

ABSTRACT
Bacteriophages are the source of many valuable tools for molecular biology and genetic manipulation. In Streptomyces, most DNA cloning vectors are based on serine integrase site-specific DNA recombination systems derived from phage. Because of their efficiency and simplicity, serine integrases are also used for diverse synthetic biology applications. Here, we present the genome of a new Streptomyces phage, ϕJoe, and investigate the conditions for integration and excision of the ϕJoe genome. ϕJoe belongs to the largest Streptomyces phage cluster (R4-like) and encodes a serine integrase. The attB site from Streptomyces venezuelae was used efficiently by an integrating plasmid, pCMF92, constructed using the ϕJoe int-attP locus. The attB site for ϕJoe integrase was occupied in several Streptomyces genomes, including that of S. coelicolor, by a mobile element that varies in gene content and size between host species. Serine integrases require a phage-encoded recombination directionality factor (RDF) to activate the excision reaction. The ϕJoe RDF was identified, and its function was confirmed in vivo. Both the integrase and RDF were active in in vitro recombination assays. The ϕJoe site-specific recombination system is likely to be an important addition to the synthetic biology and genome engineering toolbox.
IMPORTANCE Streptomyces spp. are prolific producers of secondary metabolites, including many clinically useful antibiotics. Bacteriophage-derived integrases are important tools for genetic engineering, as they enable integration of heterologous DNA into the Streptomyces chromosome with ease and high efficiency. Recently, researchers have been applying phage integrases for a variety of applications in synthetic biology, including rapid assembly of novel combinations of genes, biosensors, and biocomputing. An important requirement for optimal experimental design and predictability when using integrases, however, is the need for multiple enzymes with different specificities for their integration sites. In order to provide a broad platform of integrases, we identified and validated the integrase from a newly isolated Streptomyces phage, ϕJoe. ϕJoe integrase is active in vitro and in vivo. The specific recognition site for integration is present in a wide range of different actinobacteria, including Streptomyces venezuelae, an emerging model bacterium in Streptomyces research.
KEYWORDS: serine integrase, recombination directionality factor, integration vector, R4-like phage, Streptomyces venezuelae, Streptomyces coelicolor, mobile genetic elements, bacteriophage genetics
INTRODUCTION
Over the past few decades, serine integrases have become widely established as tools for genome engineering and synthetic biology (1, 2). Serine integrases are phage-encoded DNA site-specific recombinases that mediate recombination between two short (<50 bp) sequences. The integration reaction occurs during the establishment of lysogeny, during which the integrase causes a single crossover between the attB site on the bacterial chromosome and the attP site on the circularized phage genome, leading to the integrated phage DNA flanked by the recombinant sites attL and attR (1, 3). Integrase dimers bind to the two att sites and produce double-strand breaks with 2-bp overhangs (3, 4); the cut ends are then exchanged, and the DNA backbone is religated to produce the recombinant products (5). The attL and attR sites each contain reciprocal halves of the attP and attB sites (6). As integrases are unable to use attL and attR as substrates without an accessory protein, a recombination directionality factor (RDF), the integrated phage genome is stable until the RDF-encoding gene is expressed during prophage induction (3). Recombination between attL and attR is the excision reaction and is essentially the reverse of integration, releasing the phage genome and reforming attP and attB. While only integrase is required to mediate integration, excision requires both integrase and the RDF. Genome engineers have exploited these systems to integrate genes of interest into a specific site on the chromosome, which can either be the endogenous attB or an introduced attB or attP used as a docking site (1). The simplicity of the serine integrase-mediated site-specific recombination systems means that they are reliably portable to heterologous hosts where DNA can be integrated stably and in single copy.
The simple requirements for serine integrases make them amenable to a wide variety of applications. The earliest examples of this were to integrate an attP plasmid into a target genome containing the cognate attB (or vice versa) (7), allowing stable delivery of genes into diverse species, including bacteria (6, 8–10), mice (11), mosquitoes (12), and humans (13). More complex genetic engineering approaches use integrases in in vitro-ordered assembly of multiple DNA fragments (14, 15). In vivo genome manipulations can also be achieved either by iterative rounds of recombination (16, 17) or multiplexing orthogonal integrase/att sites (18). Integrase-mediated DNA rearrangements can also be used to provide permanent genetic memory in novel types of biosensors (19, 20). Some applications, such as post factum modifications (15) or biocomputing (19, 21), need controlled excision, and this requires integrase and its cognate RDF. The RDF binds directly to the integrase protein and is thought to induce a conformational change that allows attL and attR to be used as recombination substrates while inhibiting recombination of attB and attP (22, 23).
A limiting factor for the use of serine integrases for complex multiplexed applications is the number of well-characterized integrases and, perhaps more pressingly, RDFs. Only seven integrase-RDF pairs have been characterized to date (from phages TP901-1 [24], ϕC31 [22], ϕBT1 [25], Bxb1 [23], ϕRv1 [26], and SPBc [27], and from the excisive element of Anabaena and Nostoc cyanobacterial species [28]), but many more integrases have been studied without their RDFs (1, 2, 29–31). Integrase genes are easily identified by comparative sequence analysis, and when the integrase is prophage encoded, the attachment sites can also be predicted (31). RDFs, however, are far more difficult to predict, because known examples share little sequence homology, vary markedly in size, and differ in gene location in phage genomes (1). Expansion of the available arsenal of serine integrases and RDFs is desirable to enable advanced synthetic biology applications.
Phages that encode serine integrases are prevalent in Gram-positive bacteria, and in particular in actinobacteria. Here, we describe a newly isolated Streptomyces phage, ϕJoe, and its serine integrase (Int) that is only distantly related to characterized integrases. ϕJoe Int is active in vivo in Streptomyces and E. coli, and the integrase protein is readily purified and is able to carry out efficient in vitro recombination. We also describe the ϕJoe RDF, a 6.8-kDa protein that is able to promote excisive recombination and inhibit integration.
RESULTS AND DISCUSSION
Isolation of actinophage ϕJoe and genome sequence.
Raw soil samples were enriched for environmental phage using S. coelicolor strain M145 as a propagation host. The phage chosen for further analysis, ϕJoe, is a siphovirus with a capsid diameter of 46.5 nm (standard deviation [SD], 1.6 nm; n = 9) and a long flexible tail of 199.5 nm (SD, 12 nm; n = 8), with clear striations visible in most images (Fig. 1). ϕJoe is able to plaque on a broad range of Streptomyces hosts, producing lytic infection of seven out of nine species tested (Table 1). Saccharopolyspora erythraea (formerly Streptomyces erythraeus) and Streptomyces venezuelae were resistant to infection.
FIG 1.
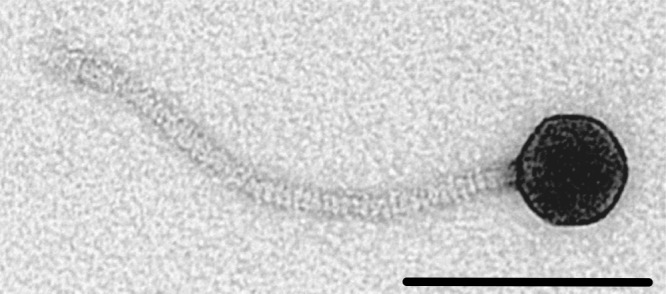
A ϕJoe virion imaged by transmission electron microscopy. Viral particles were negatively stained with uranyl acetate, and this image was taken at ×220,000 magnification. The scale bar represents 100 nm.
TABLE 1.
ϕJoe host range
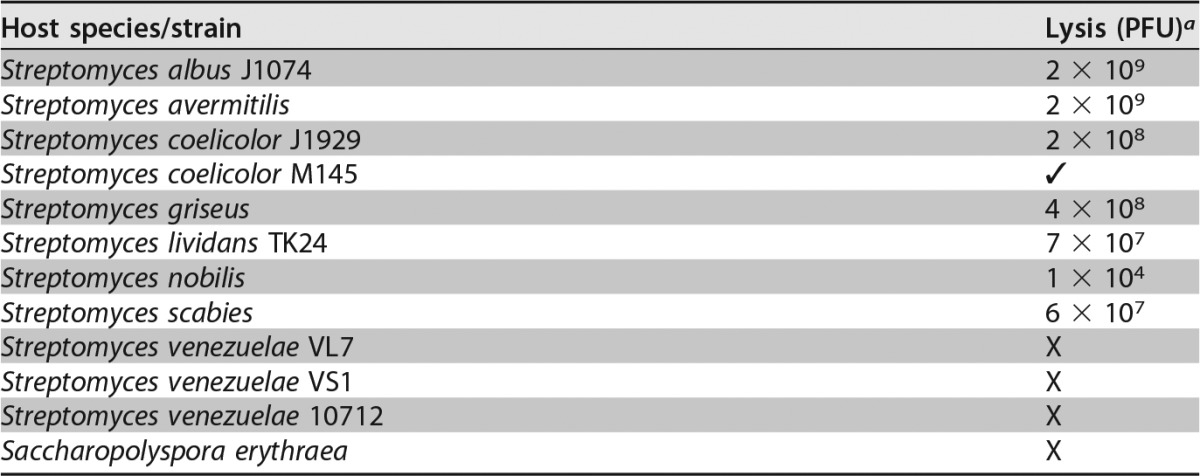
PFU per milliliter values quoted are illustrative of the relative plaquing efficiencies when challenged with the same phage stock propagated on S. coelicolor J1929. ✓, the phage could produce plaques on this strain, but the PFU/ml value was not calculated; X, we were not able to produce plaques for the indicated strain.
