

Abstract
Background
The fungal species complex Colletotrichum gloeosporioides sensu lato contains over 20 plant-interacting species. These species exhibit different life styles (e.g., endophytes, foliar and fruit pathogens) and show considerable variation in host and tissue adaptation strategies. Accurate species delimitation in C. gloeosporioides s.l. is very challenging due to nascent lineage boundaries and phenotypic plasticity, which strongly impedes studies of the complex’s host-interaction biology. In this study, we first sequenced and compared nine mitogenomes belonging to four C. gloeosporioides s.l. species lineages (C. gloeosporioides, C. fructicola, C. aenigma, and C. siamense s.l.), and evaluated the usefulness of mitogenome sequence in complementing prevailing nuclear markers for species delimitation.
Results
The C. gloeosporioides s.l. mitogenomes ranged between 52,671 and 58,666 bp in size, and each contained an identical set of genes transcribed in the same direction. Compared with previously reported Colletotrichum mitogenomes, these mitogenomes were uniquely featured by: (1) significantly larger genome size due to richer intron content and longer intergenic region; (2) striking GC content elevation at the intergenic region; and (3) considerable intron content variation among different species lineages. Compared with nuclear DNA markers commonly used in phylogeny, the mitogenome nucleotide diversity was extremely low, yet the mitogenome alignment contained the highest number of parsimony informative sites, which allowed the generation of a high-resolution phylogeny recognizing all taxonomic lineages, including ones belonging to the very nascent C. siamense s.l. complex. The tree topology was highly congruent with the phylogeny based on nuclear marker concatenation except for lineages within C. siamense s.l. Further comparative phylogenetic analysis indicated that lineage-specific rapid divergence of GS and SOD2 markers confounded concatenation-based species relationship inference.
Conclusions
This study sheds light on the evolution of C. gloeosporioides s.l. mitogenomes and demonstrates that mitogenome sequence can complement prevailing nuclear markers in improving species delimitation accuracy. The mitogenome sequences reported will be valuable resources for further genetic studies with C. gloeosporioides s.l. and other Colletotrichum species.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3480-x) contains supplementary material, which is available to authorized users.
Keywords: Evolution, Species complex, Mitochondrial genome, Concatenation, Phylogeny
Background
Colletotrichum gloeosporioides sensu lato is a fungal species complex encompassing more than 20 closely-related species [1]. These species interact with diverse herbaceous and woody plants as endophytes or foliar/fruit pathogens, and can be classified as generalists or specialists based on their host range specificity [2]. The diverse life styles within C. gloeosporioides s.l. makes it an interesting complex to study plant-microbe interactions and understand how host specificity evolves. As phytopathogens, C. gloeosporioides s.l. infect foliar as well as fruit tissues, and can cause severe field disease epidemics such as Glomerella leaf spot disease on apple (Malus domestica cv. Gala) and coffee berry disease on Arabica coffee (Coffea arabica). While many C. gloeosporioides s.l. species are economically deleterious, certain members are economically beneficial, e.g., as biocontrol agents against weeds and phytopathogens [3, 4].
Accurate species recognition in C. gloeosporioides s.l. is a complex task. Morphological characters, such as the size and shape of conidia and appressoria, are very plastic, and thus cannot be relied on for precise species identification [1, 5]. The species appear to have diverged relatively recently, so no single molecular marker commonly used in taxonomy (e.g., ITS, actin, β-tubulin 2) is informative enough to distinguish all lineages. Species recognition thus relies heavily on multi-locus phylogeny [6], and on the aid of highly polymorphic markers [7–10].
When the lineage divergence time gets very short, one critical issue for phylogeny-based species recognition is gene tree/species tree discordance due to evolutionary events such as incomplete lineage sorting, recombination, and horizontal transfer [11, 12]. Incomplete lineage sorting, or the inherently stochastic nature of gene genealogies at shallow levels of diversification, in particular, is the most common factor causing tree discordance which confounds species inference [12]; the noise/signal ratio can be strong enough to affect multi-locus phylogeny outcome, where different gene combination schemes produce well-supported yet conflicting topologies, making species inference a tricky process [13, 14]. Without a consensus on the concatenation markers and the criteria to draw species boundary lines, inconsistent species classification is unavoidable [11, 15, 16]. A striking example is C. siamense s.l., for which the number of proposed species has ranged from one to seven based on analysis with different marker combinations [1, 15, 17].
Several approaches can be taken to improve the consistency and accuracy of nascent species classifications. First, an increased sample size is helpful in teasing species-level variation apart from population-level ones. Second, different analytical approaches (e.g., concatenation-based phylogeny, genealogical concordance analysis, and coalescence-based methods) can be applied to the same set of data for cross validation purpose. Third, an improvement on the number, representativeness, and independence of analyzed genetic markers can reduce the bias effect of gene/species tree discordance.
Compared with nuclear DNA, mitochondrial DNA (mtDNA) mutate more rapidly due to a higher copy number, a greater exposure to reactive oxygen species, and a less efficient DNA repair system [18]. The mitogenome is also characterized by small size, haploidy, a shortage of recombination, and uniparental heredity. Mitochondrial DNA has been used to resolve evolutionary relationships of a wide range of eukaryotic life lineages at multiple taxonomic levels [19–22]. However, phylogenetic analysis within C. gloeosporioides s.l. has been based solely on nuclear DNA markers, whereas the suitability of mitochondrial DNA has not been tested, due in large part to a lack of available mitogenome sequence. It is worth investigating whether mitochondrial DNA, as a cytoplasmic factor independent of the prevailing nuclear markers, can provide additional insights into the genetic relationships of C. gloeosporioides s.l. lineages.
The first objective of this study was to obtain and compare mitogenomes from representative C. gloeosporioides s.l. lineages. Weir et al. [1] established Musae and Kahawae as the two main C. gloeosporioides s.l. clades, into which most recognized species can be classified. In this study, we analyzed nine isolates from four species lineages within or close to the Musae clade. Sequence comparison identified unique characteristics not observed with any previously reported Colletotrichum mitogenomes, including a significant size expansion and a striking GC content elevation at the intergenic regions. When comparing the mitogenomes belonging to the C. gloeosporioides s.l. complex, we observed considerable intron content variation among otherwise highly conserved mitogenomes. Such intron presence–absence polymorphism pattern can potentially be exploited for a rapid genetic diversity survey. Three introns were uniquely conserved among all C. gloeosporioides s.l. mitogenomes, indicating their potential value for developing C. gloeosporioides s.l. differentiation markers. We tested four primer pairs targeting these introns, and identified one primer pair that was highly efficient in differentiating C. gloeosporioides s.l. from C. actutatum s.l..
