 (shFZD9). Apoptosis defect in typical WS NPCs could be rescued by overexpression of FZD9. Morphological analysis of iPSC-derived CTIP2+ cortical neurons (layer V/VI) revealed that WS neurons had increased number of spine per neuron and longer total dendritic length, which were similarly observed in WS cortical layer V/VI neurons in postmortem brain. WS neurons in general exhibited higher calcium transient frequency compared with TDs.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the California Institute for Regenerative Medicine (CIRM) TR4–06747, the National Institutes of Health through the P01 NICHD033113, 156MH109587, U19MH107367, the “Autour des Williams” Association, and a NARSAD Independent Investigator Grant to A.R.M.
References
- [1].Urban Z, Helms C, Fekete G, Csiszar K, Bonnet D, Munnich A, Donis-Keller H, Boyd CD. 7q11.23 deletions in Williams syndrome arise as a consequence of unequal meiotic crossover. Am J Hum Genet 1996; 59:958-62; PMID:8808614 [PMC free article] [PubMed] [Google Scholar]
- [2].Morris CA, Lenhoff HM, Wang PP. Williams-Beuren syndrome: Research, evaluation, and treatment. (JHU Press, 2006). [Google Scholar]
- [3].Morris CA, Mervis CB. Williams syndrome and related disorders. Annu Rev Genomics Hum Genet 2000; 1:461-84; PMID:11701637; http://dx.doi.org/ 10.1146/annurev.genom.1.1.461 [DOI] [PubMed] [Google Scholar]
- [4].Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest 1998; 102:1783-7; PMID:9819363; http://dx.doi.org/ 10.1172/JCI4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ozonoff S, Heung K, Byrd R, Hansen R, Hertz-Picciotto I. The onset of autism: patterns of symptom emergence in the first years of life. Autism Res 2008; 1:320-8; PMID:19360687; http://dx.doi.org/ 10.1002/aur.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kaufmann WE, Tierney E, Rohde CA, Suarez-Pedraza MC, Clarke MA, Salorio CF, Bibat G, Bukelis I, Naram D, et al.. Social impairments in Rett syndrome: characteristics and relationship with clinical severity. J Intellect Disabil Res 2012; 56:233-47; PMID:21385260; http://dx.doi.org/ 10.1111/j.1365-2788.2011.01404.x [DOI] [PubMed] [Google Scholar]
- [7].Chapman RS, Hesketh LJ. Behavioral phenotype of individuals with Down syndrome. Ment Retard Dev Disabil Res Rev 2000; 6:84-95; PMID:10899801; http://dx.doi.org/ [DOI] [PubMed] [Google Scholar]
- [8].Bellugi U, Bihrle A, Jernigan T, Trauner D, Doherty S. Neuropsychological, neurological, and neuroanatomical profile of Williams syndrome. Am J Med Genet Suppl 1990; 6:115-25; PMID:2144426 [DOI] [PubMed] [Google Scholar]
- [9].Zitzer-Comfort C, Doyle T, Masataka N, Korenberg J, Bellugi U. Nature and nurture: Williams syndrome across cultures. Dev Sci 2007; 10:755-62; PMID:17973792; http://dx.doi.org/ 10.1111/j.1467-7687.2007.00626.x [DOI] [PubMed] [Google Scholar]
- [10].Williams JC, Barratt-Boyes BG, Lowe JB. Supravalvular aortic stenosis. Circulation 1961; 24:1311-8; PMID:14007182; http://dx.doi.org/ 10.1161/01.CIR.24.6.1311 [DOI] [PubMed] [Google Scholar]
- [11].Ewart AK, Morris CA, Atkinson D, Jin WS, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the Elastin Locus in a Developmental Disorder, Williams-Syndrome. Nat Genet 1993; 5:11-6; PMID:7693128; http://dx.doi.org/ 10.1038/ng0993-11 [DOI] [PubMed] [Google Scholar]