Genomic DNA was extracted from high-titer ϕJoe suspensions (>1010 PFU/ml) and sequenced on the Illumina MiSeq platform with 2,542× coverage. The phage genome is 48,941 bases (accession no. KX815338), with a G+C content slightly lower than that of the host bacteria, at 65.5% compared to ∼72% for most Streptomyces species. BLASTn was used to measure nucleotide identity for the closest relatives to ϕJoe; the generalized transducing phage ϕCAM (32) and two newly sequenced Streptomyces phages, Amela and Verse (Fig. S2), are 73, 76, and 76% identical, respectively, in global alignments. The ϕJoe genome contains 81 predicted open reading frames (Fig. 2), the majority of which have amino acid sequences similar to those of the three phages mentioned above and the well-characterized R4 phage (33). Notably, similarity to ϕJoe integrase (gp53) is absent from each of the closest genome matches but is instead present in several more distantly related phages (Fig. 3), indicative of phage mosaicism (34). Specifically, ϕJoe integrase is homologous to the uncharacterized integrases from five complete phages: Lannister (78% amino acid identity), Zemyla (74%), Danzina (73%), Lika (73%), and Sujidade (73%) (Fig. 3). Comparison to known integrases suggests that the catalytic serine is likely to be at position 46 in the protein sequence (VRLSVFT).
FIG 2.

Schematic of the ϕJoe genome. The genome is 48,941 bp in length. ORFs were predicted using GeneMark and Glimmer and then manually curated. ORFs are labeled and color-coded based on their predicted function. Orange, recombination; cyan, metabolism and DNA processing/replication; green, structural proteins; purple, lysis; black, regulatory; gray, hypothetical proteins with no known function; red, candidate RDF genes. Genes marked with an asterisk encode structural proteins that were detected by MS/MS. The histogram below the genome contains purple bars to indicate below-average G+C content (65.5%) and green bars to indicate above-average G+C content (1,000-nt window size, 20-nt step).
FIG 3.

Circos plot of the ϕJoe genome versus nine related phages. A BLASTN comparison was carried out for ϕJoe, the five sequenced phages with a ϕJoe-like integrase, the three closest whole-genome matches, and the well-characterized R4 phage. The E value cutoff was set to 1 × 10−100 and the high-scoring segment pairs (HSPs) to 100; ribbons are colored by genomic regions as defined in Fig. 1 and depicted above the Circos plot. The histograms above each genome are colored to reflect relative homology to the ϕJoe sequence based on BLAST score (red > orange > green > blue).
Purified phage particles were submitted for shotgun liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis to determine the structural proteome. At least one peptide match was detected from 14 ϕJoe gene products, five of which have predicted functions: portal, capsid, tail tape measure, scaffold, and head-tail adaptor (Fig. 2 and Table S1). The remaining nine gene products have no known function, but all cluster close to the predicted structural genes within a region of the genome spanning ∼21 kbp.
Characterization of ϕJoe integrase and attachment sites.
For most phage-encoded integration systems, the attP site lies adjacent to the int gene encoding the integrase. The attP sites for serine integrases are characteristically about 45 to 50 bp in length and contain inverted repeat sequences flanking a spacer of approximately 20 bp (3, 35). Examination of the ϕJoe genome identified a candidate attP site located 18 bp upstream of the int gene. A plasmid, pCMF92, was constructed by replacing the ϕC31 int-attP locus from the widely used integrating vector pSET152 with the ϕJoe int-attP locus (Fig. S1). Integration of pCMF92 would confirm whether the integrase is functional and the nature of the attP site, and, by rescuing the DNA flanking the integrated plasmid, the identity of the attB site could be deduced (Fig. 4). pCMF92 was introduced into S. coelicolor J1929 and S. lividans TK24 by conjugation, and apramycin-resistant colonies were obtained, but the frequencies were low (10−4 to 10−5 exconjugants/CFU) compared to other integrating vectors (10−2 to 10−3 exconjugants/CFU) (9, 18). To test whether integration was site specific, four S. coelicolor::pCMF92 cell lines were amplified from independent exconjugants, and the genomic DNAs were analyzed by Southern blotting using a probe derived from the ϕJoe int gene. In the four cell lines, pCMF92 had integrated into one of two different integration sites, as revealed by hybridization of the probe to two different restriction fragments (data not shown).
FIG 4.

ϕJoe attachment sites and integration sites. (A) Diagram of ϕJoe attP showing the central dinucleotides (blue) and imperfect inverted repeats (orange and arrows). (B) Schematic of the genomic context of the two S. coelicolor integration sites (attLsc and attRsc, red boxes) used by the ϕJoe-integrating plasmid pCMF92. The location of the PstI sites used for identification of the att sites are shown. The DNA between the attLsc and attRsc sites is an apparent mobile genetic element, with homologous integrase and RDF genes (orange arrows) to those of ϕJoe. (C) Alignment of S. venezuelae attB (attBsv) with the two S. coelicolor att sites (attRsc and attLsc) and the reconstituted attB site (attBsc) that would be produced by excision of the DNA between attRsc and attLsc. (D) Alignment of closely related attB sites identified by a BLASTN search against the nonredundant GenBank database. Hits were first filtered for matches of at least 80% and then for an E value of <1 × 1010 and a bit score of >75. (C and D) Nucleotide positions are shown as distance from the crossover dinucleotides (XX).
We then sought to characterize the two integration sites for pCMF92 in S. coelicolor by rescuing the integrated plasmids along with flanking DNA into E. coli. In pCMF92, there is 3.9 kbp of DNA between the ϕJoe attP site and the PstI cleavage site that contains the plasmid origin of replication and the apramycin resistance gene (Fig. S1). Genomic DNA from two S. coelicolor::pCMF92 cell lines, each containing pCMF92 integrated into one of the two different integration sites, was digested with PstI endonuclease, self-ligated, and introduced into E. coli DH5α by transformation. The rescued plasmids were sequenced over the recombination sites to validate the nature of the ϕJoe attP site and to identify the chromosomal positions of the two S. coelicolor integration sites. The ϕJoe attP site was confirmed to be ≤50 bp, and the 5′-GG dinucleotide at the center of an imperfect inverted repeat is predicted to be where the crossover occurs (Fig. 4A).
The two S. coelicolor integration sites for pCMF92 are located 3.9 kbp apart, separated by an apparent mobile genetic element comprising sco2603, encoding a putative serine integrase with 68% identity to ϕJoe integrase, and two further genes (Fig. 4B). Its product, SCO2603, is 68% identical to ϕJoe integrase. We hypothesized that the ϕJoe-integrating plasmid is inefficient in S. coelicolor because an ancestral and optimal attB site is occupied by the SCO2603-encoding element. The two integration sites for pCMF92 in S. coelicolor were therefore called attLsc and attRsc to reflect the provenance of the sites containing the mobile element. To test this hypothesis, the sequence of the ancestral attB site, attBsc, was predicted by removing the sequence between attLsc and attRsc, including the attP moieties that would have originated from the inserted mobile element (Fig. 4C). The reconstituted attBsc was used to interrogate the GenBank Streptomyces database for closely related extant sequences. Three species were chosen from the top 10 hits returned (S. avermitilis, S. albus, and S. venezuelae; Fig. 4D) and assayed for in vivo integration efficiency. S. venezuelae was the only host to support highly efficient integration after conjugation with pCMF92, at 160-fold greater frequency than S. coelicolor and 1,600-fold greater frequency than S. lividans (Fig. 5A). The integration frequencies for pCMF92 into S. venezuelae are similar to those reported for other characterized serine integrases (9, 18), and we demonstrate below that the attB site from S. venezuelae, attBsv, is indeed used efficiently by ϕJoe integrase. Plasmid pCMF92 could therefore be used as a new integrating vector for use in this newly emerging model system for Streptomyces research.