With complete mitogenome sequences available, we also tested the usefulness of mitogenome-based phylogeny in helping resolving the genetic relationships of C. gloeosporioides s.l. lineages. Our results demonstrated that mitogenome-based phylogeny recognizes nascent species divergence and is an important complement to nuclear markers in understanding C. gloeosporioides complex evolution.
Methods
Fungal isolates
Diseased plant samples used for fungal isolation were collected from different geographic regions in China in accordance with local regulations. Details about the taxonomy, host, locality, and DNA marker accessions of fungal isolates are listed in Additional file 1: Table S1. Isolates used for genome sequencing were isolated from diseased apple (Malus domestica) leaf or fruit tissues. C. fructicola isolates used for intron content survey were isolated from fruit or leaf lesions of apple, kiwifruit (Actinidia spp.), chili pepper (Capsicum annuum), pear (Pyrus spp.), papaya (Carica papaya), and nectarine (Prunus persica var. nucipersica). All fungal cultures were single-spore purified, maintained on potato dextrose agar (PDA), and stored as glycerol stock (15%) at −80 °C in the Fungal Laboratory of Northwest A&F University, Yangling, Shaanxi Province, China.
Genome sequencing and assembly
Fungal mycelia were produced and harvested from liquid potato dextrose broth (PDB) shake culture. Total genomic DNA was extracted based on a modified cetyl trimethylammonium bromide (CTAB) protocol [23], which was then used to prepare sequencing libraries with a mean insertion size of 350 bp. The libraries were subjected to 100-bp pair-end sequencing with the Illumina HiSeq 2000 platform at the Novogene Genomic Sequencing Center, Beijing, China. Raw data (raw reads) of fastq format were first processed through in-house perl scripts. In this step, clean data (clean reads) were obtained by removing reads containing adapter, reads contains ploy-N (N > 10%), and low-quality reads (sQ < = 5) from raw data. Clean reads were then de novo assembled into scaffolds using the AbySS assembler version 1.3.5 [24]. Contigs representing mitochondrial genomic DNAs were identified by BLAST search. Gaps were filled by PCR and standard Sanger sequencing, with the used primers listed in Additional file 1: Table S2.
Sequence annotation and comparison
Coding genes, introns, novel ORFs, rRNAs, and tRNAs were identified based on analysis with the online server MFannot (http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl), RNAweasel (http://megasun.bch.umontreal.ca/cgi-bin/RNAweasel/RNAweaselInterface.pl), and tRNAscan-SE 1.2.1 [25]. The annotations were manually inspected by comparing with Colletotrichum mitogenomes using the Mauve software version 2.4.0 [26]. To predict the functions of identified novel ORFs (mostly homing endonuclease genes), the coding proteins were searched against the NCBI nr and InterProScan databases. All measures of genetic diversity were calculated by DNASP version 5.10.1 [27].
Phylogenetic analysis
To accurately define the phylogenetic placement of the nine isolates used for mitogenome comparison, a Bayesian phylogenetic analysis was performed, including 33 additional C. gloeosporioides s.l. isolates along with two C. alatae isolates as outgroup. All 35 isolates were derived from a previous study by Weir et al. [1]. Inferences were based on seven combined nuclear loci, including actin (ACT), calmodulin (CAL), chitin synthase (CHS), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), glutamine synthetase (GS), manganese-superoxide dismutase (SOD2), and β-tubulin 2 (TUB2). DNA sequences for the seven loci were obtained from local draft genome assemblies, whereas DNA sequences for the referent isolates were downloaded from the NCBI database (please refer to Weir et al. [1] for accession numbers). For each gene, the sequences from different isolates were aligned with MEGA version 7 [28] and manually adjusted if necessary. Individual alignments were concatenated using SequenceMatrix version 1.8 [29]. MrModeltest version 2.3 [30] was used to identify the AIC best-fit nucleotide substitution model for each gene partition, which were GTR + I for GADPH, GTR + G for CAL, HKY for ACT, SYM + I for CHS, GTR + G for GS, HKY + G for TUB2, and HKY + I + G for SOD2. Phylogeny construction was based on Bayesian inference (BI) performed with MrBayes version 3.2.1 [31]. The Bayesian analysis included two separate runs for 5 × 107 generations, each sampled every 1000 generations, convergence of all parameters was checked using the internal diagnostics. To construct the 50% major rule consensus tree, the first 25% generations were discarded as burn-in. The same procedure was used to construct a six-locus phylogeny based on a concatenated alignment excluding GS, and a five-locus phylogeny excluding GS as well as SOD2.
Maximum likelihood phylogenetic trees used for comparative phylogenetic analysis were constructed with MEGA7. Best substitution model for each alignment was identified with either ProtTest version 3.2 (for amino acids) [32] or MrModeltest version 2.3 (for nucleotides). For aligning the whole mitogenome, the Mauve software version 2.4.0 [26] was used to, and the conserved sites were further extracted with Gblocks version 0.91b to obtain the final Gblocks alignment dataset [33]. Shimodaira-Hasegawa (SH) test was performed with the PAUP* test-version 4.0a150 (www.paup.scs.fsu.edu). For each test, the null distribution was generated by 10,000 bootstrap re-sampling via RELL approximation.
Survey of intron content polymorphism
To survey intron content diversity in C. fructicola, 14 isolates in addition to 1104–7 were selected (Additional file 1: Table S1). The species identity of these isolates was confirmed by ApMAT-locus sequencing. In total, 10 intron positions within five mitochondrial genes (cob, cox1, nad1, cox3, nad2, nad5) were surveyed. Primer pairs (Additional file 1: Table S2) were designed so that for a specific intron position, PCR amplicons of different sizes would be obtained between isolates containing a given intron and isolates lacking that intron. Four primer pairs targeting the three introns that were uniquely conserved among all C. gloeosporioides s.l. mitogenomes (cob-1, cox1-4, and cox1-5) were designed and their amplification efficiency and specificity were tested with a set of C. gloeosporioides s.l. and C. actutatum isolates (Isolates listed in Additional file 1: Table S1; primer sequences listed in Additional file 1: Table S2). Primer designs were based on sequence alignments of all known Colletotrichum mitogenomes. The PCRs were performed in a Bio-Rad thermal cycler in a total volume of 25 μL. The PCR mixtures contained 19 μL of double deionized water, 2.5 μL of 10 × PCR buffer (with 20 mM MgCl2), 0.5 μL of dNTPs (each 10 mM), 1 μL of each primer (10 μM), 1 μL of genomic DNA, and 0.2 μL (1U) of EasyTaq DNA polymerase (Transgen Biotech, Beijing, China). For primers detecting the intraspecific intron content diversity of C. fructicola, the PCR condition was 4 min at 95 °C, then 20 cycles of 95 °C for 30 s, 60 °C for 30 s (decrease 0.5 °C per cycle), 72 °C for 2 min, then 30 cycles of 95 °C for 30 s, 50 °C for 30 s, 72 °C for 2 min, and then 7 min at 72 °C. The PCR condition for primers aimed at detecting C. gloeosporioides s.l. was the same as above except that the stage-II annealing temperature ranged between 65 °C and 55 °C and the stage-III annealing temperature was 55 °C. For PCR aimed at detecting C. gloeosporioides s.l., the reaction for each primer-isolate combination was conducted three times to assess repeatability. PCR products were analyzed with 1.2% agarose electrophoresis.