- [12].Fusco C, Micale L, Augello B, Teresa Pellico M, Menghini D, Alfieri P, Cristina Digilio M, Mandriani B, Carella M, Palumbo O, et al.. Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits. Eur J Hum Genet 2014; 22:64-70; PMID:23756441; http://dx.doi.org/ 10.1038/ejhg.2013.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Frangiskakis JM, Ewart AK, Morris CA, Mervis CB, Bertrand J, Robinson BF, Klein BP, Ensing GJ, Everett LA, Green ED, et al.. LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell 1996; 86:59-69; PMID:8689688; http://dx.doi.org/ 10.1016/S0092-8674(00)80077-X [DOI] [PubMed] [Google Scholar]
- [14].Merla G, Howald C, Henrichsen CN, Lyle R, Wyss C, Zabot MT, Antonarakis SE, Reymond A. Submicroscopic deletion in patients with Williams-Beuren syndrome influences expression levels of the nonhemizygous flanking genes. Am J Hum Genet 2006; 79:332-41; PMID:16826523; http://dx.doi.org/ 10.1086/506371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fujiwara T, Mishima T, Kofuji T, Chiba T, Tanaka K, Yamamoto A, Akagawa K. Analysis of knock-out mice to determine the role of HPC-1/syntaxin 1A in expressing synaptic plasticity. J Neurosci 2006; 26:5767-76; PMID:16723534; http://dx.doi.org/ 10.1523/JNEUROSCI.0289-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yoshimura K, Kitagawa H, Fujiki R, Tanabe M, Takezawa S, Takada I, Yamaoka I, Yonezawa M, Kondo T, Furutani Y, et al.. Distinct function of 2 chromatin remodeling complexes that share a common subunit, Williams syndrome transcription factor (WSTF) (Retracted article. See vol. 111, pg. 2398, 2014). P Natl Acad Sci USA 2009; 106:9280-5; http://dx.doi.org/ 10.1073/pnas.0901184106 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [17].Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature 1998; 393:276-80; PMID:9607766; http://dx.doi.org/ 10.1038/30522 [DOI] [PubMed] [Google Scholar]
- [18].Hoogenraad CC, Koekkoek B, Akhmanova A, Krugers H, Dortland B, Miedema M, van Alphen A, Kistler WM, Jaegle M, Koutsourakis M, et al.. Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nat Genet 2002; 32:116-27; PMID:12195424; http://dx.doi.org/ 10.1038/ng954 [DOI] [PubMed] [Google Scholar]
- [19].Crackower MA, Kolas NK, Noguchi J, Sarao R, Kikuchi K, Kaneko H, Kobayashi E, Kawai Y, Kozieradzki I, Landers R, et al.. Essential role of Fkbp6 in male fertility and homologous chromosome pairing in meiosis. Science 2003; 300:1291-5; PMID:12764197; http://dx.doi.org/ 10.1126/science.1083022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li HH, Roy M, Kuscuoglu U, Spencer CM, Halm B, Harrison KC, Bayle JH, Splendore A, Ding F, Meltzer LA, et al.. Induced chromosome deletions cause hypersociability and other features of Williams-Beuren syndrome in mice. EMBO Mol Med 2009; 1:50-65; PMID:20049703; http://dx.doi.org/ 10.1002/emmm.200900003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131:861-72; PMID:18035408; http://dx.doi.org/ 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- [22].Andrade LN, Nathanson JL, Yeo GW, Menck CF, Muotri AR. Evidence for premature aging due to oxidative stress in iPSCs from Cockayne syndrome. Hum Mol Genet 2012; 21:3825-34; PMID:22661500; http://dx.doi.org/ 10.1093/hmg/dds211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X, Nunez Y, Walker MF, Murdoch JD, Sanders SJ, et al.. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry 2015; 20:1350-65; PMID:25385366; http://dx.doi.org/ 10.1038/mp.2014.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria KC, Beltrao-Braga PC, Trujillo CA, Mendes AP, Padmanabhan K, et al.. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry 2016. http://dx.doi.org/ 10.1038/mp.2016.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010; 143:527-39; PMID:21074045; http://dx.doi.org/ 10.1016/j.cell.2010.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mitne-Neto M, Machado-Costa M, Marchetto MC, Bengtson MH, Joazeiro CA, Tsuda H, Bellen HJ, Silva HC, Oliveira AS, Lazar M, et al.. Downregulation of VAPB expression in motor neurons derived from induced pluripotent stem cells of ALS8 patients. Hum Mol Genet 2011; 20:3642-52; PMID:21685205; http://dx.doi.org/ 10.1093/hmg/ddr284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chailangkarn T, Trujillo CA, Freitas BC, Hrvoj-Mihic B, Herai RH, Yu DX, Brown TT, Marchetto MC, Bardy C, McHenry L, et al.. A human neurodevelopmental model for Williams syndrome. Nature 2016; 536:338-43; PMID:27509850; http://dx.doi.org/ 10.1038/nature19067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chiang MC, Reiss AL, Lee AD, Bellugi U, Galaburda AM, Korenberg JR, Mills DL, Toga AW, Thompson PM. 3D pattern of brain abnormalities in Williams syndrome visualized using tensor-based morphometry. Neuroimage 2007; 36:1096-109; PMID:17512756; http://dx.doi.org/ 10.1016/j.neuroimage.2007.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, et al.. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med 2011; 17:1657-U1176; PMID:22120178; http://dx.doi.org/ 10.1038/nm.2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, et al.. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013; 503:267-71; PMID:24132240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huttenlo PR. Dendritic Development in Neocortex of Children with Mental Defect and Infantile Spasms. Neurology 1974; 24:203-10; PMID:4130661; http://dx.doi.org/ 10.1212/WNL.24.3.203 [DOI] [PubMed] [Google Scholar]
- [32].Purpura DP. Dendritic Spine Dysgenesis and Mental-Retardation. Science 1974; 186:1126-8; PMID:4469701; http://dx.doi.org/ 10.1126/science.186.4169.1126 [DOI] [PubMed] [Google Scholar]
- [33].Marin-Padilla M. Pyramidal cell abnormalities in the motor cortex of a child with Down's syndrome. A Golgi study. J Comp Neurol 1976; 167:63-81; PMID:131810; http://dx.doi.org/ 10.1002/cne.901670105 [DOI] [PubMed] [Google Scholar]
- [34].Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective Dendritic Alterations in the Cortex of Rett-Syndrome. J Neuropath Exp Neur 1995; 54:195-201; PMID:7876888; http://dx.doi.org/ 10.1097/00005072-199503000-00006 [DOI] [PubMed] [Google Scholar]
- [35].Armstrong DD, Dunn K, Antalffy B. Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J Neuropath Exp Neur 1998; 57:1013-7; PMID:9825937; http://dx.doi.org/ 10.1097/00005072-199811000-00003 [DOI] [PubMed] [Google Scholar]
- [36].Mariani J, Simonini MV, Palejev D, Tomasini L, Coppola G, Szekely AM, Horvath TL, Vaccarino FM. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci U S A 2012; 109:12770-5; PMID:22761314; http://dx.doi.org/ 10.1073/pnas.1202944109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O'Rourke NA, Nguyen KD, et al.. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Met 2015; 12:671-8; PMID:26005811; http://dx.doi.org/ 10.1038/nmeth.3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JL, Guimaraes KP, Benazzato C, Almeida N, Pignatari GC, Romero S, et al.. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016; 534:267-71; PMID:27279226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al.. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016; 165:1238-54; PMID:27118425; http://dx.doi.org/ 10.1016/j.cell.2016.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sakurai T, Dorr NP, Takahashi N, McInnes LA, Elder GA, Buxbaum JD. Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res 2011; 4:28-39; PMID:21328569; http://dx.doi.org/ 10.1002/aur.169 [DOI] [PubMed] [Google Scholar]