FIG 5.

Activity of ϕJoe integrase in vivo and in vitro. (A) Conjugation efficiency of an integrating vector, containing ϕJoe int and attP, into five recipient species: Streptomyces coelicolor (Sc), S. lividans (Sl), S. venezuelae (Sv), S. albus (Sal), and S. avermitilis (Sav). Levels of significance for S. venezuelae versus all other species in a one-way analysis of variance (ANOVA) were a P value of <0.001 (3 asterisks); all other comparisons were nonsignificant (n.s.). Error bars are standard deviation (Sc, n = 5; Sv and Sl, n = 3; Sal and Sav, n = 2). (B) Representative image of an in vivo integration assay to assess attBsv-attP recombination by ϕJoe integrase (pCMF107) and a negative control (pBAD-HisA). Recombination leads to deletion of an intervening lacZα gene and white colonies, and inactivity produces blue colonies. Integration efficiency is shown in parentheses (n = 3). (C) Representative image of in vitro recombination of two substrate plasmids, attP (pCMF91) and attBsv (pCMF95), to produce the cointegrant plasmid pCMF98. The concentration of ϕJoe integrase and incubation time for each reaction are indicated above the gel. (D) Time course for the integration reaction shown in panel C.
The S. venezuelae attBsv site was used as a BLASTn query to estimate the prevalence of potential ϕJoe insertion sites in sequenced species. In many instances, each half of the query sequence matched separate locations in the target genome, suggesting that ϕJoe-like attB sites are frequently occupied by either a prophage or a mobile element similar to that observed in S. coelicolor J1929. Hits were subsequently filtered for matches of at least 80% coverage, with an E value of <1 × 10−10 and a bit score of >75, which revealed numerous apparently unoccupied ϕJoe attB sites in diverse Streptomyces, Kitasatospora, and Dermacoccus species (Fig. 4D). Generally, the attB site for ϕJoe and the SCO2603 integrase-encoding elements are located 74 bp from the end of an open reading frame (ORF) encoding an SCO2606-like predicted B12 binding domain-containing radical S-adenosylmethionine (SAM) protein. Insertions this close to the end of an ORF may not necessarily cause loss of function of the gene product, and this could explain the prolific number of mobile elements that use this locus as an insertion site. Other than the recombination genes, the genetic content of the mobile elements located here varies markedly in different bacterial species (Fig. S2). Some Streptomyces strains have an SCO2603-containing genetic element almost identical to that of S. coelicolor J1929 (e.g., WM6391), others have no genes other than the recombination genes (e.g., NRRLF-5123), and some contain up to 40 kbp between the predicted attL and attR sites (Fig. S2).
ϕJoe integrase catalyzes efficient in vivo and in vitro integration.
In order for an integrase to have broad appeal as a bioengineering tool, it must be functional in heterologous hosts. As a proof of principle, we tested the activity of ϕJoe integrase in E. coli by cloning the integrase gene into an arabinose-inducible expression vector, pBAD-HisA, to produce pCMF107. Meanwhile, we constructed a reporter plasmid, pCMF116, containing the E. coli lacZα gene flanked by ϕJoe attBsv and attP sites in head to tail orientation (Fig. S3). Both plasmids were introduced into E. coli TOP10 cells (Invitrogen) by cotransformation and plated on selective agar plates containing 0.2% l-arabinose and 80 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). pBAD-HisA lacking an insert was used as a negative control. All of the transformants were white in the presence of ϕJoe int, indicating efficient recombination between the attBsv and attP sites, leading to loss of the lacZα gene (Fig. 5B and S3). ϕJoe integrase and its cognate attBsv and attP sites are, therefore, active in E. coli.
Another key application for serine integrases is for in vitro combinatorial assembly of genes for optimizing the expression of metabolic pathways (14, 15). In this application, different integrases are used to join (by recombination) specific pairs of DNA fragments tagged with their cognate attachment sites. In theory, this procedure can be multiplexed to assemble many DNA fragments together using different orthogonally acting integrases. The aim is to generate artificial operons with defined or random order. To test the suitability of ϕJoe Int for in vitro recombination reactions, the integrase gene was cloned into the His tag expression vector pEHISTEV and purified after overexpression in E. coli. In vitro recombination assays were carried out with ϕJoe attP (pCMF91) versus each of attBsc, attLsc, attRsc, and attBsv (pCMF97, pCMF90, pCMF94, and pCMF95, respectively) and using a range of ϕJoe integrase concentrations. Successful recombination between attachment sites produces a cointegrant plasmid, which can be distinguished from the substrate plasmids by restriction digestion and agarose gel electrophoresis (Fig. S3). In this assay, recombination was undetectable when attLsc (pCMF90) or attRsc (pCMF94) was used with attP (pCMF91) as the substrate. A small amount of recombination was observed (≤2%, Fig. S4) when the reconstituted attBsc (pCMF97) was used with attP (pCMF91). However, consistent with the observations in E. coli and in Streptomyces, the S. venezuelae attBsv site (pCMF95) was a highly efficient substrate for recombination with the ϕJoe attP site. ϕJoe integrase was effective over a broad range of concentrations (50 to 1,000 nM) (Fig. 5C and S4). Using 200 nM integrase, detectable recombination product was produced after ∼10 to 15 min, and after 2 h, approximately 70% of the substrate molecules were converted to product (Fig. 5C and D).
There are only 6 bp that differ between attBsc and attBsv, and all the differences are on the left-hand arm of the attB sites (Fig. 4C). Previously, a mutational analysis of the ϕC31 attB site showed that mutationally sensitive bases occur 2, 15, and 16 bases to either side of the crossover dinucleotide (36). As two of the differences between attBsc and attBsv are also 2 and 16 bases from the putative crossover 5′-GG (Fig. 4C), these base pair differences might account for the poor activity of attBsc in the in vitro assays.
Identification and validation of the ϕJoe RDF protein, gp52.
Although there are dozens of serine integrases that have been described in the literature, there are only seven published RDFs for serine integrases (ϕC31 gp3 [22], ϕBT1 gp3 [25], Bxb1 gp47 [23], TP901 ORF7/Xis [24], Anabaena/Nostoc XisI [28], SPBc SprB [27], and ϕRv1 Rv1584c/Xis [26]). The Bxb1 and ϕC31 RDFs are among the largest of these RDF proteins (approximately 27.5 kDa and 250 amino acids), and their genes are located in proximity to the phage DNA replication genes. Both RDFs have functions during phage replication in addition to acting as RDFs, but they are evolutionarily unrelated (25, 37). The RDFs from ϕBT1 and another ϕC31-like phage, TG1, are close relatives of the ϕC31 RDF at the sequence level (85% and 59% identical, respectively); furthermore, the ϕBT1-encoded RDF acts on ϕC31 integrase and vice versa (25). The ϕRv1 and SPBc RDFs are located within 1 or 2 ORFs of the int gene, a feature which is reminiscent of the xis genes that act with tyrosine integrases. ϕRv1, SPBc, TP901, and Anabaena/Nostoc RDFs are much smaller proteins than ϕC31 gp3 or Bxb1 gp47 (58 and 110 amino acids). Given the variation in RDF size, sequence, and genomic location, there are no sound generalizations yet for identifying new RDFs in phage genomes.
A list of four candidate genes (g40, g43, g49, and g52) for the ϕJoe RDF was drawn up based on comparable size to known small RDFs and genomic location (i.e., not located among the late/structural genes) (Fig. 2). One of the potential RDF genes (g52) is adjacent to int in the ϕJoe genome, but it is transcribed divergently, with the attP site situated between int and g52 (Fig. 2). Unlike the other candidate RDFs, gp52 homologues are only found in those phages with ϕJoe-like integrases (Fig. 3), and phylogenetic analysis of gp52 and the integrase indicated that the two proteins have followed a parallel evolutionary path (Fig. S5). Pairwise alignment of the 6.8-kDa (62 amino acids) gp52 protein with other known small RDFs revealed homology with ϕRv1 RDF (25.7% identity and 35.1% similarity; Fig. 6A). Also, examination of the mobile elements that have inserted into the attB sites in S. coelicolor and other Streptomyces spp. revealed that they also contain a gene encoding a gp52 homologue in a similar genetic context, i.e., the int and g52 genes are adjacent to the attL and attR sites, respectively, and would flank attP after excision (Fig. 4B and S2). The predicted secondary structure of ϕJoe gp52 contains an alpha-helix in the N-terminal region, a beta-sheet in the C-terminal region, and an unstructured region between (Fig. S6). Alignment of the ϕJoe-like RDFs found in intact phages and the RDFs found in the SCO2603-encoding mobile elements indicated that both of the structured regions are well conserved, particularly the putative alpha-helix, but the center of the protein is variable (Fig. S6).