Results
Phylogenetic placement of the nine C. gloeosporioides s.l. isolates selected for mitogenome sequencing
The combined nuclear gene alignment dataset comprised 3,642 sites, among which 391 were parsimony-informative. The 50% majority rule consensus Bayesian tree (Fig. 1) was identical in topology to the one reported by Weir et al. [1]. Based on the dendrogram, we classified the nine isolates (shaded in brown) into four lineages, including C. fructicola (1104–7), C. aenigma (XY-15), C. gloeosporioides sensu stricto (LQ33, hereafter C. gloeosporioides), and C. siamense s.l. (SQ01, LQ22, ZH01, ZH02, ZH03, and YT02). C. fructicola has been reported to contain two clades indistinguishable in morphology and cultural appearance, both being widely distributed and associated with diverse hosts [1]. Isolate 1104–7 fell into the clade containing the ex-holotype ICMP 18581 (Fig. 1). The isolate XY-15 was classified as C. aenigma, a sister species of C. fructicola. Worldwide, C. aenigma has been known only from two collections, ICMP 18608 from Persea americana in Israel and ICMP 18686 from Pyrus pyrifolia in Japan (Fig. 1). On the phylogenetic tree, ICMP 18608 and ICMP 18686 were much closer to each other than to XY-15. Six isolates in this study (SQ01, LQ22, ZH01, ZH02, ZH03, and YT02) belonged to C. siamense s.l., a taxonomically controversial lineage being regarded as either a single species [1, 15] or a species complex [17]. As a species complex, ICMP 18642 and ICMP 19118 had been treated as independent species, being C. hymenocallidis [34] and C. jasmini-sambac [35] respectively. On the phylogenetic tree, the six C. siamense s.l. isolates located on three separate clades. SQ01, LQ22 and ZH03 were close to ICMP 18642 (C. hymenocallidis), ZH01 and ZH02 were close to ICMP 18574; YT02 was distal from all other C. siamense s.l. isolates, forming an independent branch with100% posterior probability, which supported the interpretation that YT02 represents a novel species if C. siamense s.l. is seen as a species complex. LQ33 was classified as C. gloeosporioides based on its close affinity to the epitype culture ICMP 17821 (Fig. 1).
Fig. 1.
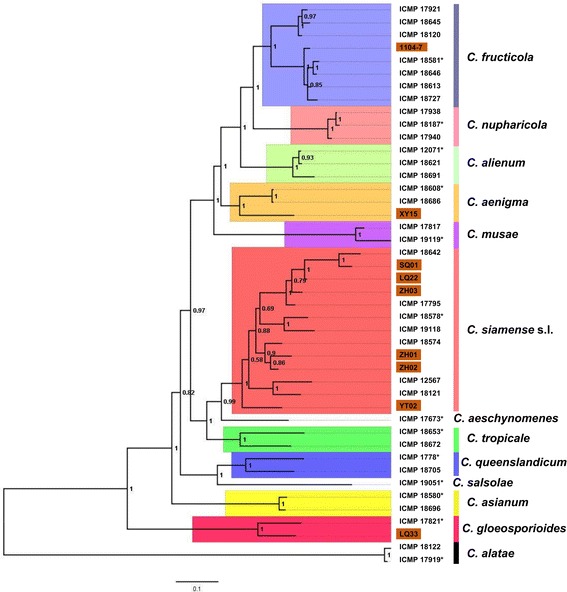
Phylogenetic placement of the Colletotrichum gloeosporioides s.l. isolates selected for mitogenome sequencing. The fifty percent majority rule consensus Bayesian tree was based on a 7-gene combined dataset (GADPH, CAL, SOD2, ACT, CHS, GS, TUB2). The alignment was partitioned, and the best DNA substitution model for each gene was determined by MrModelTest. Colletotrichum alatae isolates were used as the outgroup and the tree was mid-point rooted. Ex-type cultures are marked with ‘*’. For each node, posterior probability is displayed
Introns and intergenic regions drive the significant size expansion of C. gloeosporioides s.l. mitogenomes
Prior to this study, five complete Colletotrichum mitogenomes were available in the GenBank database: three within the C. acutatum species complex (C. acutatum, C. tamarilloi, and C. lupini), one within the C. orbiculare complex (C. lindemuthianum), and one within the C. graminicola complex (C. graminicola). Through illumina DNA sequencing and assembly, we obtained the complete mitogenome sequences of the nine aforementioned C. gloeosporioides s.l. isolates. We compared these 14 mitogenomes in detail regarding their sizes, GC contents, gene contents, codon usage patterns and intron characteristics (Additional file 1: Table S3 to S7).
The sizes of the nine C. gloeosporioides s.l. mitogenomes ranged between 52,671 and 58,666 bp (55,234 ± 1,863 bp), the sizes of the five mitogenomes outside of the complex ranged between 30,824 and 39,649 bp (34,975 ± 3,942 bp) (Additional file 1: Table S3, Fig. 2a); the average size of the former group was 1.58 times larger than the latter group. The average coding region lengths for mitogenomes within and outside of C. gloeosporioides s.l. were very close: 21,974 ± 46 and 21,509 ± 103 bp respectively. On the other hand, the corresponding values for the intergenic regions differed by 1.62 fold, being 20,228 ± 679 and 12,452 ± 3,551 bp respectively. C. gloeosporioides s.l. mitogenomes also contained significantly more introns. The three C. acutatum s.l. mitogenomes (C. acutatum, C. lupini, C. tamarilloi) contained no intron, C. lindemuthianum and C. graminicola contained three and one intron, with total length of 4,071 and 1,000 bp, respectively; the nine C. gloeosporioides s.l. mitogenomes, on the other hand, contained from six to 10 introns (Additional file 1: Table S4), with total length ranging from 11,057 to 16,199 bp.
