

ABSTRACT
Herpes simplex virus (HSV) entry into a subset of cells requires endocytosis and endosomal low pH. Preexposure of isolated virions to mildly acidic pH of 5 to 6 partially inactivates HSV infectivity in an irreversible manner. Acid inactivation is a hallmark of viruses that enter via low-pH pathways; this occurs by pretriggering conformational changes essential for fusion. The target and mechanism(s) of low-pH inactivation of HSV are unclear. Here, low-pH-treated HSV-1 was defective in fusion activity and yet retained normal levels of attachment to cell surface heparan sulfate and binding to nectin-1 receptor. Low-pH-triggered conformational changes in gB reported to date are reversible, despite irreversible low-pH inactivation. gB conformational changes and their reversibility were measured by antigenic analysis with a panel of monoclonal antibodies and by detecting changes in oligomeric conformation. Three-hour treatment of HSV-1 virions with pH 5 or multiple sequential treatments at pH 5 followed by neutral pH caused an irreversible >2.5 log infectivity reduction. While changes in several gB antigenic sites were reversible, alteration of the H126 epitope was irreversible. gB oligomeric conformational change remained reversible under all conditions tested. Altogether, our results reveal that oligomeric alterations and fusion domain changes represent distinct conformational changes in gB, and the latter correlates with irreversible low-pH inactivation of HSV. We propose that conformational change in the gB fusion domain is important for activation of membrane fusion during viral entry and that in the absence of a host target membrane, this change results in irreversible inactivation of virions.
IMPORTANCE HSV-1 is an important pathogen with a high seroprevalence throughout the human population. HSV infects cells via multiple pathways, including a low-pH route into epithelial cells, the primary portal into the host. HSV is inactivated by low-pH preexposure, and gB, a class III fusion protein, undergoes reversible conformational changes in response to low-pH exposure. Here, we show that low-pH inactivation of HSV is irreversible and due to a defect in virion fusion activity. We identified an irreversible change in the fusion domain of gB following multiple sequential low-pH exposures or following prolonged low-pH treatment. This change appears to be separable from the alteration in gB quaternary structure. Together, the results are consistent with a model by which low pH can have an activating or inactivating effect on HSV depending on the presence of a target membrane.
KEYWORDS: conformational changes, glycoprotein B, herpes simplex viruses, herpesviruses, inactivation, low pH, membrane fusion
INTRODUCTION
Herpes simplex virus (HSV) causes a wide array of human infections resulting in significant morbidity and mortality worldwide. HSV is an enveloped virus and therefore must fuse with a host cell membrane to initiate entry and infection. HSV can surmount the plasma membrane by endocytosis or via direct penetration. Endocytic entry can be low pH dependent or pH independent (1–3). Entry into human epithelial cells, which represent the primary site of infection, occurs via a low-pH endocytic mechanism (4). Conformational changes in gB triggered by mildly acidic pH are proposed to be critical for fusion during HSV entry into physiologically relevant cells.
Viruses that enter cells by a pH-triggered fusion mechanism are typically inactivated by pretreatment of isolated virions with mildly acidic pH (pH 5.0 to 6.0) (5, 6), which mimics the intraendosomal milieu. In the absence of a target membrane, low pH prematurely activates the fusion protein, thereby rendering the virion incompetent for fusion and entry into cells (7, 8). HSV-1 is inactivated by mildly acidic pH in a rapid and temperature-dependent manner. A 10-min preexposure of HSV-1 to low pH results in only partial inactivation (9, 10), while other viruses, such as influenza virus, are completely inactivated within minutes (5). For influenza virus, low-pH preexposure triggers the fusion machinery by a mechanism similar to the membrane fusion reaction (5, 11). A better understanding of inactivation can inform the more physiologically relevant process of activation during fusion and entry. For HSV, the mechanism and target of low-pH inactivation are unclear. HSV mutants individually lacking viral envelope protein gC, gE, gG, gI, gJ, gM, pUL45, or pUS9 are able to successfully infect cells via pH-dependent endocytosis and are susceptible to low-pH inactivation (9, 10, 12). Thus, these proteins are not the virion target of low-pH inactivation.
Inactivation can be the result of viral envelope proteins failing to function at one or more steps of HSV entry. These include viral attachment to cell surface heparan sulfate; virion gD binding to a cognate host cell receptor; or fusion mediated by the cooperation of gB, gH/gL, and gD (13–15). Receptor binding is likely not affected by low-pH inactivation, as HSV gD binding to nectin-1 or herpesvirus entry mediator (HVEM) receptors is not sensitive to low-pH exposure (16). The core fusion protein gB is a potential target of inactivation.
Low-pH preexposure of virions prematurely triggers conformational changes that normally occur during the membrane fusion reaction (11, 17). Understanding conformational changes that occur during inactivation will enhance understanding of the changes that activate fusion. Exposure to mildly acidic pH triggers reversible conformational changes in prefusion gB present in virions (16, 18–20). Reversibility may prevent premature triggering and unwanted fusion during viral egress (21). The reversible changes in gB detected to date are seemingly incompatible with the irreversible inactivation of virion infectivity.
Here, we investigated the effect of low-pH treatment of HSV virions on attachment, membrane fusion, viral infectivity, reversibility of infectivity, and gB conformational changes and their reversibility. We found that irreversible low-pH inactivation is caused by a defect in membrane fusion activity of virions and identify gB as a target of inactivation. Following sequential or prolonged low-pH exposures, irreversible antigenic changes in gB were detected, including domain I of gB, which contains the hydrophobic fusion loops. Other antigenic changes and oligomeric changes remained reversible. This work represents the first irreversible conformational changes detected in gB and also suggests that the pH-induced change in gB oligomeric structure is distinct from the conformational change in the fusion domain. We propose that low pH activates the gB fusion domain during HSV-1 entry, and in the absence of a host membrane, a similar change results in inactivation of virion fusion activity.
RESULTS
Irreversibility of mildly acidic inactivation of HSV-1.
The infectivity of HSV is reduced when treated with mildly acidic pH of 5.0 to 6.0 for 10 min and then returned to neutral pH for 10 min (2, 9, 10, 20). Thus, low-pH inactivation of HSV is irreversible by this measure (22–24). To verify the irreversibility of inactivation, we determined whether prolonged periods of neutralization with pH 7.4 caused low-pH inactivation to become reversible. HSV-1 was exposed to pH 7.4 or 5.0 for 20 min at 37°C. Virions were neutralized to pH 7.4 for 10, 60, or 120 min and assayed for infectivity (Fig. 1). A 20-min inactivation by pH 5.0 followed by a 10-min neutral-pH treatment resulted in ∼40% inhibition of infectivity, consistent with previous results (2, 9). A prolonged neutralization period of 60 or 120 min did not result in additional recovery of infectivity (Fig. 1), although there was an overall decrease of infectivity following the extended neutralizations. The fraction of virus that is no longer infectious represents irreversible inactivation. Thus, HSV-1 inactivation by mildly acidic pH remained irreversible even when virus was allowed extended time to recover at neutral pH.