FIG 6.

Identification of the ϕJoe RDF, gp52. (A) Alignment of ϕJoe and RV1 RDFs, colored using the BLOSUM62 scheme. (B) Representative agarose gel showing in vitro inhibition of integration by ϕJoe RDF. The concentrations of ϕJoe integrase and RDF for each reaction are indicated above the image. Reactions were stopped after 2 h and linearized using XhoI. (C) Representative agarose gel showing in vitro excision reactions catalyzed by ϕJoe integrase and RDF. The concentrations of ϕJoe integrase and RDF for each reaction are indicated above the image. Reactions were stopped after 2 h and linearized using XhoI. (D) In vivo excision assay to assess attLsv × attRsv recombination by ϕJoe integrase alone, ϕJoe RDF alone, and ϕJoe integrase coexpressed with the RDF. Recombination leads to deletion of an intervening lacZα gene and white colonies, and inactivity produces blue colonies. Expression from the T7 promoter successfully achieved almost complete excision activity for ϕJoe Int + RDF.
RDFs are able to influence integrase-catalyzed recombination in two ways; they activate the attL × attR reaction to regenerate attP and attB (excision), and they inhibit the attB × attP integration reaction (22, 23). We were unable to produce sufficient soluble gp52 protein for in vitro assays when expressed with a simple histidine tag; however, a maltose-binding protein (MBP)-gp52 fusion protein was more soluble. We tested the ability of MBP-gp52 to inhibit integration by titrating the protein against a fixed concentration of integrase at MBP-gp52/Int ratios of 1:2 to 22.5:1. When the MBP-gp52 was in excess, integration was repressed to less than 10%; however, at less-than-equimolar concentrations, recombination was equivalent to the control in which no MBP-gp52 was added (Fig. 6B). These results are similar to observations for ϕC31 and Bxb1 integrases and their cognate RDFs, gp3 and gp47 (22, 23).
To test the ability of gp52 to activate an excision reaction, a plasmid containing the cognate attLsv and attRsv sites was produced, pCMF98 (Fig. S3). The MBP-gp52 protein was unable to promote efficient excision under any conditions tested (not shown). Removal of the MBP tag using 3c protease increased excision activity, but the reaction was still inefficient after 2 h of incubation (Fig. 6C). Longer incubations of 5 to 20 h further increased the amount of substrates converted to product up to 45%, but it also led to significant amounts of excision products (10 to 20%) by the integrase alone. Thus, in comparison to the activity of other RDFs, gp52 has rather poor activity; ϕC31 gp3 activates approximately 60 to 80% conversion of the attL × attR substrates to products (22), and similar results are obtained with other RDFs (23, 25, 26).
To test the excision ability of ϕJoe gp52 in vivo, a g52-int coexpression operon was designed in which int and g52 were located directly downstream of the T7 promoter and ribosome binding site (RBS) in the expression vector pEHISTEV to produce pCMF117. A reporter plasmid, pCMF103, was produced containing the lacZα gene flanked by ϕJoe attLsv and attRsv sites (Fig. S3). pCMF117 and pCMF103 were introduced into E. coli BL21(DE3) cells by cotransformation and plated onto LB agar supplemented with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to induce expression of the g52-int operon (30). The reporter plasmid was then extracted from the BL21(DE3) transformants and introduced into E. coli DH5α to determine the percentage of plasmids that had undergone attLsv × attRsv recombination and had lost the lacZα gene. As controls, plasmids expressing either only integrase (pCMF87) or only gp52 (pCMF100) were also introduced together with the reporter (pCMF103) into BL21(DE3), and the assay was repeated using the same procedure. When ϕJoe integrase alone was expressed, excision occurred at a frequency of 37.6% (SD, 5.1%; n = 5), but when coexpressed with gp52, the frequency rose to 96.8% (SD, 1.3%; n = 5) (Fig. 6D). Expression of gp52 without integrase led to no detectable excision events (Fig. 6D). Although overall recombination in vivo was higher than that in vitro, the relative levels of attLsv × attRsv recombination by ϕJoe integrase alone and ϕJoe integrase with gp52 were comparable. Taken together, the in vivo and in vitro data indicate that ϕJoe gp52 has RDF activity.
The observation that ϕJoe integrase has a basal level of excision activity in the absence of its RDF is highly unusual for a phage-encoded integrase, and further study may provide novel insights into the mechanism and evolution of the serine integrases. Streptomyces phage ϕBT1 integrase was shown to catalyze bidirectional recombination, albeit at extremely low levels (38). The archetypal ϕC31 integrase is only able to mediate attL × attR recombination in the absence of gp3 when certain mutations are introduced just upstream or within a motif, the coiled-coil motif, required for subunit-subunit interactions during synapsis of DNA substrates (39). The coiled-coil motifs are also thought to play a role in inhibiting recombination between attL and attR in the absence of the RDF; the ϕC31 IntE449K mutation or its RDF, gp3, relieves this inhibition (35, 39–41). Three independent structural predictions indicate the presence of a coiled-coil domain in the ϕJoe Int C-terminal domain (A395-T453, Fig. S7). The high basal excision activity of ϕJoe integrase could be due to incomplete inhibition of synapsis by the coiled-coil motif when integrase is bound to attL and attR, reminiscent of the hyperactive ϕC31 mutant IntE449K (39). Natural bidirectional large serine recombinases include the transposases TnpX (42) and TndX (43) from clostridial integrated conjugative elements (ICEs); ϕJoe integrase could be an evolutionary intermediate between these bidirectional recombinases and the highly directional recombinases, such as ϕC31 and Bxb1 integrases. Our data show that, under the in vitro conditions used, gp52 was highly effective at inhibiting integration by ϕJoe integrase but only weakly activated excision. It remains to be seen whether this system, with its unusual properties, is sufficiently robust to regulate phage genome integration and excision according to the developmental choices of ϕJoe.
The properties of the ϕJoe integrase and gp52 are compatible with some of the existing applications for serine integrases, but they could also present opportunities for new applications. ϕJoe integrase is highly efficient in integration assays in vivo and in vitro, and in in vivo excision when the RDF is present. In attB × attP integration assays, the yield of products by ϕJoe integrase was comparable to that of well-established integrases, such as that of ϕC31 or Bxb1. Furthermore, ϕJoe integrase is active in buffers compatible with other characterized integrases, indicating that it could be used in DNA assembly procedures in combination with other integrases. Although yet to be tested, assemblies generated with ϕJoe integrase could later be used as substrates for modification by ϕJoe integrase in a single step. The innate excision activity of ϕJoe integrase could excise a fragment flanked by attLsv or attRsv sites and, in the same reaction, replace it via an integration reaction. ϕJoe integrase could therefore provide a more streamlined tool than the existing requirement for two steps by the more directional integrases, such as those from ϕC31 and Bxb1 (15). Furthermore, given that ϕJoe Int can mediate basal levels of excision in the absence of RDF, integrating plasmids based on ϕJoe int-attP may display a degree of instability. Selection for the plasmid marker would ensure plasmid maintenance when desired, but if the plasmid is easily lost without selection, this trait could be desirable if there is a need to cure the strain of the plasmid or during studies on synthetic lethality.
Conclusions.
On the basis of sequence and genome organization, phage ϕJoe is a member of a large cluster of R4-like Streptomyces phages. Its closest relatives at the nucleotide level are Streptomyces phages Amela and Verse, with very high levels of nucleotide identity in the regions containing essential early and structural genes. However, ϕJoe integrase is more closely related to the integrases from five other R4-like cluster phages: Lannister, Danzina, Zemlya, Lika, and Sujidade. At this time, the majority of Streptomyces phages belong to the R4-like cluster phages, but there is a continuum of relatedness throughout the cluster; for example, R4 is a more distant relative to ϕJoe than any of the other phages mentioned above.
We identified the RDF for ϕJoe integrase on the basis of its gene location, small size, and distant similarity to another known RDF, Rv1584c. Although this identification was relatively straightforward, it is not clear yet how general such an approach might be. The activities of ϕJoe integrase and RDF contribute to the growing number of complete serine integrase site-specific recombination systems that are available for use in synthetic biology applications. The ϕJoe int-attP plasmid, pCMF92, also adds to the number of useful integrating vectors for use in Streptomyces species. However, and unusually for a phage integrase, ϕJoe Int displays a significant level of excisive recombination in the absence of its RDF while still being efficient at mediating integration. This bidirectional property could be applied in new ways in future applications of serine integrases.
MATERIALS AND METHODS
Growth media.