Fig. 2.
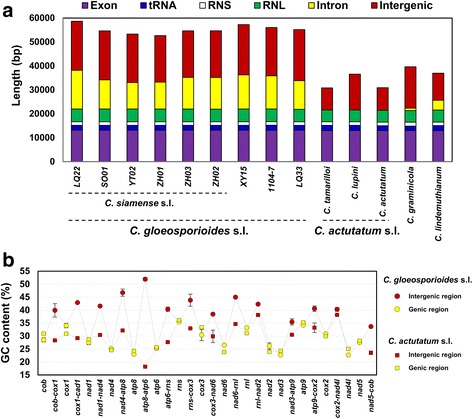
Size expansions and intergenic GC content elevations of the Colletotrichum gloeosporioides s.l. mitogenomes. a Size and content variations of Colletotrichum mitogenomes. Exon refers to oxidative phosphorylation protein-coding genes. RNS, small ribosomal RNA; RNL, large ribosomal RNA. b Differences in average GC contents between the genic and intergenic regions of C. gloeosporioides s.l. and C. actutatum s.l.. Plotted values are mean ± S.D., n = 9 for C. gloeosporioides s.l., n = 3 for C. actutatum s.l
When considering all 14 isolates, the summed length of intronic and intergenic regions closely predicted the total length of the mitogenome (r 2 = 0.999). When introns and intergenic regions were considered separately, the r 2 values were 0.954 and 0.900 respectively. Taken together, the C. gloeosporioides s.l. mitogenomes were much larger in size than those of any previously reported Colletotrichum groups, and the size expansion was attributable to length increases of both introns and intergenic regions, but not the coding regions.
The C. gloeosporioides s.l. mitogenomes have significantly higher GC content compared with other Colletotrichum spp. due specifically to striking intergenic GC content elevation
The GC content of the nine C. gloeosporioides s.l. mitogenomes ranged between 33.8 and 34.6% (34.24 ± 0.21%), whereas the values for members not belonging to the complex ranged between 29.9 and 30.9% (30.34 ± 0.43%). The average value for the former group was significantly higher than the latter (t [12] = 22.01, p < 0.001). In the genic regions (exons, introns, rRNAs), the average GC content values for members in and out of C. gloeosporioides s.l. were close: 31.07 ± 0.17% and 30.2 ± 0.05% respectively. In contrast, the corresponding values for the intergenic regions were 39.72 ± 0.25% and 30.76 ± 1.23% respectively, differing by almost 10%.
The C. gloeosporioides s.l. and C. acutatum s.l. mitogenomes were identical in gene order, which allowed us to compare their genic/intergenic GC content dynamics in more detail. Average GC content values for 17 genic and 14 intergenic regions were plotted (Fig. 2b). In all genic regions, the values of the two groups were very similar, but in all intergenic regions except rnl-nad2 and cox2-nad4l, the values for C. gloeosporioides s.l. were much higher than for C. acutatum s.l. (Fig. 2b, Additional file 1: Table S5). Moreover, the GC content for all but one (nad3-atp9) C. gloeosporioides s.l. intergenic region was much higher than the neighboring genic regions, which however, was only observed with three out of 14 intergenic regions in C. acutatum s.l. (nad4-atp8, rnl-nad2, and cox2-nad4l).
Comparative analysis of the sequence, codon usage, and gene organization features of the C. gloeosporioides s.l. mitogenomes
DNA sequences of the nine C. gloeosporioides s.l. mitogenomes were highly conserved. The mitogenome GBlocks alignment dataset contained 44,699 characters, among which merely 573 were polymorphic (12.8 per kb on average). Alignment of the protein-coding regions produced a dataset containing14,688 characters, among which merely 64 were polymorphic (4.36 per kb on average). The mitogenome nucleotide diversity value (π) was lower than all compared nuclear markers (Fig. 3a), indicating extremely slow overall evolution rate. Yet, due to length advantage, the mitogenome GBlocks alignment dataset contained the largest number of phylogenetically informative sites (212 in total, Fig. 3b), which was 2.6 times more than that of the most polymorphic nuclear marker, ApMAT (81 in total).
Fig. 3.
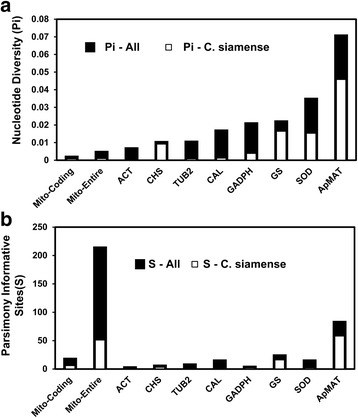
A comparison of the phylogenetic information conveyed by the mitogenome sequences and that of commonly used nuclear markers. a Nucleotide diversity value (π). b Parsimony informative sites (S). The calculations of π and s were either based on all C. gloeosporioides s.l. isolates (− All) or based on the C. siamense s.l. isolates alone (− C. siamense)
Each of the nine C. gloeosporioides s.l. mitogenomes encoded 44 identical genes transcribed in the same direction (Fig. 4a), including 14 oxidative phosphorylation proteins (three subunits of cytochrome oxidase [cox1-3], apocytochrome b [cob], three subunits of ATP synthetase [atp6, 8, 9], and seven subunits of NADH: ubiquinone oxidoreductase [nad1-6 and nad4L]), a ribosomal protein (rps3), large (rnl) and small (rns) rRNAs, and 27 tRNAs (Additional file 1: Table S4). The 27 tRNAs recognized all 20 standard amino acids, with three for valine, three for methionine, two for arginine, two for serine, and two for leucine. The order of the 44 genes was identical among all mitogenomes.
Fig. 4.