FIG 1.
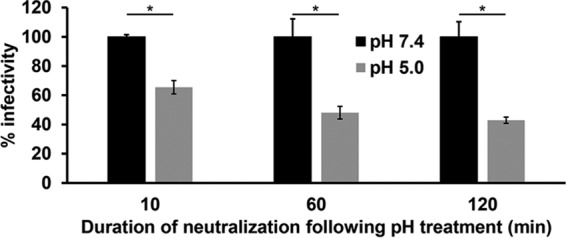
Effect of extending neutralization times on reversibility of low-pH inactivation of HSV-1 infectivity. Extracellular HSV-1 KOS virions were treated with pH 7.4 or 5.0 for 20 min at 37°C. Samples were neutralized to pH 7.4 for the time indicated, and titers were determined on Vero cells. At 18 to 24 h postinfection, titers were determined by plaque assay. Infectivity of pH 7.4-treated virions was set to 100%. Data are the means from four replicates with standard errors. The P values were determined using Student's t test (*, P < 0.002). Results are representative of three independent experiments.
Low-pH pretreatment of HSV results in unaltered attachment to cells.
The step in HSV-1 entry that is targeted by low pH and responsible for inactivation of infectivity is not known. To probe the step(s) in the HSV entry process that is sensitive to low-pH inactivation, first the cell attachment of low-pH-treated virions was assessed. HSV-1 KOS virions were treated at pH 7.4 or 5.0 for 10 min and then neutralized to pH 7.4 for 10 min (Fig. 2). Virions were bound to cells for 1 h at 4°C, and cell-associated virus was quantitated by quantitative PCR (qPCR) as a measure of attachment. HSV-1 pretreated with pH 5.0 attached to cells in a manner similar to pH 7.4-treated particles (Fig. 2). Control heparin treatment reduced HSV attachment by 1 log as expected (25). These results suggest that low-pH inactivation of HSV-1 occurs at a step of viral entry subsequent to attachment.
FIG 2.
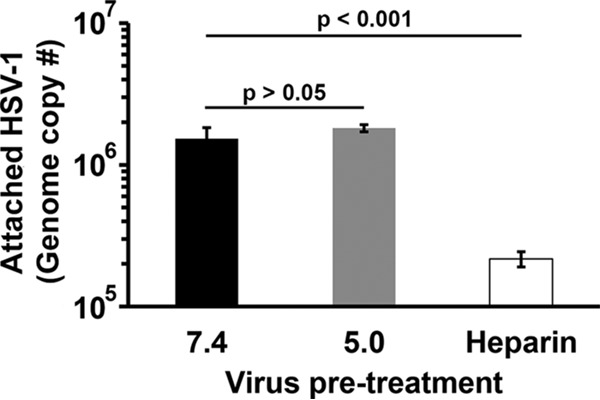
Effect of low-pH pretreatment of HSV-1 on attachment to cells. Extracellular HSV-1 KOS virions were treated with pH 7.4 or 5.0 for 10 min and then neutralized to pH 7.4 for 10 min. Heparin (2 μg/ml) was used as a positive control to inhibit attachment. Treated virions (40 genome copies per cell) were bound to Vero cells for 1 h at 4°C. Cells were washed three times with PBS, and cell-associated virions were quantitated by qPCR. Data are the means from four replicates with standard errors. The P values were determined using Student's t test. Results are representative of three independent experiments.
Mildly acidic pH pretreatment reduces fusion activity of herpesvirus virions.
Low-pH inactivation of HSV-1 is not due to defects in attachment (Fig. 2) or gD receptor binding as previously reported (16). We next investigated the sensitivity of membrane fusion function to low-pH pretreatment using fusion-from-without (FFWO) activity. FFWO is the induction of target cell fusion by addition of virions to the monolayer surface in the absence of viral protein expression. FFWO is a surrogate assay for virus-cell fusion during entry as the two processes share several common features. Both FFWO and entry into Vero cells occur at the plasma membrane, require an appropriate gD receptor, and are blocked by neutralizing antibodies. A subset of syncytial strains of HSV-1, including the ANG path strain, have FFWO activity (26–30). The HSV-1 ANG path strain is inactivated by mildly acidic pH in a manner similar to the non-FFWO HSV-1 strain KOS (20). ANG path virions were treated with pH 5.0 for 10 min, neutralized for 10 min, and then assayed for FFWO. Virions pretreated with pH 7.4 (Fig. 3A) and then added to cells for 3 h triggered fusion of cell membranes as indicated by clustering of nuclei. The pH 5.0-pretreated virions (Fig. 3B) had reduced FFWO activity, mediating an intermediate level of fusion between that of pH 7.4-treated virions and that of uninfected cells (Fig. 3C). The pH 7.4 pretreatment resulted in a baseline fusion activity of 71%, while pH 5.0 pretreatment reduced fusion to 43% (Fig. 3D). Together, the results suggest that a defect in fusion function is responsible for mildly acidic-pH inactivation of HSV-1. The FFWO results are consistent with a model by which low pH acts as a trigger or activator of HSV-1 membrane fusion.
FIG 3.
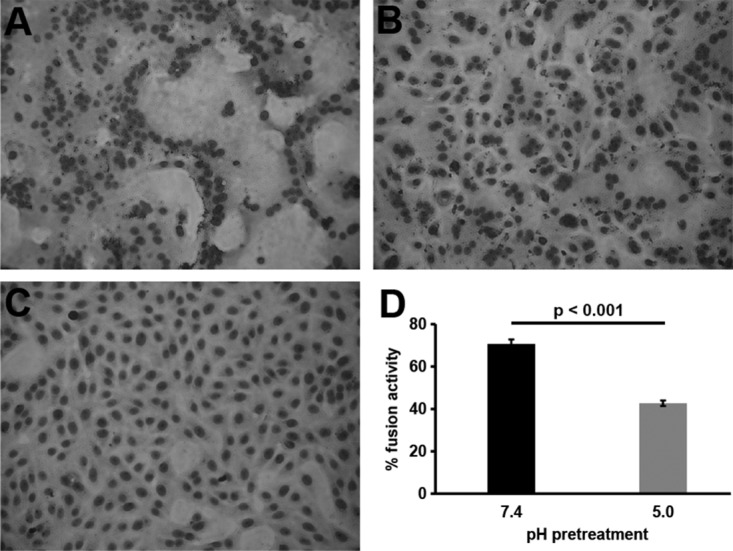
Effect of low-pH pretreatment of HSV-1 on fusion-from-without activity. HSV-1 ANG path virions were pretreated with pH 7.4 (A) or pH 5.0 (B) for 10 min at 37°C and bound to cells (MOI, ∼50) at neutral pH for 1 h at 4°C. Cells shifted to 37°C in the presence of cycloheximide for 3 h were fixed and stained with Giemsa stain. Control uninfected cells were identically treated in parallel (C). Three or more adjacent nuclei in contact were divided by total nuclei in the field to yield percent fusion activity (D). More than 300 nuclei were counted in each of 4 replicate wells. The P value was determined using Student's t test. Results are the means with standard errors and are representative of three independent experiments.