Escherichia coli strains were generally grown in LB, except where otherwise noted. Antibiotics were added for selection where appropriate (apramycin, 50 μg/ml; chloramphenicol, 50 μg/ml; kanamycin, 50 μg/ml; ampicillin, 100 μg/ml). Preparation of competent cells and transformation of E. coli were performed as described by Sambrook et al. (44). Streptomyces strains were grown on mannitol soya agar (45) supplemented with 10 mM MgCl2, for plating conjugation mixtures, and antibiotics, where required (apramycin, 50 μg/ml; nalidixic acid, 25 μg/ml).
Phage isolation.
The procedures for isolation, plating, and titration of phage with Streptomyces as the isolation host are described in detail by Kieser et al. (45). Raw soil samples were enriched for environmental phage using S. coelicolor M145 as a propagation host (46). Briefly, 3 g of soil was added to 9 ml of Difco nutrient (DN) broth (BD Diagnostics, Oxford, UK) supplemented with 10 mM CaCl2, 10 mM MgSO4, and 0.5% glucose. Streptomyces spores were added to a concentration of 106 CFU/ml and incubated at 30°C with agitation for 16 h. Soil and bacteria were removed by centrifugation and filtration through a 0.45-μm-pore filter. A dilution series of the filtrate in SM buffer (100 mM NaCl, 8.5 mM MgSO4, 50 mM Tris-HCl [pH 7.5], 0.01% gelatin) was plated with S. coelicolor spores to isolate single plaques. Phage were recovered from single well-isolated plaques by single-plaque soak-outs in DN broth and replated with the host strain for three rounds of plaque purification. A high-titer phage preparation was generated from plates inoculated with sufficient PFU to generate almost-confluent lysis (45). The phage suspensions were filtered, pelleted by ultracentrifugation, and resuspended in 0.5 ml of SM buffer (47). The concentrated phages were further purified by cesium chloride isopycnic density gradient centrifugation (48).
Next-generation sequencing.
Phage DNA was extracted by phenol-chloroform purification (44), and the presence of pure phage DNA was confirmed by restriction digestion. Phage DNA was sequenced and assembled in collaboration with Darren Smith at NU-OMICS (Northumbria University, Newcastle, UK). DNA was prepared for next-generation sequencing on the Illumina MiSeq platform using the Nextera XT library preparation kit (Illumina, Saffron Walden, UK). Samples were loaded and run using a 2 × 250 cycle version 2 kit. DNA samples were diluted to 0.2 ng/μl, prior to normalization and pooling. Paired-end sequencing reads were provided as FASTQ files (NU-OMICS) and subjected to downstream analysis. ORF prediction and annotations were assigned using DNA Master (Lawrence lab, Pittsburgh, PA), Glimmer (49), and GeneMark (50).
Electron microscopy.
Purified phage were negatively stained with uranyl acetate (51) and imaged in an FEI Tecnai 12 G2 transmission electron microscope fitted with a charge-coupled-device (CCD) camera.
Mass spectrometry.
Whole-phage samples were run into a 7-cm NuPAGE Novex 10% Bis-Tris gel (Life Technologies) at 200 V for 6 min. The total protein band was excised and digested in-gel with 0.5 μg of trypsin, overnight at 37°C. Peptides were extracted, concentrated, and loaded onto a nanoACQUITY ultraperformance liquid chromatography (UPLC) system (Waters) equipped with a nanoACQUITY symmetry C18, 5-μm trap (180 μm by 20 mm; Waters) and a nanoACQUITY HSS T3 1.8-μm C18 capillary column (75 μm by 250 mm; Waters). The nanoLC system was interfaced with a maXis HD LC-MS/MS system (Bruker Daltonics) with CaptiveSpray ionization source (Bruker Daltonics). Positive electrospray ionization-MS (ESI-MS) and MS/MS spectra were acquired using AutoMSMS mode. Instrument control, data acquisition, and processing were performed using the Compass 1.7 software (micrOTOF control, HyStar, and DataAnalysis; Bruker Daltonics). The collision energy and isolation width settings were automatically calculated using the AutoMSMS fragmentation table, with an absolute threshold of 200 counts, preferred charge states of 2 to 4, and singly charged ions excluded. A single MS/MS spectrum was acquired for each precursor, and former target ions were excluded for 0.8 min unless the precursor intensity increased 4-fold. Protein identification was performed by searching tandem mass spectra against the NCBI nr database using the Mascot search program. Matches were filtered to accept only peptides with expect scores of 0.05 or better.
Plasmid construction.
Plasmids used in this study are listed in Table 2 and oligonucleotides in Table 3. General molecular biology techniques, including plasmid DNA preparation, genomic DNA preparation, restriction endonuclease digestion, and agarose gel electrophoresis, were performed as described by Sambrook et al. (44). In-fusion cloning technology (Clontech) was generally used for construction of plasmids. PCR-amplified DNA was generated using primers with infusion tags for insertion into plasmid vectors, which had been cut with restriction endonucleases. The ϕJoe integrating plasmid, pCMF92, was created by infusion cloning of the ϕJoe int gene and attP region, obtained by PCR with Joe Int-attP F/R primers and ϕJoe genomic DNA as the template, into the 3.1-kbp EcoRI-SphI fragment from pSET152. Plasmid pCMF91 was generated by inserting the amplified attP site prepared using ϕJoe genomic DNA as the template and primers Joe attP F/R into EcoRI-linearized pSP72. The integration sites in S. coelicolor were named attLsc and attRsc and were amplified from S. coelicolor genomic DNA (gDNA) using Joe attB1 F/R and Joe attB2 F/R. The attB site from S. venezuelae (attBsv) was amplified using S. venezuelae gDNA with Joe attB Sv F/Joe attB R primers. All three attachment sites were inserted into EcoRI-linearized pGEM7 to produce pCMF90, pCMF94, and pCMF95, respectively. The reconstituted S. coelicolor attB sequence (attBsc) was prepared from two complementary oligonucleotides, Joe attB Recon F and Joe attB Recon R (Ultramer primers; IDT) that were annealed and inserted into EcoRI-linearized pGEM7 to produce pCMF97. pCMF98 contains the ϕJoe attLsv and attRsv sites in head-to-tail orientation and was isolated by transformation of an in vitro recombination reaction between pCMF91 (containing ϕJoe attP) and pCMF95 (containing attBsv) into E. coli. The attLsv and attRsv sites in pCMF98 were confirmed by Sanger sequencing (GATC Biotech Ltd., London, UK). The recombination reporter plasmid pCMF116 was constructed by PCR amplification of lacZα using E. coli MG1655 gDNA (52) as the template and Joe BzP forward and reverse primers containing the ϕJoe attBsv and ϕJoe attP sequences, respectively; this resulted in the attBsv and attP sites flanking the lacZα gene in head-to-tail orientation. The amplified DNA was inserted into XmnI-linearized pACYC184. pCMF103 was constructed in the same way as pCMF116 except that Joe LzR F/R primers containing the ϕJoe attLsv and attRsv sites were used.
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Resistancea | Reference or source |
|---|---|---|---|
| pSET152 | ϕC31 int + attP integrating vector | Apra | 64 |
| pEHISTEV | Expression vector, T7 promoter, C-terminal His6, TEV cleavage site | Kan | 65 |
| pETFPP_2 | Expression vector; His6-MBP-3c cleavage site | Kan | 66 |
| pBAD-HisA | Expression vector, araBAD inducible promoter | Amp | Invitrogen |
| pCMF87 | pEHISTEV + ϕJoe int (gp53) | Kan | This study |
| pCMF90 | pGEM7 + S. coelicolor attRsc (274 bp) | Amp | This study |
| pCMF91 | pSP72 + ϕJoe attP (354 bp) | Amp | This study |
| pCMF92 | ϕJoe int + attP integrating vector; pSET152 | Apra | This study |
| pCMF94 | pGEM7 + S. coelicolor attLsc (419 bp) | Amp | This study |
| pCMF95 | pGEM7 + S. venezuelae attBsv (462 bp) | Amp | This study |
| pCMF96 | pETFPP_2 + ϕJoe MBP-RDF (gp52) | Kan | This study |
| pCMF97 | pGEM7 + S. coelicolor reconstituted attBsc (152 bp) | Amp | This study |
| pCMF98 | ϕJoe attLsv-attRsv; pCMF91 integrated into pCMF95 | Amp | This study |
| pCMF100 | pEHISTEV + ϕJoe RDF | Kan | This study |
| pCMF103 | pACYC184 + ϕJoe attLsv-lacZα-attRsv | Cm | This study |
| pCMF107 | pBAD + ϕJoe int | Amp | This study |
| pCMF108 | pBAD + ϕJoe RDF + int coexpression | Amp | This study |
| pCMF116 | pACYC184 + ϕJoe attBsv-lacZα-attP | Cm | This study |
| pCMF117 | pEHISTEV + ϕJoe RDF + int coexpression | Kan | This study |
| pGEM7 | General cloning vector | Amp | Promega |
| pSP72 | General cloning vector; accession no. X65332 | Amp | Promega |
| pACYC184 | General cloning vector; accession no. X06403 | Cm | 67 |
| pUZ8002 | Conjugation helper plasmid; RK2 derivative with defective oriT | Kan | 68 |
Apra, apramycin; Kan, kanamycin; Amp, ampicillin; Cm, chloramphenicol.