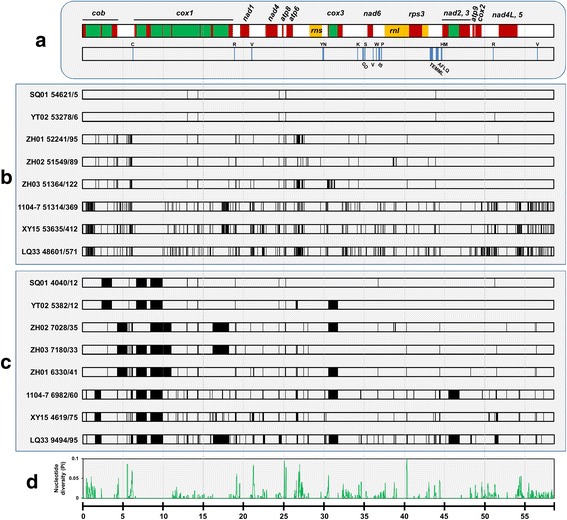
Sequence comparison of the Colletotrichum gloeosporioides s.l. mitogenomes. The features of LQ22 (C. siamense s.l.) and its comparison with other members are shown. a The LQ22 mitogenome gene features. Proteins (exon in red, intron in green) and rRNAs (yellow) are shown on top whereas tRNAs (amino acid abbreviations) are shown in bottom. b Distributions of LQ22 mismatch sites compared with indicated mitogenomes. Following isolate ID, total matched and mismatched sites (separated by dash line) are shown. c Distribution of LQ22 insertion sites compared with indicated mitogenomes. Following isolate ID, total insertion length and insertion sites are shown. d Sliding window analysis of nucleotide diversity (π) based on the nine mitogenomes (window length 50 bp, step size 5 bp, gaps omitted). Plots in all panels (a to d) share the same coordinate (in the bottom), the unit for which is kilo base (kb)
Pairwise comparison identified several characteristics associated with the C. gloeosporioides s.l. mitogenomes: (1) mitogenomes within C. siamense s.l. were more similar to each other than to mitogenomes from different species lineages (Fig. 4b); (2) within C. siamense s.l., SQ01 and YT02 were nearly identical in DNA sequences (Additional file 2: Figure S1) despite falling far apart on the nuclear gene tree (Fig. 1); the mitogenomes of SQ01 and YT02 were identical except for eight InDel sites located either in introns or in intergenic regions, among which were six small-sized ones (one to five bp) and two large-sized ones (207 bp and 1,131 bp [cox3 gene intron]); all six small-sized InDels were within single-nucleotide short DNA repeats; (3) nucleotide insertion/deletion events were less frequent compared with mutation events, and most large insertion/deletion events were due to intron gain/losses (Fig. 4c); (4) polymorphic sites distributed unevenly along the mitogenome, with the frequency being much higher in the intergenic and intronic regions than the coding regions (Fig. 4d); (5) a small coding region of the cox3 gene showed strong signal of lineage-specific rapid divergence. Among the 64 polymorphic sites present in the mitogenome protein-coding regions, 36 were synonymous and 28 were nonsynonymous; these sites were distributed in eight of 15 genes (one in nad2, three in rps3, five in nad4, five in nad5, six in cox1, eight in cox2, nine in cob, and 27 in cox3). Strikingly, up to 18 of the 28 nonsynonymous sites were specifically located within a 142-bp long region of cox3 (76 to 218 bp in the ORF), and most were singletons conferred by ZH03 (C. siamense s.l.) (Additional file 3: Figure S2). Further PCR and Sanger resequencing confirmed the nucleotide identity of ZH03 in this region (data not shown).
Codon usage frequencies were indistinguishable among the nine mitogenomes (Additional file 1: Table S6). All protein-encoding genes began with the typical AUG start codon except cox3 with UUG, and all ended with the UAA stop codon except cox3 and nad6 with UAG. The most-frequently used codons were the same: UUA (L, 12.07–12.13%), AUA (I, 7–7.02%), UUU (F, 5.62–5.64%), and AAU (N, 4.29–4.33%). On the other hand, UGC (C), CGC (R), CGG (R), AGG (R) were universally absent. In addition, a number of codons were under-represented (relative frequency < 1%), e.g. CGA (R, 0.2%), CUC (L, 0.2%), ACG (T, 0.2–0.4%), AAG (K, 0.2–0.7%), CAG (Q, 0.4%), CCC (P, 0.4%), GGC (G, 0.4–0.7%), GUC (V, 0.7%), ACC (T, 0.7%), UGG (W, 0.7%), and CCG (P, 0.9%). Such codon usage patterns were similarly observed in other Colletotrichum mitogenomes (Additional file 1: Table S6). Among all Colletotrichum mitogenomes, the CGC and AGG codons coding for lysine were absent; moreover, codons present at higher frequency tended to favor A and T nucleotides, in accordance with reports in other fungi [22].
The nine C. gloeosporioides s.l. mitogenomes shared identical orders of protein-coding genes, rRNAs and tRNAs. We further examined the extent of such order conservation in the Hypocreomycetidae subclass. For protein-coding genes and rRNAs (Fig. 5, left), the C. gloeosporioides s.l. order was conserved among all C. actutatum s.l. members and all Hypocreales members except Acremonium chrysogenum, suggesting that this order represents the ancestral status of Hypocreomycetidae. The orders of C. graminicola, C. lindemuthianum, Verticillium nonalfalfae, V. dahlia and A. chrysogenum were slightly different, involving one or two DNA rearrangement events (Fig. 5, left).
Fig. 5.

Mitogenome gene order evolution in the Hypocreomycetidae subclass. Dotted line indicates identical gene order compared with the reference (LQ22). Gene rearrangement events are highlighted in color. For tRNAs, the corresponding amino acid abbreviations are shown. GenBank accessions for Colletotrichum mitogenomes are listed in Additional file 1: Table S3. GenBank accessions for additional mitogenomes are as follows. Verticillium nonalfalfae, KR704425; V. dahliae, DQ351941; Acremonium chrysogenum, KF757229; Fusarium solani, NC_016680; F. graminearum, NC_009493; F. oxysporum, AY945289; F. verticillioides, NC_016687; Beauveria bassiana, EU371503; Cordyceps brongniartii, NC_011194; Pochonia chlamydosporia, KF479445; Metarhizium anisopliae, AY884128; Trichoderma asperellum, KR952346
In each of the nine C. gloeosporioides s.l. mitogenomes, 22 of 27 tRNAs clustered together and formed four groups, which were between rns and cox3 (cluster I, trnY, trnN), between cox3 and nad6 (cluster II, trnK, trnG, trnD, trnS), between nad6 and rnl (cluster III, trnV, trnW, trnI, trnS, trnP), and between rnl and cox1 (cluster IV, trnT, trnE, trnM, trnM, trnL, trnA, trnF, trnL, trnQ, trnH, trnM). We identified and compared tRNAs belonging to these four clusters in different Hypocreomycetidae mitogenomes (Fig. 5, right). In general, the extent of tRNA content and organization variation correlated well with taxonomic distance. For instance, in cluster I, all Verticillium and Colletotrichum members had ‘Y N’ whereas Hypocreales members had ‘Y D S’ or ‘Y D S N’; in cluster II, all Verticillium and Colletotrichum members had at least three genes, ‘G D S’, whereas Hypocreales members only had a ‘G’ or none at all; and in cluster IV, all Hypocreales members had a trnK (‘K’) gene, which was absent in both Verticillium and Colletotrichum.