Multiple, sequential low-pH exposures drastically reduce HSV-1 infectivity.
A single 20-min exposure to pH 5.0 irreversibly inactivated HSV-1 by ∼40% (Fig. 1). From the time of assembly and egress of a newly synthesized virion to its entry into a new target cell, a single enveloped HSV particle may encounter multiple acidic-pH environments (2, 31). We next assessed the effect of multiple low-pH exposures on HSV-1 infectivity. Virions were treated at pH 5.0 for 10 min and then neutralized to pH 7.4 for an additional 10 min (Fig. 4; 1×). This process was also repeated for a total of two or four times, representing 2× or 4× sequential treatments, respectively. The infectivity of virions subjected to one cycle of acidification and neutralization was decreased by ∼50% (Fig. 4). Following two sequential treatments, there was a 1-log reduction in infectivity. Four sequential treatments resulted in an ∼2.5-log loss in infectivity. Virions in the 4× sample were subjected to pH 5.0 for a total of 40 min. To rule out whether the reduction in infectivity was due to the prolonged exposure to pH 5.0 rather than the sequential pH treatments, virions were treated continuously with pH 5.0 for 40 min followed by 40 min at pH 7.4 (Fig. 4; nonsequential). The virions treated nonsequentially lost infectivity in a manner similar to the 1×-treated sample, suggesting that the multiple, sequential low-pH treatments were responsible for the more deleterious effect on infectivity. Thus, low pH markedly reduces HSV-1 infectivity, and multiple low-pH exposures during the HSV replication cycle may ultimately decrease virion infectivity.
FIG 4.
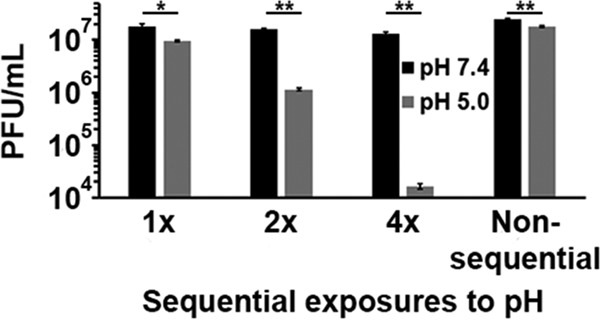
Effect of multiple, sequential low-pH exposures on HSV-1 infectivity. Extracellular HSV-1 KOS virions were treated for 10 min with pH 5.0 and then neutralized to pH 7.4 for 10 min (1×). This treatment was repeated for a total of two (2×) or four (4×) times. Virus titers were determined on Vero cells. Nonsequential samples were treated with pH 5.0 for 40 min and neutralized to pH 7.4 for 40 min. Data are the means from four replicates with standard errors. The P values were determined using Student's t test (*, P < 0.01; **, P < 0.001). Results are representative of three independent experiments.
Low-pH-triggered change in the gB oligomer is exquisitely reversible.
Mildly acidic pH reversibly alters features of the gB quaternary structure as measured by multiple assays (16, 18). Here, we utilized one such assay that measures the sensitivity of gB to SDS and low pH. Oligomers of gB are stable in the presence of 1% SDS; however, treatment with low pH renders the gB oligomer susceptible to disruption by 1% SDS, resulting in the disappearance of a slow-migrating gB species when resolved under native PAGE conditions. We refer to this disappearance of slower-migrating gB species as oligomeric change. Low-pH-treated virions neutralized to pH 7.4 are not sensitive to 1% SDS destabilization, consistent with reversibility of the oligomeric change. To determine if this change could occur multiple times, virions were exposed to pH 7.4 or pH 5.1 for 10 min (Fig. 5). Following pH treatment, either the samples were neutralized to pH 7.4 (5.1→7.4) or virions were treated with 1% SDS. Sequential treatments ensued, resulting in virions being exposed to four cycles of acidification and neutralization.
FIG 5.

Effect of sequential, low-pH exposures on conformation of gB oligomer. Extracellular HSV-1 KOS virions (105 PFU) were treated with pH 5.1 for 10 min and neutralized back to pH 7.4 for 10 min for the indicated number of times. Samples were treated with 1% SDS and separated by “native” PAGE (54). Western blots were probed with monoclonal antibody H1359 to gB. Each blot shown is representative of at least three independent experiments. Number at left is molecular mass in kilodaltons.
Following 10 min of low-pH treatment and treatment with 1% SDS, the slower-migrating gB species disappeared (Fig. 5), consistent with previous results (18). Ten minutes of neutralization resulted in the reappearance of the slower-migrating band, suggesting that the conformational change in gB oligomer was reversible under these conditions. The oligomeric change and its reversibility occurred four times sequentially. Less gB was detected following 4× treatment. Overall, the conformation change in gB measured by this approach is freely reversible following either sequential, low-pH exposures (Fig. 5) or prolonged exposures (see Fig. 7B below).
FIG 7.

Effect of prolonged low-pH exposure on HSV-1 infectivity and gB conformation. (A) Extracellular KOS virions were incubated for 10 min or 3 h at pH 7.4 or pH 5. Virions were neutralized for 10 min, and infectivity was determined by plaque assay on Vero cells. Infectivity of the sample treated with pH 7.4 was set to 100%. Data are the means from four replicates with standard errors. The P value was determined using Student's t test. (B) Virions were treated with pH 7.4 or 5.0 for 10 min or 3 h, and virions were neutralized for 10 min if reversibility was being tested, treated with 1% SDS, and analyzed as described for Fig. 5. Number at left is molecular mass in kilodaltons. (C) Virions were treated with pH 7.4 or 5.0 for 10 min or 3 h and then neutralized for 10 min if reversibility was being tested, and antigenic reactivity was determined as described for Fig. 6. Results are representative of three independent experiments.
gB undergoes irreversible changes in antigenic conformation following sequential low-pH exposures.
In addition to low-pH-triggered conformational changes in the quaternary structure of gB, the prefusion conformation of gB undergoes low-pH-triggered changes in domains I and V. These changes are at least partially reversible and are thought to be important for membrane fusion. Since infectivity is severely and irreversibly impacted by multiple sequential low-pH exposures (Fig. 4) and yet the gB oligomeric change remains reversible, we tested the effect of sequential treatments on the pH-triggered changes in the antigenic conformation of gB and their reversibility.