TABLE 3.
Primers used in this study
| Primer | Sequence (5′ to 3′) |
|---|---|
| Joe Int-attP F | CCGTCGACCTGCAGGCATGCCGTTCCCGCAGGTCAGAGC |
| Joe Int-attP R | ACATGATTACGAATTCTGTGGATCAGAACGTCTCGG |
| Joe H6-Int F | TTTCAGGGCGCCATGATGAGTAACCGACTACATG |
| Joe H6-Int R | CCGATATCAGCCATGTCAGAACGTCTCGGCGAAG |
| Joe attP F | TACCGAGCTCGAATTAAGACCGTCTCAGCCAGG |
| Joe attP R | TATCATCGATGAATTTCAGTGAAGACGGACAGG |
| Joe attB1 F | CCGGGGTACCGAATTTGTGACGTCAGCCACAGC |
| Joe attB1 R | TAGACTCGAGGAATTGACAAGGAGTGGCTCTGG |
| Joe attB2 F | CCGGGGTACCGAATTGACTGCGTGCCGTCAGCC |
| Joe attB2 R | TAGACTCGAGGAATTCGTCGTGTCGTCTGTCAG |
| Joe attB Sv F | CCGGGGTACCGAATTACCAGGTGGTGGATGAGC |
| Joe attB Recon F | TAGACTCGAGGAATTACCTTGATCTCGGTGTCCATCGCCGGGCAGACGCCGCAGTCGAAGCACGG |
| Joe attB Recon R | CCGGGGTACCGAATTGACAAGGAGTGGCTCTGG |
| Joe MBP-gp52 F | TCCAGGGACCAGCAATGAACGGACAGATCCTGG |
| Joe MBP-gp52 R | TGAGGAGAAGGCGCGCTACACCCAGCGCACCGA |
| CleF | CGCGCCTTCTCCTCACATATGGCTAGC |
| CleR | TTGCTGGTCCCTGGAACAGAACTTCC |
| Joe H6-gp52 F | TTTCAGGGCGCCATGAACGGACAGATCCTGGAG |
| Joe H6-gp52 R | CCGATATCAGCCATGCTACACCCAGCGCACCGA |
| Joe pBAD Int F | GAGGAATTAACCATGAGTAACCGACTACATG |
| Joe pBAD Int R | TGAGAACCCCCCATGTCAGAACGTCTCGGCGAAG |
| Joe pBAD gp52 F | GAGGAATTAACCATGAACGGACAGATCCTGGAG |
| Joe pBAD Int Co-Ex F | AGTGGTAGGTTCCTCGCCATG |
| Joe pBAD gp52 R | GAGGAACCTACCACTCTACACCCAGCGCACCGA |
| Joe LzR F | GGGTGTCAGTGAAGTAGTTGTGGCCATGTGTCCATCTGGGGGCAGACGCCGCAGTCGAAGCACGGCGATTTCGGCCTATTGGT |
| Joe LzR R | CCTGCCACATGAAGCGGATGTGACCCCGTCTCCATCTGCCCGCAGATGGACACCCACATCCAGATAATACGCAAACCGCCTCT |
| Joe BzP F | GGGTGTCAGTGAAGTATCTGGATGTGGGTGTCCATCTGCGGGCAGACGCCGCAGTCGAAGCACGGCGATTTCGGCCTATTGGT |
| Joe BzP R | CCTGCCACATGAAGCGGATGTGACCCCGTCTCCATCTGCCCCCAGATGGACACATGGCCACAACTAATACGCAAACCGCCTCT |
| SPBc H6-sprA F | CCGATATCAGCCATGGAGTTAAAAAACATTGTT |
| SPBc H6-sprA R | TTTCAGGGCGCCATGCTTACTACTTTTCTTAGTGG |
| SPBc MBP-sprB F | TCCAGGGACCAGCAATGGAACCTTACCAACGT |
| SPBc MBP-sprB R | TGAGGAGAAGGCGCGAAGCTTACTCTGCCTTCC |
| SPBc LZR F | GGGTGTCAGTGAAGTAGTGCAGCATGTCATTAATATCAGTACAGATAAAGCTGTATATTAAGATACTTACTACATATCTACGATTTCGGCCTATTGGT |
| SPBc LZR R | CCTGCCACATGAAGCTGGCACCCATTGTGTTCACAGGAGATACAGCTTTATCTGTTTTTTAAGATACTTACTACTTTTCTAATACGCAAACCGCCTCT |
The integrase expression plasmid for protein purification, pCMF87, was constructed by insertion of a PCR fragment containing the ϕJoe int gene, amplified from ϕJoe gDNA using primers Joe H6-Int F/R, into NcoI-linearized pEHISTEV expression vector. ϕJoe g52, encoding the RDF, was PCR amplified from ϕJoe gDNA using primers Joe MBP-g52 F/R and inserted into pETFPP_2 MBP-tag expression vector linearized by PCR with CleF/R to create pCMF96. For in vivo recombination assays, the integrase expression plasmid pCMF107 was constructed by insertion of a PCR fragment containing the ϕJoe int gene and amplified from ϕJoe gDNA using primers Joe pBAD Int F/R into NcoI-linearized pBAD-HisA expression vector. A ϕJoe gp52 and integrase coexpression plasmid, pCMF108, was created by amplification of each gene using Joe pBAD gp52 F/R and Joe pBAD Int Co-Ex F/Joe pBAD Int R primers, respectively, and insertion of both PCR products simultaneously into pBAD-HisA. The coexpression insert from pCMF108 was subsequently PCR amplified using Joe H6-gp52 F/Joe H6-Int R primers and transferred to NcoI-linearized pEHISTEV to produce an alternative expression vector, pCMF117.
Conjugation and integration of plasmids in Streptomyces.
Transfer of plasmids into Streptomyces strains was performed according to the procedures described by Kieser et al. (45). Conjugation donors were produced by introduction of plasmids into the nonmethylating E. coli strain ET12567, containing an RP4 derivative plasmid (pUZ8002), by transformation. Recipient Streptomyces spores were used at a concentration of 108/ml, mixed with the E. coli donors, plated onto mannitol soya agar supplemented with 10 mM MgCl2 with no antibiotic selection, and incubated at 30°C overnight. Plates containing the donor cells were overlaid with 1 ml of water containing 0.5 mg of nalidixic acid (for E. coli counterselection) and antibiotic for selection of exconjugants (apramycin) before further incubation of all plates at 30°C for 3 days. Integration efficiency was calculated as the number of apramycin-resistant colonies/108 CFU (8).
Protein purification.
E. coli BL21(DE3) containing the relevant expression plasmid was grown (37°C with agitation) in 500 ml of 2YT medium (1.6%, [wt/vol] tryptone, 1.0%, [wt/vol] yeast extract, 0.5 [wt/vol] NaCl) to mid-exponential-growth phase. The cultures were rapidly chilled on ice for 15 min, IPTG was added (final concentration, 0.15 mM), and the cultures were further incubated (17°C for 16 h, with agitation). Cells were harvested by centrifugation, resuspended in 20 ml of lysis buffer (1 M NaCl, 75 mM Tris [pH 7.75], 0.2 mg/ml lysozyme, 500 U of BaseMuncher endonuclease; Expedeon Ltd.), and incubated on ice (30 min). The cells were lysed by sonication, and debris was removed by centrifugation (18,000 × g, 5 min, 4°C). The supernatant was applied to a 5-ml HisTrap FF crude column that had been preequilibrated with binding buffer (20 mM sodium phosphate, 0.5 M NaCl, 20 mM imidazole [pH 7.4]) on an Äkta pure 25 chromatography system (GE Healthcare). Bound His-tagged protein was eluted with a step gradient of binding buffer containing 125 mM and 250 mM imidazole. Imidazole was removed from the eluted fractions by pooling the fractions containing the desired protein and applying the pooled solutions to a HiPrep 26/10 desalting column (GE Healthcare) equilibrated with imidazole-free binding buffer. Finally, the protein extracts were subjected to size exclusion chromatography on a HiLoad 16/60 Superdex column. Purified protein fractions were concentrated in a Vivaspin sample concentrator (GE Healthcare) and quantified by absorbance at 280 nm on a NanoDrop spectrophotometer (Thermo Scientific). Protein analysis was performed by denaturing acrylamide gel electrophoresis using premade gels (4 to 12% gradient acrylamide; Expedeon Ltd.); gels were stained with InstantBlue (Expedeon, Ltd.). For storage, an equal volume of 100% glycerol was added to protein samples before freezing at −80°C.