Within the genus Colletotrichum, tRNA organization of C. graminicola and C. lindemuthianum was identical to that of C. gloeosporioides s.l. members. The three species belonging to the C. actutatum complex, on the other hand, had different tRNA organization in cluster II. Compared with C. gloeosporioides s.l. (‘K G D S’), the tRNA organization for C. tamarilloi and C. actutatum was ‘G D S’ whereas that for C. lupini was ‘G D S S’. Sequence comparison indicated that the trnS duplication in C. lupini was due to a lineage-specific 5,708 bp DNA insertion event (data not shown).
Significant intron content variation among the C. gloeosporioides s.l. mitogenomes
In total, seventy-one introns were identified among the nine C. gloeosporioides s.l. mitogenomes (Fig. 6, Additional file 1: Table S7). These introns ranged between 997 and 3,516 bp in size, and were located at 16 conserved positions within six oxidative phosphorylation protein-encoding genes, which included nine in cox1, two in cob, two in nad2, one in nad1, one in cox3, and one in nad5. All identified introns belonged to the group I intron family except for cox1-4, which lacked distinguishable features for classification. The group I introns could be further subdivided into type I-A (cob-2, seven in total), type I-B (cox1-1, cox1-3, cox1-5, cox1-6, cox1-8, cox1-9, nad1-1, cox3-1, nad5-1; 35 in total), type I-C2 (cox1-2, one in total), and type I-D (cob-1, nad2-1, nad2-2; 17 in total). The cox1-7 intron, present in ZH02 and ZH03, belonged to type I but could not be further subdivided. Except for cox1-4, introns at all positions harbored homing endonuclease genes (HEGs) known to direct intron mobility. Intronic HEGs could be further divided into LAGLIDADG and GIY-YIG families. Forty-five introns at 10 positions harbored LAGLIDADGs whereas 17 introns at another five positions harbored GIY-YIGs.
Fig. 6.
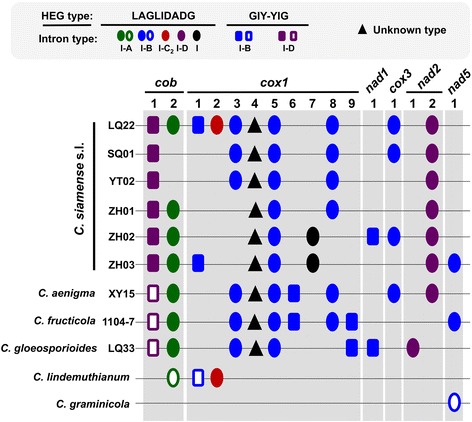
The Colletotrichum gloeosporioides s.l. mitogenome introns. The 71 type I introns (further classified into type I-A, I-B, I-C2, and I-D) locate at 16 conserved positions in six genes, and most harbor homing endonuclease genes (classified into ‘LAGLIDADG’ and ‘GIY-YIG’ types). Filled and open symbols mark introns with slightly different features. For cob-1, the intron sizes of XY15, 1104–7 and LQ33 are smaller. For cox1-1 and nad5-1, the introns in C. lindemuthianum and C. graminicola have different intron phases. Please refer to Additional file 1: Table S7 for detailed information about the positions, codon phases and lengths of introns
In general, introns at the same position were highly similar in terms of length and sequence. However, the cob-1 site was an exception, where the intron sizes of XY15, 1104–7, and LQ33 (1,259 to 1,274 bp) were much smaller than the C. siamense s.l. members (LQ22, SQ01, YT02, ZH01, ZH02, ZH03, 1909 to 1912 bp) (Additional file 1: Table S7). Sequence comparison showed the size difference was due to non-homologous sequences at the 3′ region (data not shown).
High-level intron content variation was observed among the nine C. gloeosporioides s.l. mitogenomes. Each mitogenome contained a unique set of introns. Among the 16 intron positions, cob-1, cox1-4, and cox1-5 were the only three positions conserved among all genomes (Fig. 6). Considerable intron presence-absence polymorphism was also observed within C. siamense s.l., for which nine out of 12 positions were polymorphic. The presence-absence polymorphism was also observed between SQ01 and YT02, two isolates being almost identical in mitogenome sequence. These result demonstrated rapid evolutionary dynamics of introns in C. gloeosporioides s.l.
We further surveyed the intraspecific intron content variation in another C. gloeosporioides s.l. species, C. fructicola, for which we analyzed 10 intron positions and 15 isolates (including 1104–7) (Fig. 7). These C. fructicola isolates were isolated from different hosts and from different locations (Additional file 1: Table S1), and contained five ApMAT haplotypes, and were thus genetically different. The intron content patterns of these 15 isolates could be classified into three types. Eleven isolates, containing all five ApMAT haplotypes, had completely identical intron content. Three other isolates (MWG_02, PGYSQ03, LZCQ01) had slightly different intron contents. The ApMAT nucleotide diversity value calculated from the 15 C. fructicola isolates was 0.00126, much lower than that of C. siamense s.l. (based on LQ22, SQ01, YT02, ZH01, ZH02, ZH03, π = 0.0458). Thus, both intron content and ApMAT locus sequence data supported a lower level of intraspecific genetic diversity of C. fructicola.
Fig. 7.