Domain I of gB contains internal hydrophobic fusion loops that are critical for membrane fusion (32, 33). The domain I epitope recognized by monoclonal antibody (MAb) H126 underwent two low-pH-induced conformational changes and subsequent reversibility (Fig. 6). Following a third low-pH exposure, there was a third conformational change detected by a loss of H126 reactivity. However, following a third neutralization, notably H126 reactivity did not return, suggesting that an irreversible change in gB had occurred. MAb SS55 (domain I) detected reversible conformational changes throughout these exposures. The results suggest that following each cycle of acidification and neutralization, the reactivity of a given MAb is similarly reduced. Whether sequential, different changes in gB conformation are occurring concurrently is not clear.
FIG 6.
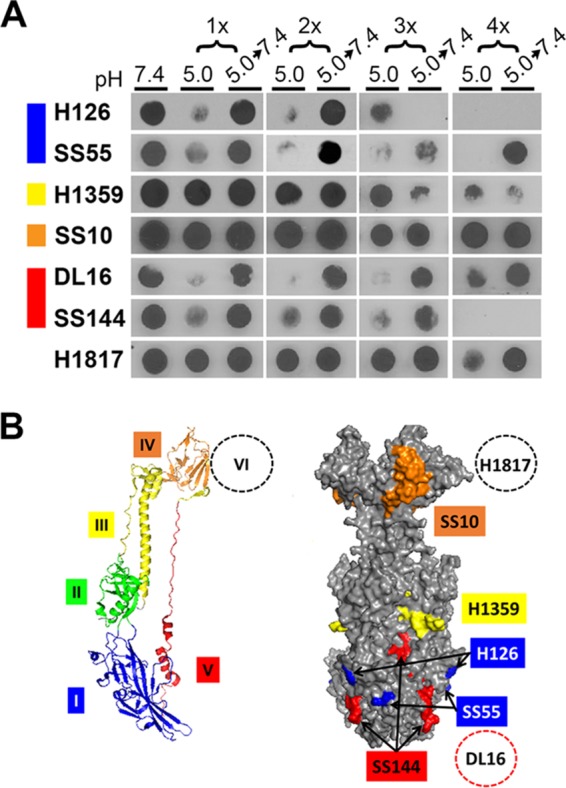
Conformational change in gB from virions that have undergone a series of low-pH treatment reneutralization cycles. (A) Extracellular HSV-1 KOS virions (106 PFU) were treated with pH 5.0 for 10 min and neutralized back to pH 7.4 for 10 min for the indicated number of times. Samples were immediately blotted to nitrocellulose and probed at neutral pH with the indicated monoclonal antibody to gB. Each blot shown is representative of at least three independent experiments. (B) gB monomer with specific domains indicated by color (left) and space-filling model of gB trimer with surface-exposed epitopes detected by gB-specific monoclonal antibodies (right) (42). Domain I MAb H126 maps to residue 303 (42), and MAb SS55 maps to residues 203, 335, and 199 (51). Domain III MAb H1359 maps to residues 457 to 475 (49). Domain IV MAb SS10 maps to residues 640 to 670 (50). Domain V MAb SS144 maps to residues 697 to 725 (42), and MAb DL16 maps to residues 678 to 730 (51). Domain VI MAb H1817 maps to residues 31 to 43, which are unresolved in the crystal structure (50).
MAb H1359 (to domain III) exhibited a partial loss of reactivity following 2× or greater sequential exposures. Interestingly, the change following 4× treatment appeared to be irreversible. Low-pH-induced alterations in gB domain III have not been reported previously. The MAb to domain IV, SS10, did not exhibit loss of reactivity throughout the sequential exposures. A domain V epitope detected by DL16 underwent reversible conformational changes throughout the sequential treatments. Another domain V epitope, detected by MAb SS144, underwent three sequential conformational changes and reversals but became irreversibly altered following a fourth conformational change. MAb H1817 (domain IV), which recognizes an epitope near the gB N terminus, exhibited similar reactivities throughout the different exposures. Thus, following multiple sequential low-pH exposures, virion gB retains the ability to undergo reversible conformational changes in several of the epitopes tested, but the H126 epitope in the fusion domain and the SS144 epitope underwent irreversible conformational change following a third or fourth low-pH exposure, respectively. This contrasts with the reversibility of low-pH-triggered changes in the gB oligomer (Fig. 5). This suggests that the conformational changes in the H126 and SS144 epitopes are separable from the oligomeric alteration.
Prolonged mildly acidic-pH exposure results in virtually complete inactivation of herpesvirus virions and an irreversible change in the fusion domain of gB.
The irreversible conformational changes in gB (Fig. 6) correlate with an ∼2.5-log irreversible reduction in infectivity (Fig. 4). We tested if prolonged low-pH inactivation had a similar pronounced effect on infectivity and on the reversibility of gB conformational changes. Ten minutes of preexposure to pH 5.0 is not sufficient to inactivate the majority of virions (2) (Fig. 4). To test if prolonged low-pH exposure was sufficient to more completely ablate infectivity, we incubated HSV-1 virions for 3 h at pH 5.0. Treated virions were neutralized, and infectivity was assessed. Indeed, prolonged low-pH incubation resulted in a dramatic decrease in infectivity (>2 log; >99% inhibition) (Fig. 7A).
Virions treated under prolonged-duration conditions were subjected to gB oligomeric analysis. Following pH treatment of virions for 3 h, changes in the gB oligomer still occurred and were reversible. Low-pH pretreatment reduced the amount of slower-migrating oligomeric gB, and following 10 min of neutralization, this species returned (Fig. 7B). Altogether, the gB oligomeric change remained reversible under all test conditions.
After confirmation that gB oligomeric change is reversible following prolonged low-pH exposure, virions were subjected to gB antigenic analysis (Fig. 7C). Following 3 h of pH 5.0 treatment, MAb H126 had reduced reactivity, and reactivity was not reversible upon neutralization, indicating an irreversible change. Another domain I antibody, SS55, had reduced reactivity with virion gB that had been exposed to pH 5.0 for 10 min or 3 h. This conformational change, however, remained reversible. Domain V antibodies DL16 and SS144 yielded similar results. The reversibility of changes in the SS144 epitope differed following prolonged or sequential treatments for reasons that are not clear; perhaps the manners in which gB conformation is affected are slightly different between these two assays.
MAb H1817 exhibited a loss of reactivity following 3 h at pH 5.0, but upon neutralization, antibody reactivity returned. Interestingly, H1359 and H1817 epitopes appeared to undergo conformational changes following 3-h pH 5.0 treatment, whereas 10-min treatment had no detectable effect as reported previously (16, 18). SS10 had similar reactivities under all tested conditions.