- [41].Dai L, Bellugi U, Chen XN, Pulst-Korenberg AM, Jarvinen-Pasley A, Tirosh-Wagner T, Eis PS, Graham J, Mills D, Searcy Y, et al.. Is it Williams syndrome? GTF2IRD1 implicated in visual-spatial construction and GTF2I in sociability revealed by high resolution arrays. Am J Med Genet A 2009; 149A:302-14; PMID:19205026; http://dx.doi.org/ 10.1002/ajmg.a.32652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Danoff SK, Taylor HE, Blackshaw S, Desiderio S. TFII-I, a candidate gene for Williams syndrome cognitive profile: parallels between regional expression in mouse brain and human phenotype. Neurosci 2004; 123:931-8; PMID:14751286; http://dx.doi.org/ 10.1016/j.neuroscience.2003.08.038 [DOI] [PubMed] [Google Scholar]
- [43].Kim DW, Cheriyath V, Roy AL, Cochran BH. TFII-I enhances activation of the c-fos promoter through interactions with upstream elements. Mol Cell Biol 1998; 18:3310-20; PMID:9584171; http://dx.doi.org/ 10.1128/MCB.18.6.3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Caraveo G, van Rossum DB, Patterson RL, Snyder SH, Desiderio S. Action of TFII-I outside the nucleus as an inhibitor of agonist-induced calcium entry. Science 2006; 314:122-5; PMID:17023658; http://dx.doi.org/ 10.1126/science.1127815 [DOI] [PubMed] [Google Scholar]
- [45].Wingate M, Kirby RS, Pettygrove S, Cunniff C, Schulz E, Ghosh T, Robinson C, Lee LC, Landa R, Constantino J, et al.. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 Sites, United States, 2010. Mmwr Surveill Summ 2014; 63:1-21. [PubMed] [Google Scholar]
- [46].Piven J, Palmer P, Jacobi D, Childress D, Arndt S. Broader autism phenotype: evidence from a family history study of multiple-incidence autism families. Am J Psychiatry 1997; 154:185-90; PMID:9016266; http://dx.doi.org/ 10.1176/ajp.154.2.185 [DOI] [PubMed] [Google Scholar]
- [47].Ronald A, Happe F, Bolton P, Butcher LM, Price TS, Wheelwright S, Baron-Cohen S, Plomin R. Genetic heterogeneity between the three components of the autism spectrum: a twin study. J Am Acad Child Adolesc Psychiatry 2006; 45:691-9; PMID:16721319; http://dx.doi.org/ 10.1097/01.chi.0000215325.13058.9d [DOI] [PubMed] [Google Scholar]
- [48].Velleman SL, Mervis CB. Children with 7q11.23 Duplication Syndrome: Speech, Language, Cognitive, and Behavioral Characteristics and their Implications for Intervention. Perspect Lang Learn Edu 2011; 18:108-16; PMID:22754604; http://dx.doi.org/ 10.1044/lle18.3.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhao C, Aviles C, Abel RA, Almli CR, McQuillen P, Pleasure SJ. Hippocampal and visuospatial learning defects in mice with a deletion of frizzled 9, a gene in the Williams syndrome deletion interval. Development 2005; 132:2917-27; PMID:15930120; http://dx.doi.org/ 10.1242/dev.01871 [DOI] [PubMed] [Google Scholar]
- [50].Ranheim EA, Kwan HCK, Reya T, Wang YK, Weissman IL, Francke U. Frizzled 9 knock-out mice have abnormal B-cell development. Blood 2005; 105:2487-94; PMID:15572594; http://dx.doi.org/ 10.1182/blood-2004-06-2334 [DOI] [PubMed] [Google Scholar]