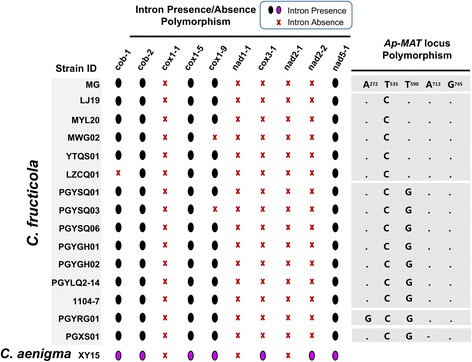
Intraspecific intron content polymorphism in Colletotrichum fructicola. The intron presence-absence patterns of 15 surveyed isolates, together with the nucleotides of these isolates at five polymorphic sites within a 762 bp long alignment of ApMAT locus DNAs are shown. The intron content of the C. aenigma isolate XY15 is shown as a comparison. GenBank accessions of the ApMAT locus sequences are listed in Additional file 1: Table S1
The unique conservation of cob-1, cox1-4, and cox1-5 introns in C. gloeosporioides s.l. isolates indicated their potential value for molecular identification of taxa within C. gloeosporioides s.l.. We designed four primer pairs targeting these introns (primer design strategy shown in Additional file 4: Figure S3). The primer pair P1/P2, P7/P8 targeted cob-1 and cox1-5 respectively; the four primer binding sites were in exons and were highly conserved among at least four divergent Colletotrichum species complexes (C. gloeosporioides s.l., C. actutatum s.l., C. orbiculare s.l., C. graminicola s.l.; Additional file 4: Figure S3b). The primer pairs P3/P4 and P5/P6 recognized two separate regions inside cox1-4, supposed to be unique to C. gloeosporioides s.l.. The designed primer pairs were tested against eight C. gloeosporioides s.l. isolates and five C. actutatum s.l. isolates (results shown in Additional file 5: Figure S4). All primer pairs produced strong and specific bands when using the C. gloeosporioides s.l. DNAs as templates, indicating high amplification efficiencies. When using the C. actutatum s.l. DNAs as templates, P3/P4 and P5/P6 generated amplifications from time to time, the size of which matched that expected for C. gloeosporioides s.l., indicating poor primer specificity. P7/P8 produced ~1,440 bp amplicons for all C. gloeosporioides s.l. isolates and ~120 bp amplicons for all C. actutatum s.l. isolates, the ~120 bp amplicons were relatively weak and indistinctive from primer dimers, so this can be problematic for identification purposes. For P1/P2, all C. actutatum s.l. isolates produced an expected ~ 360 bp band whereas all C. gloeosporioides s.l. isolates produced bands larger than 1,500 bp (~1650 bp for XY15, 1104–7, LQ33; and ~ 2300 bp for other isolates), amplicon size differences among the C. gloeosporioides s.l. isolates perfectly matched their cob-1 intron sizes as reported above. In repeated experiments, P1/P2 always produced strong and specific bands for both C. actutatum s.l. and C. gloeosporioides s.l., providing clear distinction between these groups.
Implications of mitogenome-based phylogeny
We first constructed maximum likelihood (ML) phylogenetic trees based on either mitogenome proteins or mitogenome protein-coding DNAs. On both trees, all nine C. gloeosporioides s.l. isolates clustered into a well-support monophyletic clade neighbored by C. lindemuthianum, C. actutatum s.l. and C. graminicola (Additional file 6: Figure S5). These species together represented the Colletotrichum genus. On the protein tree, the genus Colletotrichum was neighbored by Verticillium spp.; the two genera together were further neighbored by Hypocreales. Such a topological pattern was in accordance with the genetic relationships of Glomerellales, Plectosphaerellaceae and Hypocreales [36, 37], and supported the previous phylogenetic placement of C. gloeosporioides s.l. based on nuclear markers. No phylogenetic structure was detected within C. gloeosporioides s.l. members on either tree, due to an exceptionally low level of sequence variations.
A ML phylogenetic tree was further constructed based on the 44,699 bp long mitogenome Gblocks alignment (Fig. 8a). The nine isolates formed four well-separated clades, corresponding to C. siamense s.l. (green color), C. aenigma (XY15), C. fructicola (1104–7), and C. gleosporioides (LQ33) respectively. Such topological pattern correlates well with their distributions on the nuclear marker-based phylogenetic tree (Figs. 1 and 8b). Within C. siamense s.l., however, a strong discordance was observed between the mitogenome and nuclear trees. The ‘Mito’ tree contained two distinctive clades, one clade included ZH01, ZH02, and ZH03 whereas the other clade included SQ01, YT02, and LQ22 (Fig. 8a), SQ01, YT02, and LQ22 were almost indistinguishable. On the nuclear tree (Fig. 8b), the six C. siamense s.l. isolates formed two groups with YT02 being separated from all other isolates. The striking different phylogenetic positions of YT02 highlighted a discordance between mitogenome and nuclear markers in C. siamense s.l.. Preliminary analysis showed that the phylogenetic discordance could be eliminated by excluding the GS and SOD2 markers from analysis (Fig. 8 c, d, g, h). The ApMAT marker-based phylogeny (Fig. 8f) was highly similar in topology compared with that based on mitogenome.
Fig. 8.
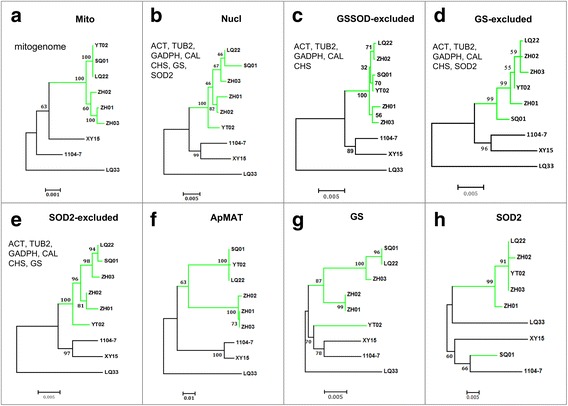
Maximum likelihood phylogenetic trees constructed from different datasets. For concatenation tree, markers used for concatenation are shown. Best nucleotide substitution models for ‘Mito’, ‘Nucl’, ‘GSSOD-excluded’, ‘GS-excluded’, ‘SOD2-excluded’, ‘ApMAT’, ‘GS’, and ‘SOD’ phylogenetic trees were determined by MrModelTest, which were ‘GTR + G + I’, ‘HKY + G + I’, ‘HKY + I’, ‘HKY + I’, ‘HKY + G + I’, ‘HKY + I’, ‘HKY’, and ‘HKY + G’ respectively. Numbers at the nodes are bootstrap values based on 1,000 replicates. Green color indicates C. siamense s.l
Shimodaira-Hasegawa (SH) test was further performed to test marker incongruence (Table 1). The mitogenome-based tree (‘Mito’ tree, Fig. 8a) was compared with the tree based on concatenated nuclear loci (‘Nucl’ tree, Fig. 8b). As a control, the ‘Mito’ tree was also compared with the ‘GSSOD-excluded’ tree (Fig. 8c), the ‘GS-excluded’ tree (Fig. 8d), the ‘SOD-excluded’ tree (Fig. 8e), and the ‘ApMAT’ tree (Fig. 8f). SH tests were carried out against different nuclear marker datasets. As shown in Table 1, at 0.01 significance level, all nuclear markers equally supported the tested trees when comparing the ‘Mito’ tree with either the ‘ApMAT’ tree or with the ‘GSSOD-excluded’ tree. When comparing the ‘Mito’ tree with the ‘Nucl’ tree, ACT, TUB2, CAL, GADPH, CHS and SOD2 equally supported the two trees whereas GS strongly favored the ‘Nucl’ tree (P = 0, ‘Diff -ln L’ = −145.73). Such strong topological bias of the GS marker, however, was not observed when comparing the ‘Mito’ tree with the ‘GS-excluded’ tree (GS marker excluded for concatenation). In actual fact, the GS marker favored the ‘Mito’ tree slightly more than the ‘GS-excluded’ tree (P = 0.0201, ‘Diff -ln L’ = 17.18). Thus, the topological incongruence between the ‘Mito’ tree and the ‘Nucl’ tree was mainly caused by GS. The SOD2 marker strongly favored the ‘GS-excluded’ tree over the ‘Mito’ tree (P = 0.0076, ‘Diff -ln L’ = −39.27), while equally supporting the ‘Mito’ tree and the ‘Nucl’ tree (P = 0.203, ‘Diff -ln L’ = −2.01), indicating that the effect of GS masked that of SOD2.