Three hours of low-pH pretreatment of HSV-1 irreversibly inactivates >99% of virions, and the H126 domain I change remains irreversible following this treatment. The results (Fig. 6A and 7C) represent the first irreversible conformational changes in gB identified to date. We propose that mildly acidic pH inactivates HSV at least in part due to irreversible changes induced in the gB fusion domain.
DISCUSSION
Endosomal low pH is required for HSV entry into a subset of pathophysiologically relevant cell types. In a related manner, mildly acidic pH reduces the infectivity of isolated virions. Our results reveal the mechanism of low-pH inactivation of HSV: defective fusion activity. We also demonstrate that irreversible conformational changes in the class III fusion protein gB correlate with irreversible inactivation of infectivity. The conformational changes that govern herpesvirus membrane fusion during entry are not easily captured. Changes concurrent with low-pH inactivation of isolated virions may reflect those that drive pH-dependent fusion during HSV entry. The data support a model in which the functional region of gB that contains the fusion domain confers sensitivity of HSV to mildly acidic-pH inactivation. We propose that similar gB conformational changes in this domain are critical for membrane fusion.
Low-pH inactivation of HSV is irreversible.
Short exposures of HSV to mildly acidic pH result in irreversible and partial inactivation of infectivity (2, 9, 10, 20, 22, 23). Virtually complete, irreversible inactivation of HSV requires hours of pretreatment with mildly acidic pH (Fig. 7A). Similarly, 6-h pretreatment of rabies virus with low pH completely inactivates fusion activity. This loss of fusion activity correlates with irreversible conformational changes in the rabies virus class III fusion protein G (6). In contrast, irreversible low-pH inactivation of viruses bearing class I or class II fusion proteins occurs within minutes (5, 8). Following low-pH treatment of HSV-1, inactivation is irreversible, even if virions are permitted to recover at neutral pH for hours. In contrast to HSV-1, when low-pH-treated rabies virus is allowed to recover at neutral pH, inactivation becomes reversible with fusion activity restored (21). This discrepancy may be explained by the more complex fusion mechanism of HSV-1, which requires the action of at least four distinct glycoproteins. Further, low pH alone does not induce HSV-1 fusion (34), whereas it is sufficient for fusion mediated by rhabdovirus and baculovirus class III fusion proteins (21, 35). Low-pH treatment of cells expressing HSV-1 gD and gB mutants and an HSV-2 gH/gL mutant enhances cell-cell fusion in the absence of a gD receptor (36).
Irreversible change in gB following low-pH exposure.
Reversible conformational changes are unique to the class III fusion protein family and have been hypothesized to permit the virus to travel through acidic compartments during egress without pretriggering the fusion machinery (1, 18, 21, 37). To avoid pretriggering, some enveloped viruses maintain their fusion protein in an inactive precursor form until it is cleaved posttranslationally by a host cell endoprotease (38). During the HSV replication cycle, HSV particles can encounter low pH multiple times. Newly synthesized virions may egress through acidic compartments (31, 39, 40). Released progeny virions that subsequently infect a naive epithelial cell are thought to again encounter endosomal low pH during entry (2). Following in vitro simulation of these exposures, infectivity was recovered after a single round of low-pH exposure but was increasingly inactivated by additional exposures. Cell-associated virions, some of which have presumably been only minimally subjected to acidic compartments, were similarly susceptible to low-pH inactivation (data not shown). Together, our data indicate that for HSV, reversibility of conformational change does not protect the virus from premature exposure to low pH, despite gB being a class III fusion protein.
Both sequential and prolonged pH treatments of HSV-1 resulted in previously undetected irreversible changes in gB. Switching HSV-1 back and forth between neutral and low pHs may be harsher on the virus and on gB than a single prolonged exposure to low pH (Fig. 4 to 7). Previously, MAbs to a given gB domain, such as H126 and SS55 to domain I, all reacted similarly to HSV-1 gB treated for 10 min. Here, conformational changes suggested by the lack of MAb reactivity are reversible for SS55 but irreversible for H126, suggesting that the gB subdomains do not change en masse in response to low pH. Rather, mildly acidic pH induces distinct conformational changes within a particular domain. Likewise, pH-induced changes in gB domains I and V were previously thought to be similar (16, 18, 20), particularly since these domains are in close proximity in the postfusion structure, but the results described here suggest that domain I conformational changes occur independently of the changes in domain V.
MAb H126 has complement-independent, virus-neutralizing activity and binds to gB domain I, the fusion domain (41–43). The H126 epitope becomes less accessible upon exposure of gB to low pH both in vitro and during viral entry (18). H126 blocks virion-induced FFWO, and a hyperfusogenic, FFWO form of virion gB bears an H126 epitope with altered accessibility (27). Thus, the conformation of low-pH-treated wild-type gB is similar to the antigenic conformation of a mutant gB with enhanced fusion activity, supporting the notion that low pH triggers gB fusion activity. Also, H126 neutralizes HSV entry into cells to similar extents regardless of whether the cells support low-pH entry or pH-independent entry (10, 27). We propose that MAb H126 neutralizes HSV infection by inhibiting fusion, possibly by blocking gB conformational change or by preventing contact of fusion loops with the target membrane. Interestingly, the H126 epitope is less accessible in the structure of a full-length membrane-bound gB determined by cryo-electron tomography than in the postfusion ectodomain structure (44). Thus, low-pH-treated gB may resemble this available cryostructure.
This work distinguishes changes in gB antigenic structure from changes in the quaternary structure of gB. Low-pH-triggered changes in both the gB antigenic and oligomeric structure were previously reported to be reversible, to have similar pH thresholds, and to occur over similar pH ranges (16, 18, 45). A possible explanation for these results is that the approaches employed were measuring aspects of the same conformational change. Results reported here suggest that this is not the case. Oligomeric changes as detected by oligomer-specific MAb DL16 or the SDS stability assay are exquisitely reversible. However, under the same treatment conditions, the reduction of reactivity of MAbs H126 and SS144 becomes irreversible. This suggests that these two approaches are measuring distinct acid-induced conformational changes in gB. HSV gB is an oligomer, and a truncated ectodomain form is trimeric (42, 46). Several lines of evidence suggest that low pH triggers gB to assume a lower-density oligomeric conformation (18). The precise identity of the distinct gB-containing high-molecular-weight complexes is not known. Whether they represent, for example, distinct trimeric conformations or hetero-oligomeric complexes with another protein(s) is under investigation.
Virions exposed to low pH either for 3 h or for 4 sequential times were robustly and irreversibly inactivated. In both cases, there was an irreversible change in gB, which we propose is partly responsible for inactivation (Fig. 5 and 7B). However, the irreversible effect on infectivity following 10 min of low-pH exposure does not appear to correlate with changes in the H126 epitope of gB, which are reversible under these same conditions. The irreversible 10-min inactivation of virions, albeit limited, may be due to changes other than those detected here. Alternately, the dot blot approach may not be sensitive enough to detect a similarly limited reduction in H126 reactivity.