In vitro assays.
Recombination reactions (final volume, 20 μl) were carried out in ϕC31 RxE buffer (10 mM Tris [pH 7.5], 100 mM NaCl, 5 mM dithiothreitol [DTT], 5 mM spermidine, 4.5% glycerol, 0.5 mg/ml bovine serum albumin [BSA]) (53), Bxb1 RxE buffer (20 mM Tris [pH 7.5], 25 mM NaCl, 1 mM DTT, 10 mM spermidine, 10 mM EDTA) (23), or TG1 RxE (as Bxb1 RxE plus 0.1 mg/ml BSA) (54). Integrase and RDF proteins were added at the concentrations indicated for each experiment. Plasmids containing the recombination substrates were used at 100 ng per reaction. Reactions were either incubated at 30°C for 2 h (to reach steady state) or for specified times. Reactions were stopped by heat (10 min, 75°C), the buffer was adjusted to be compatible with restriction enzymes, and the plasmids were digested with XhoI (NEB). The linearized reaction mixtures were run on a 0.8% agarose gel, and the relative band intensities were measured to assess activity. Recombination efficiencies were calculated as the intensity of product band(s)/sum intensity of all bands.
Bioinformatics.
The ϕJoe genome was visualized using DNAPlotter (55). The attB DNA alignment and logo consensus sequence were created with Jalview (56). Protein sequence alignments for visual presentation were produced using the Clustal W (57) program within the BioEdit suite (58). Protein alignments for phylogenetic analysis were produced using Clustal Omega (59), and maximum likelihood trees were created in MEGA6 (60). The BLOSUM62 similarity matrix was used for protein alignment and annotation (61). Structural alignment of the small RDF proteins was carried out with Promals3D (62). Band densities for in vitro assays were measured using the Fiji GelAnalyzer module (63).
Accession number(s).
The annotated genome sequence of ϕJoe was submitted to GenBank (accession number: KX815338). Accession numbers for all sequences used here are also provided in Table S2.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Darren Smith (NU-OMICS, Northumbria University) for phage genome sequencing and assembly, and to the York Biosciences Technology Facility for proteomics and electron microscopy.
This research was performed with funding from the Biotechnology and Biological Sciences Research Council (project grant BB/K003356/1) and the Microbiology Society (formerly Society for General Microbiology) with a Harry Smith Vacation Scholarship for J.A.H.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02767-16.
REFERENCES
- 1.Fogg PCM, Colloms S, Rosser S, Stark M, Smith MCM. 2014. New applications for phage integrases. J Mol Biol 426:2703–2716. doi: 10.1016/j.jmb.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Groth AC, Calos MP. 2004. Phage integrases: biology and applications. J Mol Biol 335:667–678. doi: 10.1016/j.jmb.2003.09.082. [DOI] [PubMed] [Google Scholar]
- 3.Smith M. 2015. Phage-encoded serine integrases and other large serine recombinases. Microbiol Spectr 3:MDNA3-0059-2014. doi: 10.1128/microbiolspec.MDNA3-0059-2014. [DOI] [PubMed] [Google Scholar]
- 4.Smith MCA, Till R, Smith MCM. 2004. Switching the polarity of a bacteriophage integration system. Mol Microbiol 51:1719–1728. doi: 10.1111/j.1365-2958.2003.03942.x. [DOI] [PubMed] [Google Scholar]
- 5.Olorunniji FJ, Buck DE, Colloms SD, McEwan AR, Smith MCM, Stark WM, Rosser SJ. 2012. Gated rotation mechanism of site-specific recombination by ϕC31 integrase. Proc Natl Acad Sci U S A 109:19661–19666. doi: 10.1073/pnas.1210964109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorpe HM, Smith MC. 1998. In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc Natl Acad Sci U S A 95:5505–5510. doi: 10.1073/pnas.95.10.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuhstoss S, Richardson MA, Rao RN. 1991. Plasmid cloning vectors that integrate site-specifically in Streptomyces spp. Gene 97:143–146. doi: 10.1016/0378-1119(91)90022-4. [DOI] [PubMed] [Google Scholar]
- 8.Fayed B, Ashford DA, Hashem AM, Amin MA, El Gazayerly ON, Gregory MA, Smith MCM. 2015. Multiplexed integrating plasmids for engineering of the erythromycin gene cluster for expression in Streptomyces spp. and combinatorial biosynthesis. Appl Environ Microbiol 81:8402–8413. doi: 10.1128/AEM.02403-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregory MA, Till R, Smith MCM. 2003. Integration site for Streptomyces phage ϕBT1 and development of site-specific integrating vectors. J Bacteriol 185:5320–5323. doi: 10.1128/JB.185.17.5320-5323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong Y, Hondalus MK. 2008. Site-specific integration of Streptomyces ϕC31 integrase-based vectors in the chromosome of Rhodococcus equi. FEMS Microbiol Lett 287:63–68. doi: 10.1111/j.1574-6968.2008.01298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chavez CL, Keravala A, Chu JN, Farruggio AP, Cuéllar VE, Voorberg J, Calos MP. 2012. Long-term expression of human coagulation factor VIII in a tolerant mouse model using the φC31 integrase system. Hum Gene Ther 23:390–398. doi: 10.1089/hum.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meredith JM, Basu S, Nimmo DD, Larget-Thiery I, Warr EL, Underhill A, McArthur CC, Carter V, Hurd H, Bourgouin C, Eggleston P. 2011. Site-specific integration and expression of an anti-malarial gene in transgenic Anopheles gambiae significantly reduces Plasmodium infections. PLoS One 6:e14587. doi: 10.1371/journal.pone.0014587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groth AC, Olivares EC, Thyagarajan B, Calos MP. 2000. A phage integrase directs efficient site-specific integration in human cells. Proc Natl Acad Sci U S A 97:5995–6000. doi: 10.1073/pnas.090527097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Zhao G, Ding X. 2011. Tandem assembly of the epothilone biosynthetic gene cluster by in vitro site-specific recombination. Sci Rep 1:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colloms SD, Merrick CA, Olorunniji FJ, Stark WM, Smith MCM, Osbourn A, Keasling JD, Rosser SJ. 2014. Rapid metabolic pathway assembly and modification using serine integrase site-specific recombination. Nucleic Acids Res 42:e23. doi: 10.1093/nar/gkt1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dafhnis-Calas F, Xu Z, Haines S, Malla SK, Smith MCM, Brown WRA. 2005. Iterative in vivo assembly of large and complex transgenes by combining the activities of ϕC31 integrase and Cre recombinase. Nucleic Acids Res 33:e189. doi: 10.1093/nar/gni192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Z, Lee NCO, Dafhnis-Calas F, Malla S, Smith MCM, Brown WRA. 2008. Site-specific recombination in Schizosaccharomyces pombe and systematic assembly of a 400kb transgene array in mammalian cells using the integrase of Streptomyces phage ϕBT1. Nucleic Acids Res 36:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fayed B, Younger E, Taylor G, Smith MCM. 2014. A novel Streptomyces spp. integration vector derived from the S. venezuelae phage, SV1. BMC Biotechnol 14:51. doi: 10.1186/1472-6750-14-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siuti P, Yazbek J, Lu TK. 2013. Synthetic circuits integrating logic and memory in living cells. Nat Biotechnol 31:448–452. doi: 10.1038/nbt.2510. [DOI] [PubMed] [Google Scholar]
- 20.Bonnet J, Subsoontorn P, Endy D. 2012. Rewritable digital data storage in live cells via engineered control of recombination directionality. Proc Natl Acad Sci U S A 109:8884–8889. doi: 10.1073/pnas.1202344109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonnet J, Yin P, Ortiz ME, Subsoontorn P, Endy D. 2013. Amplifying genetic logic gates. Science 340:599–603. doi: 10.1126/science.1232758. [DOI] [PubMed] [Google Scholar]
- 22.Khaleel T, Younger E, Mcewan AR, Varghese AS, Smith MCM. 2011. A phage protein that binds ϕC31 integrase to switch its directionality. Mol Microbiol 80:1450–1463. doi: 10.1111/j.1365-2958.2011.07696.x. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh P, Wasil LR, Hatfull GF. 2006. Control of phage Bxb1 excision by a novel recombination directionality factor. PLoS Biol 4:e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Breüner A, Brøndsted L, Hammer K. 1999. Novel organization of genes involved in prophage excision identified in the temperate lactococcal bacteriophage TP901-1. J Bacteriol 181:7291–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang L, Zhu B, Dai R, Zhao G, Ding X. 2013. Control of directionality in Streptomyces phage ϕBT1 integrase-mediated site-specific recombination. PLoS One 8:e80434. doi: 10.1371/journal.pone.0080434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bibb LA, Hancox MI, Hatfull GF. 2005. Integration and excision by the large serine recombinase ϕRv1 integrase. Mol Microbiol 55:1896–1910. doi: 10.1111/j.1365-2958.2005.04517.x. [DOI] [PubMed] [Google Scholar]