- [51].Ashe A, Morgan DK, Whitelaw NC, Bruxner TJ, Vickaryous NK, Cox LL, Butterfield NC, Wicking C, Blewitt ME, Wilkins SJ, et al.. A genome-wide screen for modifiers of transgene variegation identifies genes with critical roles in development. Genome Biol 2008; 9:R182; PMID:19099580; http://dx.doi.org/ 10.1186/gb-2008-9-12-r182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. P Natl Acad Sci USA 2004; 101:7281-6; http://dx.doi.org/ 10.1073/pnas.0401516101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fujiwara T, Mishima T, Kofuji T, Chiba T, Tanaka K, Yamamoto A, Akagawa K. Analysis of knock-out mice to determine the role of HPC-1/syntaxin 1A in expressing synaptic plasticity. J Neurosci 2006; 26:5767-76; PMID:16723534; http://dx.doi.org/ 10.1523/JNEUROSCI.0289-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].McRory JE, Rehak R, Simms B, Doering CJ, Chen L, Hermosilla T, Duke C, Dyck R, Zamponi GW. Syntaxin 1A is required for normal in utero development. Biochem Bioph Res Co 2008; 375:372-7; http://dx.doi.org/ 10.1016/j.bbrc.2008.08.031 [DOI] [PubMed] [Google Scholar]
- [55].Curran ME, Atkinson DL, Ewart AK, Morris CA, Leppert MF, Keating MT. The elastin gene is disrupted by a translocation associated with supravalvular aortic-stenosis. Cell 1993; 73:159-68; PMID:8096434; http://dx.doi.org/ 10.1016/0092-8674(93)90168-P [DOI] [PubMed] [Google Scholar]
- [56].Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, et al.. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 2002; 35:121-33; PMID:12123613; http://dx.doi.org/ 10.1016/S0896-6273(02)00758-4 [DOI] [PubMed] [Google Scholar]
- [57].Volna P, Lebduska P, Draberova L, Simova S, Heneberg P, Boubelik M, Bugajev V, Malissen B, Wilson BS, Horejsi V, et al.. Negative regulation of mast cell signaling and function by the adaptor LAB/NTAL. J Exp Med 2004; 200:1001-13; PMID:15477348; http://dx.doi.org/ 10.1084/jem.20041213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].van Hagen JM, van der Geest JN, van der Giessen RS, Lagers-van Haselen GC, Eussen HJ, Gille JJ, Govaerts LC, Wouters CH, de Coo IF, Hoogenraad CC, et al.. Contribution of CYLN2 and GTF2IRD1 to neurological and cognitive symptoms in Williams Syndrome. Neurobiol Dis 2007; 26:112-24; PMID:17270452; http://dx.doi.org/ 10.1016/j.nbd.2006.12.009 [DOI] [PubMed] [Google Scholar]
- [59].Tassabehji M, Hammond P, Karmiloff-Smith A, Thompson P, Thorgeirsson SS, Durkin ME, Popescu NC, Hutton T, Metcalfe K, Rucka A, et al.. GTF2IRD1 in craniofacial development of humans and mice. Science 2005; 310:1184-7; PMID:16293761; http://dx.doi.org/ 10.1126/science.1116142 [DOI] [PubMed] [Google Scholar]
- [60].Young EJ, Lipina T, Tam E, Mandel A, Clapcote SJ, Bechard AR, Chambers J, Mount HT, Fletcher PJ, Roder JC, et al.. Reduced fear and aggression and altered serotonin metabolism in Gtf2ird1-targeted mice. Genes Brain Behav 2008; 7:224-34; PMID:17680805; http://dx.doi.org/ 10.1111/j.1601-183X.2007.00343.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Antonell A, Del Campo M, Magano LF, Kaufmann L, de la Iglesia JM, Gallastegui F, Flores R, Schweigmann U, Fauth C, Kotzot D, et al.. Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile. J Med Genet 2010; 47:312-20; PMID:19897463; http://dx.doi.org/ 10.1136/jmg.2009.071712 [DOI] [PubMed] [Google Scholar]