Low-pH preexposure of virions reduces HSV infectivity into cells that support diverse pathways of entry regardless of a low-pH requirement, suggesting that inactivation targets a feature that is conserved. Our results suggest that the mechanism of inactivation lies in a perturbation of the fusion function and not at the level of cell attachment or engagement of gD receptor (Fig. 2) (16). Low-pH pretreatment of virions also reduces entry into Vero cells, which support direct fusion of HSV with the plasma membrane, consistent with mildly acidic pH inactivating virus-cell fusion. A longstanding assay of HSV-1 entry kinetics relies on inactivation of noninternalized, attached virions by pH 3.0 buffer (24, 47). It is not yet clear whether inactivation by pH 3.0 and that by the more physiologically relevant mildly acidic pH share a similar mechanism. Low-pH-triggered changes have also been detected in herpesvirus gH (19, 48). It remains to be determined whether gH is an additional target of acid inactivation. We propose that changes in the region of gB containing fusion loops are responsible for low-pH inactivation and that, correspondingly, similar changes are critical for the activation of the HSV fusion machinery during viral entry.
MATERIALS AND METHODS
Cells and viruses.
Vero cells (American Type Culture Collection, Manassas, VA) were propagated in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals. Atlanta, GA). HSV-1 strains KOS (a gift from Priscilla Schaffer, Harvard University) and ANG path (a gift from Thomas Holland, Wayne State University) were propagated on Vero cells, and their titers were determined.
Preparation of HSV-1.
Vero cells were infected with HSV-1 at a multiplicity of infection (MOI) of 0.05. At 72 h postinfection (p.i.), infected-cell supernatant was centrifuged at 27,000 × g for 45 min through a 5% sucrose–phosphate-buffered saline (PBS) cushion. Pellets were resuspended overnight at 4°C in 20 mM HEPES (Thermo Fisher Scientific)-buffered DMEM supplemented with 10% FBS. Concentrated virions were sonicated and stored at −80°C.
Antibodies.
Anti-HSV-1 gB mouse monoclonal antibodies H126 (41) (domain I [42]), H1359 (domain III [49]), and H1817 (49) (domain VI [50]) were purchased from Virusys, Taneytown, MD. Anti-gB monoclonal antibodies DL16 (domain V [51]), SS10 (domain IV [50]), SS55 (52) (domain I [51]), and SS144 (50) (domain V [42]) were provided by G. Cohen and R. Eisenberg, University of Pennsylvania. MAbs H126, SS10, SS55, and SS144 neutralize HSV-1 infectivity (41, 50).
Prolonged neutralization of low-pH inactivation.
HSV-1 KOS was diluted in fusion medium: serum-free, sodium bicarbonate-free DMEM, containing 5 mM HEPES, 5 mM 2-(N-morpholino)ethanesulfonic acid (MES, Sigma, St. Louis, MO), 5 mM sodium succinate (Sigma), and 0.2% bovine serum albumin (BSA). Medium containing virions was adjusted to pH 7.4 or 5.0 with HCl at 37°C for 20 min. The pH 5.0-treated virions were then neutralized to pH 7.4 with a pretitrated amount of NaOH for 10, 60, or 120 at 37°C and added to Vero cells. At 2 h p.i., inoculum was replaced with DMEM containing 10% FBS. At 18 to 24 h p.i., cells were fixed with 2:1 methanol-acetone. Plaques were visualized with HSV-1 polyclonal antibody HR50 (Fitzgerald Industries, Acton, MA) and protein A conjugated with horseradish peroxidase (Thermo Fisher Scientific).
Attachment of HSV to cells.
HSV-1 KOS virions were treated with Turbo DNAfree (Thermo Fisher Scientific) to remove any extravirion DNA. Treated virions were diluted in fusion medium, treated for 10 min at 37°C with pH 5.0, and neutralized to pH 7.4 for 10 min. Heparin (2 μg/ml; Sigma) was added to virions as an inhibition of attachment control. Vero cells grown on coverslips were chilled, and ice-cold virions were added (40 genome copies per cell) followed by spinoculation at 200 × g for 1 h at 4°C (53). Cells were washed three times with ice-cold phosphate-buffered saline (PBS; Thermo Fisher Scientific) and then trypsinized. DNA was harvested from single-cell suspensions using the QIAamp DNA blood minikit (Qiagen, Germany), and ICP22 copy number was determined using qPCR (9, 34).
Fusion-from-without (FFWO) activity.
Confluent Vero cells were pretreated with 0.5 mM cycloheximide (Sigma) for 15 min. Cell-free HSV-1 ANG path virions were diluted in fusion medium adjusted to pH 7.4 or 5.0 for 10 min at 37°C, neutralized for 10 min, and bound to cells (MOI, ∼50) via spinoculation at 200 × g for 1 h at 4°C. FBS (10%) was added, and cells were shifted to 37°C for 3 h in the presence of cycloheximide. Cells were washed with PBS and fixed in 100% methanol. Monolayers were air dried and stained with Giemsa stain (Sigma). Micrographs were captured with a Zeiss Axiovert 40C microscope equipped with a Canon PowerShot G6 digital camera. Random fields of >300 nuclei were scored. The number of nuclei in contact with two or more nuclei divided by the total number of nuclei and then multiplied by 100% yielded the percent fusion.
Infectivity of HSV sequentially exposed to low pH.
HSV-1 KOS virions were diluted in pH 7.4 fusion medium, adjusted to pH 5.0 for 10 min at 37°C with HCl, and then neutralized to pH 7.4 using equivalent NaOH. This represents “1×” sequential treatment. This process was repeated twice (2×) or four times (4×). Nonsequentially exposed virions were treated at pH 5.0 for 40 min and neutralized to 7.4 for 40 min. Sample titers were then determined on Vero cells.
Analysis of gB oligomeric structure by PAGE.
HSV-1 KOS virions were diluted in fusion medium and adjusted to pH 7.4 or pH 5.0 for 10 min at 37°C. To test reversibility, samples were neutralized for 10 min, representing 1× sequential treatment. Virions were exposed to low pH sequentially 2 times or 4 times. For prolonged neutralization, virions were incubated at pH 7.4 or 5.0 for 3 h and neutralized for 10 min when testing reversibility. Following pH exposures, 1% SDS was added. Samples were subjected to “native” (nonreducing, semidenaturing) PAGE essentially as described by Cohen et al. (54). Modified Laemmli buffer containing 0.2% SDS and no reducing agent was added to samples. Unheated samples were resolved via PAGE and transferred to nitrocellulose. Membranes were blocked and probed with gB MAb H1359. Following incubation with horseradish peroxidase-conjugated secondary antibody, SuperSignal West Dura extended-duration substrate (Thermo Fisher Scientific) was added, and membranes were exposed to X-ray film (Genesee Scientific).