- 27.Abe K, Kawano Y, Iwamoto K, Arai K, Maruyama Y, Eichenberger P, Sato T. 2014. Developmentally-regulated excision of the SPβ prophage reconstitutes a gene required for spore envelope maturation in Bacillus subtilis. PLoS Genet 10:e1004636. doi: 10.1371/journal.pgen.1004636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramaswamy KS, Carrasco CD, Fatma T, Golden JW. 1997. Cell-type specificity of the Anabaena fdxN-element rearrangement requires xisH and xisI. Mol Microbiol 23:1241–1249. doi: 10.1046/j.1365-2958.1997.3081671.x. [DOI] [PubMed] [Google Scholar]
- 29.Xu Z, Brown WRA. 2016. Comparison and optimization of ten phage encoded serine integrases for genome engineering in Saccharomyces cerevisiae. BMC Biotechnol 16:13. doi: 10.1186/s12896-016-0241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu Z, Thomas L, Davies B, Chalmers R, Smith M, Brown W. 2013. Accuracy and efficiency define Bxb1 integrase as the best of fifteen candidate serine recombinases for the integration of DNA into the human genome. BMC Biotechnol 13:87. doi: 10.1186/1472-6750-13-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang L, Nielsen AAK, Fernandez-Rodriguez J, McClune CJ, Laub MT, Lu TK, Voigt CA. 2014. Permanent genetic memory with >1-byte capacity. Nat Methods 11:1261–1266. doi: 10.1038/nmeth.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monson R, Salmond GP. 2012. Genome sequence of a new Streptomyces coelicolor generalized transducing bacteriophage, ϕCAM. J Virol 86:13860. doi: 10.1128/JVI.02681-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald JE, Smith DL, Fogg PCM, McCarthy AJ, Allison HE. 2010. High-throughput method for rapid induction of prophages from lysogens and its application in the study of Shiga toxin-encoding Escherichia coli strains. Appl Environ Microbiol 76:2360–2365. doi: 10.1128/AEM.02923-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. 1999. Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc Natl Acad Sci U S A 96:2192–2197. doi: 10.1073/pnas.96.5.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rutherford K, Yuan P, Perry K, Sharp R, Van Duyne GD. 2013. Attachment site recognition and regulation of directionality by the serine integrases. Nucleic Acids Res 41:8341–8356. doi: 10.1093/nar/gkt580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta M, Till R, Smith MCM. 2007. Sequences in attB that affect the ability of phiC31 integrase to synapse and to activate DNA cleavage. Nucleic Acids Res 35:3407–3419. doi: 10.1093/nar/gkm206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savinov A, Pan J, Ghosh P, Hatfull GF. 2012. The Bxb1 gp47 recombination directionality factor is required not only for prophage excision, but also for phage DNA replication. Gene 495:42–48. doi: 10.1016/j.gene.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Ou X, Zhao G, Ding X. 2008. Highly efficient in vitro site-specific recombination system based on Streptomyces phage ϕBT1 integrase. J Bacteriol 190:6392–6397. doi: 10.1128/JB.00777-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rowley PA, Smith MCA, Younger E, Smith MCM. 2008. A motif in the C-terminal domain of ϕC31 integrase controls the directionality of recombination. Nucleic Acids Res 36:3879–3891. doi: 10.1093/nar/gkn269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rutherford K, Van Duyne GD. 2014. The ins and outs of serine integrase site-specific recombination. Curr Opin Struct Biol 24:125–131. doi: 10.1016/j.sbi.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hwang WC, Golden JW, Pascual J, Xu D, Cheltsov A, Godzik A. 1 September 2014. Site-specific recombination of nitrogen-fixation genes in cyanobacteria by XisF-XisH-XisI complex: structures and models. Proteins doi: 10.1002/prot.24679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lyras D, Adams V, Lucet I, Rood JI. 2004. The large resolvase TnpX is the only transposon-encoded protein required for transposition of the Tn4451/3 family of integrative mobilizable elements. Mol Microbiol 51:1787–1800. doi: 10.1111/j.1365-2958.2003.03950.x. [DOI] [PubMed] [Google Scholar]
- 43.Wang H, Mullany P. 2000. The large resolvase TndX is required and sufficient for integration and excision of derivatives of the novel conjugative transposon Tn5397. J Bacteriol 182:6577–6583. doi: 10.1128/JB.182.23.6577-6583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sambrook J, Fritsch EF, Maniatis T. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Press, Cold Spring Harbor, NY. [Google Scholar]
- 45.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. John Innes Centre Ltd., Norwich, United Kingdom. [Google Scholar]
- 46.Bentley SD, Chater KF, Cerdeño-Tárraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang C-H, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream M, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 47.Fogg PCM, Hynes AP, Digby E, Lang AS, Beatty JT. 2011. Characterization of a newly discovered Mu-like bacteriophage, RcapMu, in Rhodobacter capsulatus strain SB1003. Virology 421:211–221. doi: 10.1016/j.virol.2011.09.028. [DOI] [PubMed] [Google Scholar]
- 48.Clokie MRJ, Kropinski AM. 2009. Bacteriophages: methods and protocols: volume 1: isolation, characterization, and interactions. Springer, New York, NY. [Google Scholar]
- 49.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Besemer J, Borodovsky M. 2005. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res 33:W451–W454. doi: 10.1093/nar/gki487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Booth DS, Avila-Sakar A, Cheng Y. 2011. Visualizing proteins and macromolecular complexes by negative stain EM: from grid preparation to image acquisition. J Vis Exp 2011:e3227. doi: 10.3791/3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 53.McEwan AR, Raab A, Kelly SM, Feldmann J, Smith MCM. 2011. Zinc is essential for high-affinity DNA binding and recombinase activity of ϕc31 integrase. Nucleic Acids Res 39:6137–6147. doi: 10.1093/nar/gkr220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morita K, Morimura K, Fusada N, Komatsu M, Ikeda H, Hirano N, Takahashi H. 2012. Site-specific genome integration in alphaproteobacteria mediated by TG1 integrase. Appl Microbiol Biotechnol 93:295–304. doi: 10.1007/s00253-011-3545-3. [DOI] [PubMed] [Google Scholar]
- 55.Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J. 2009. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics 25:119–120. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, Valentin F, Wallace I, Wilm A, Lopez R, Thompson J, Gibson T, Higgins D. 2007. ClustalW and ClustalX version 2. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 58.Hall T. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser (Oxf) 41:95–98. [Google Scholar]
- 59.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pearson WR. 2013. Selecting the right similarity-scoring matrix. Curr Protoc Bioinformatics 43:3.5.1–3.5.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pei J, Grishin NV. 2014. PROMALS3D: multiple protein sequence alignment enhanced with evolutionary and three-dimensional structural information. Methods Mol Biol 1079:263–271. doi: 10.1007/978-1-62703-646-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilkinson CJ, Hughes-Thomaz ZA, Martin CJ, Bohm I, Mironenko T, Deacon M, Wheatcraft M, Wirtz G, Stanton J, Leadlay PF. 2002. Increasing the efficiency of heterologous promoter in actinomycetes. J Mol Microbiol Biotechnol 4:417–426. [PubMed] [Google Scholar]
- 65.Liu H, Naismith JH. 2009. A simple and efficient expression and purification system using two newly constructed vectors. Protein Expr Purif 63:102–111. doi: 10.1016/j.pep.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fogg MJ, Wilkinson AJ. 2008. Higher-throughput approaches to crystallization and crystal structure determination. Biochem Soc Trans 36:771–775. doi: 10.1042/BST0360771. [DOI] [PubMed] [Google Scholar]
- 67.Rose RE. 1988. The nucleotide sequence of pACYC184. Nucleic Acids Res 16:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paget MSB, Chamberlin L, Atrih A, Foster SJ, Buttner MJ. 1999. Evidence that the extracytoplasmic function sigma factor σE is required for normal cell wall structure in Streptomyces coelicolor A3(2). J Bacteriol 181:204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.