Dot blot analysis of antigenic reactivity.
HSV-1 KOS was diluted in fusion medium. Virions were adjusted to pH 7.4 or 5.0 for 10 min at 37°C. Samples were either blotted directly to nitrocellulose (Minifold dot blot apparatus; Whatman, Little Chalfont, Buckinghamshire, United Kingdom) or neutralized to pH 7.4. The sequentially treated samples were then reexposed to pH 5.0 followed by neutralization. This process was repeated one, two, three, or four times. Samples subjected to prolonged low-pH treatment were incubated at pH 7.4 or 5.0 for 3 h at 37°C and either blotted immediately or first neutralized to pH 7.4 for 10 min at 37°C. Membranes were blocked with 5% milk in PBS. Anti-gB antibodies were added to membranes at neutral pH followed by horseradish peroxidase (HRP)-conjugated secondary antibody (Thermo Fisher Scientific). SuperSignal West Dura extended-duration substrate (Thermo Fisher Scientific) was added, and membranes were exposed to X-ray film (Genesee Scientific, San Diego, CA).
Infectivity of HSV exposed to low pH for a prolonged period.
HSV-1 KOS virions were diluted in pH 7.4 fusion medium and adjusted to pH 5.0 for 3 h at 37°C. Virions were neutralized to pH 7.4 for 10 min prior to determining titer on Vero cells.
ACKNOWLEDGMENTS
We thank Gary Cohen, Roselyn Eisenberg, and Thomas Holland for generous gifts of reagents.
This work was supported by Public Health Service grants AI119159, AI122059, and AI096103 (A.V.N.) and GM008336 (D.J.W.) and National Science Foundation Graduate Research Fellowship DGE 1347973 (D.J.W.).
REFERENCES
- 1.Nicola AV. 2016. Herpesvirus entry into host cells mediated by endosomal low pH. Traffic 17:965–975. doi: 10.1111/tra.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol 77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J Virol 79:6655–6663. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicola AV, Hou J, Major EO, Straus SE. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J Virol 79:7609–7616. doi: 10.1128/JVI.79.12.7609-7616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stegmann T, Booy F, Wilschut J. 1987. Effects of low pH on influenza virus. Activation and inactivation of the membrane fusion capacity of the hemagglutinin. J Biol Chem 262:17744–17749. [PubMed] [Google Scholar]
- 6.Roche S, Gaudin Y. 2002. Characterization of the equilibrium between the native and fusion-inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology 297:128–135. doi: 10.1006/viro.2002.1429. [DOI] [PubMed] [Google Scholar]
- 7.Edwards J, Mann E, Brown DT. 1983. Conformational changes in Sindbis virus envelope proteins accompanying exposure to low pH. J Virol 45:1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bron R, Wahlberg JM, Garoff H, Wilschut J. 1993. Membrane fusion of Semliki Forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J 12:693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komala Sari T, Pritchard SM, Cunha CW, Wudiri GA, Laws EI, Aguilar HC, Taus NS, Nicola AV. 2013. Contributions of herpes simplex virus 1 envelope proteins to entry by endocytosis. J Virol 87:13922–13926. doi: 10.1128/JVI.02500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dollery SJ, Lane KD, Delboy MG, Roller DG, Nicola AV. 2010. Role of the UL45 protein in herpes simplex virus entry via low pH-dependent endocytosis and its relationship to the conformation and function of glycoprotein B. Virus Res 149:115–118. doi: 10.1016/j.virusres.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramalho-Santos J, Nir S, Duzgunes N, Pato de Carvalho A, Pedroso de Lima MDC. 1993. A common mechanism for influenza virus fusion activity and inactivation. Biochemistry 32:2771–2779. doi: 10.1021/bi00062a006. [DOI] [PubMed] [Google Scholar]
- 12.Nicola AV, Straus SE. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol 78:7508–7517. doi: 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heldwein E, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 65:1653–1668. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dollery SJ, Wright CC, Johnson DC, Nicola AV. 2011. Low-pH-dependent changes in the conformation and oligomeric state of the prefusion form of herpes simplex virus glycoprotein B are separable from fusion activity. J Virol 85:9964–9973. doi: 10.1128/JVI.05291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Junankar PR, Cherry RJ. 1986. Temperature and pH dependence of the haemolytic activity of influenza virus and of the rotational mobility of the spike glycoproteins. Biochim Biophys Acta 854:198–206. doi: 10.1016/0005-2736(86)90111-2. [DOI] [PubMed] [Google Scholar]
- 18.Dollery SJ, Delboy MG, Nicola AV. 2010. Low pH-induced conformational change in herpes simplex virus glycoprotein B. J Virol 84:3759–3766. doi: 10.1128/JVI.02573-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cairns TM, Whitbeck JC, Lou H, Heldwein EE, Chowdary TK, Eisenberg RJ, Cohen GH. 2011. Capturing the herpes simplex virus core fusion complex (gB-gH/gL) in an acidic environment. J Virol 85:6175–6184. doi: 10.1128/JVI.00119-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siekavizza-Robles CR, Dollery SJ, Nicola AV. 2010. Reversible conformational change in herpes simplex virus glycoprotein B with fusion-from-without activity is triggered by mildly acidic pH. Virol J 7:352–352. doi: 10.1186/1743-422X-7-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaudin Y, Tuffereau C, Segretain D, Knossow M, Flamand A. 1991. Reversible conformational changes and fusion activity of rabies virus glycoprotein. J Virol 65:4853–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitbeck JC, Zuo Y, Milne RS, Cohen GH, Eisenberg RJ. 2006. Stable association of herpes simplex virus with target membranes is triggered by low pH in the presence of the gD receptor, HVEM. J Virol 80:3773–3780. doi: 10.1128/JVI.80.8.3773-3780.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuyama AC, Cheshenko N, Carlucci MJ, Li J-H, Goldberg CL, Waller DP, Anderson RA, Profy AT, Klotman ME, Keller MJ. 2006. ACIDFORM inactivates herpes simplex virus and prevents genital herpes in a mouse model: optimal candidate for microbicide combinations. J Infect Dis 194:795–803. doi: 10.1086/506948. [DOI] [PubMed] [Google Scholar]
- 24.Huang AS, Wagner RR. 1964. Penetration of herpes simplex virus into human epidermoid cells. Exp Biol Med 116:863–869. doi: 10.3181/00379727-116-29392. [DOI] [PubMed] [Google Scholar]
- 25.Laquerre S, Argnani R, Anderson DB, Zucchini S, Manservigi R, Glorioso JC. 1998. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J Virol 72:6119–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saharkhiz-Langroodi A, Holland TC. 1997. Identification of the fusion-from-without determinants of herpes simplex virus type 1 glycoprotein B. Virology 227:153–159. doi: 10.1006/viro.1996.8327. [DOI] [PubMed] [Google Scholar]
- 27.Roller DG, Dollery SJ, Doyle JL, Nicola AV. 2008. Structure–function analysis of herpes simplex virus glycoprotein B with fusion-from-without activity. Virology 382:207–216. doi: 10.1016/j.virol.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 28.Delboy MG, Patterson JL, Hollander AM, Nicola AV. 2006. Nectin-2-mediated entry of a syncytial strain of herpes simplex virus via pH-independent fusion with the plasma membrane of Chinese hamster ovary cells. Virology J 3:1. doi: 10.1186/1743-422X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wudiri GA, Pritchard SM, Li H, Liu J, Aguilar HC, Gilk SD, Nicola AV. 2014. Molecular requirement for sterols in herpes simplex virus entry and infectivity. J Virol 88:13918–13922. doi: 10.1128/JVI.01615-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Falke D, Knoblich A, Müller S. 1985. Fusion from without induced by herpes simplex virus type 1. Intervirology 24:211–219. doi: 10.1159/000149645. [DOI] [PubMed] [Google Scholar]
- 31.Hogue IB, Bosse JB, Hu J-R, Thiberge SY, Enquist LW. 2014. Cellular mechanisms of alpha herpesvirus egress: live cell fluorescence microscopy of pseudorabies virus exocytosis. PLoS Pathog 10:e1004535. doi: 10.1371/journal.ppat.1004535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hannah BP, Cairns TM, Bender FC, Whitbeck JC, Lou H, Eisenberg RJ, Cohen GH. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J Virol 83:6825–6836. doi: 10.1128/JVI.00301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hannah BP, Heldwein EE, Bender FC, Cohen GH, Eisenberg RJ. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J Virol 81:4858–4865. doi: 10.1128/JVI.02755-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker EB, Pritchard SM, Cunha CW, Aguilar HC, Nicola AV. 2015. Polyethylene glycol-mediated fusion of herpes simplex type 1 virions with the plasma membrane of cells that support endocytic entry. Virology J 12:1. doi: 10.1186/s12985-014-0235-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blissard GW, Wenz JR. 1992. Baculovirus gp64 envelope glycoprotein is sufficient to mediate pH-dependent membrane fusion. J Virol 66:6829–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atanasiu D, Saw WT, Eisenberg RJ, Cohen GH. 2016. Regulation of herpes simplex virus glycoprotein-induced cascade of events governing cell-cell fusion. J Virol 90:10535–10544. doi: 10.1128/JVI.01501-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaudin Y. 2000. Reversibility in fusion protein conformational changes the intriguing case of rhabdovirus-induced membrane fusion. Subcell Biochem 34:379–408. [DOI] [PubMed] [Google Scholar]
- 38.Nagai Y. 1995. Virus activation by host proteinases. A pivotal role in the spread of infection, tissue tropism and pathogenicity. Microbiol Immunol 39:1–9. [DOI] [PubMed] [Google Scholar]
- 39.Hogue IB, Scherer J, Enquist LW. 2016. Exocytosis of alphaherpesvirus virions, light particles, and glycoproteins uses constitutive secretory mechanisms. mBio 7:e00820-16. doi: 10.1128/mBio.00820-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harley CA, Dasgupta A, Wilson DW. 2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J Virol 75:1236–1251. doi: 10.1128/JVI.75.3.1236-1251.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kousoulas KG, Pellett PE, Pereira L, Roizman B. 1984. Mutations affecting conformation or sequence of neutralizing epitopes identified by reactivity of viable plaques segregate from syn and ts domains of HSV-1 (F) gB gene. Virology 135:379–394. doi: 10.1016/0042-6822(84)90194-6. [DOI] [PubMed] [Google Scholar]
- 42.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 43.Pellett PE, Kousoulas K, Pereira L, Roizman B. 1985. Anatomy of the herpes simplex virus 1 strain F glycoprotein B gene: primary sequence and predicted protein structure of the wild type and of monoclonal antibody-resistant mutants. J Virol 53:243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeev-Ben-Mordehai T, Vasishtan D, Durán AH, Vollmer B, White P, Pandurangan AP, Siebert CA, Topf M, Grünewald K. 2016. Two distinct trimeric conformations of natively membrane-anchored full-length herpes simplex virus 1 glycoprotein B. Proc Natl Acad Sci U S A 113:4176–4181. doi: 10.1073/pnas.1523234113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siekavizza-Robles CR, Dollery SJ, Nicola AV. 2010. Reversible conformational change in herpes simplex virus glycoprotein B with fusion-from-without activity is triggered by mildly acidic pH. Virol J 7:1. doi: 10.1186/1743-422X-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Claesson-Welsh L, Spear PG. 1986. Oligomerization of herpes simplex virus glycoprotein B. J Virol 60:803–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson DC, Spear PG. 1982. Monensin inhibits the processing of herpes simplex virus glycoproteins, their transport to the cell surface, and the egress of virions from infected cells. J Virol 43:1102–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gillet L, Colaco S, Stevenson PG. 2008. The murid herpesvirus-4 gL regulates an entry-associated conformation change in gH. PLoS One 3:e2811. doi: 10.1371/journal.pone.0002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pereira L, Ali M, Kousoulas K, Huo B, Banks T. 1989. Domain structure of herpes simplex virus 1 glycoprotein B: neutralizing epitopes map in regions of continuous and discontinuous residues. Virology 172:11–24. doi: 10.1016/0042-6822(89)90102-5. [DOI] [PubMed] [Google Scholar]
- 50.Bender FC, Samanta M, Heldwein EE, de Leon MP, Bilman E, Lou H, Whitbeck JC, Eisenberg RJ, Cohen GH. 2007. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J Virol 81:3827–3841. doi: 10.1128/JVI.02710-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cairns TM, Fontana J, Huang Z-Y, Whitbeck JC, Atanasiu D, Rao S, Shelly SS, Lou H, de Leon MP, Steven AC. 2014. Mechanism of neutralization of herpes simplex virus by antibodies directed at the fusion domain of glycoprotein B. J Virol 88:2677–2689. doi: 10.1128/JVI.03200-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bender FC, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J Virol 79:11588–11597. doi: 10.1128/JVI.79.18.11588-11597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delboy MG, Roller DG, Nicola AV. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J Virol 82:3381–3390. doi: 10.1128/JVI.02296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cohen G, Isola V, Kuhns J, Berman P, Eisenberg R. 1986. Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: use of a nondenaturing (“native” gel) system of polyacrylamide gel electrophoresis coupled with Western blotting. J Virol 60:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]