Table 1.
The Shimodaira-Hasegawa (SH) test result
| Treea | Value typesb | ACT | TUB2 | CAL | GADPH | CHS | GS | SOD2 |
|---|---|---|---|---|---|---|---|---|
| Mito | -ln L | 435.06 | 1154.96 | 1272.82 | 512.87 | 504.86 | 1754.51 | 815.79 |
| ApMAT | Diff -ln L | 0 | −18.64 | −4.96 | 0 | 3.18 | −6.38 | −2.01 |
| P-value | / | 0.0433 | 0.1901 | / | 0.2279 | 0.1519 | 0.203 | |
| Nucl | Diff -ln L | 0 | −10.63 | −14.26 | 13.98 | 12.48 | −145.73 | −2.01 |
| P-value | / | 0.1872 | 0.0893 | 0.0798 | 0.0819 | 0* | 0.203 | |
| GSSOD-excluded | Diff -ln L | 0 | −18.64 | −4.75 | −5.24 | −2.77 | 17.18 | −2.01 |
| P-value | / | 0.0447 | 0.204 | 0.1472 | 0.276 | 0.0203 | 0.203 | |
| GS-excluded | Diff -ln L | 0 | −10.63 | −4.75 | 13.98 | −20.16 | 17.18 | −39.27 |
| P-value | / | 0.1926 | 0.204 | 0.0756 | 0.0626 | 0.0201 | 0.0076* | |
| SOD-excluded | Diff -ln L | 0 | −10.63 | −14.26 | 13.98 | 12.48 | −145.73 | −2.01 |
| P-value | / | 0.1894 | 0.0905 | 0.0776 | 0.0823 | 0* | 0.203 |
a The ‘Mito’ tree was compared with the ‘ApMAT’ tree, concatenated nuclear tree (‘Nucl’), ‘GSSOD-excluded’ tree, ‘GS-excluded’ tree, and ‘SOD-excluded’ tree based on indicated datasets. Tree topologies are shown in Fig. 8
bFor the ‘Mito’ tree, ‘-ln L’ represents the -ln(likelihood score) value. For each compared tree, ‘Diff -ln L’ represent the differences between their -ln(likelihood score) values and that of the ‘Mito’ tree. Negative value indicates that the dataset supports the tested tree better than the ‘Mito’ tree. The P-value for each test is reported and ‘*’ indicates P < 0.01. ‘/’ indicates equal -ln(likelihood score) values in which case SH test could not be performed
Given the significant biased effects of GS and SOD2, we performed further concatenation phylogenic analysis of the 42 C. gloeosporioides s.l. isolates by excluding either the GS marker alone (Additional file 7: Figure S6a), or the GS marker as well as the SOD2 marker (Additional file 7: Figure S6b) from the alignment dataset. When only GS was excluded, all species lineages still formed monophyletic clades and the tree topology was in general identical to the previous seven-locus tree (Fig. 1). The six C. siamense s.l. isolates formed two clades, and clade one includes all isolates except SQ01. When both GS and SOD2 were excluded, all C. siamense s.l. isolates clustered into one well-supported clade (posterior probability = 1), and no further genetic structure could be recognized; all other species lineages could still be recognized with high confidence except for C. fructicola, for which the two intraspecific groups formed sister clades. In general, it was clear that presence/absence of the GS and SOD2 markers strongly affected the phylogenetic positions of YT02 and SQ01; thus, lineage-specific divergences of GS and SOD2 were the main sources causing phylogenetic incongruence.
We further examined polymorphism sites in the GS and SOD2 alignments for sequence patterns potentially explaining their lineage-specific divergences (Additional file 8: Figure S7). The GS marker is made up of a large partial intron (~850 bp) and a very small coding region (8 bp). The alignment dataset contained 31 polymorphic sites for the C. siamense s.l. isolates, among which 14 were YT02-specific mutations; these 14 mutations occurred throughout the alignment, and 12 were transitions, including eight C to T (or G to A) and four T to C (or A to G). The SOD2 marker located in the coding region (374 bp in C. siamense s.l.), and the alignment contained 16 polymorphic sites among which 12 were SQ01-specific mutations, also dispersed in distribution. All SQ01-specific mutations were synonymous and located at the third codon positions except 216A to G and 217G to A, which together caused a ‘AGC’ (Ser) to ‘GAC’ (Asp) codon change. In general, we observed no obvious selection-indicative sequence pattern with either the GS or SOD2 marker, in accordance with a hypothesis that incomplete lineage sorting has driven the observed divergences.
Discussion
The fungal species complex C. gloeosporioides s.l. is economically and ecologically important, yet taxonomically challenging. In this study, mitogenomes representing nine taxonomic lineages were first determined, which provides useful resources for further genetic studies. Many features observed with the nine mitogenomes are also typical of the Hypocreomycetidae subclass and even the Sordariomycetes class; for instance, the transcription of all protein-encoding genes in the same direction, the presence of rps3 gene within the ribosomal large subunit, the conserved ordering of protein-encoding genes, tRNAs, and rRNAs, and the prevalence of HEG-containing introns. Compared with known Colletotrichum mitogenomes, however, the C. gloeosporioides s.l. mitogenomes are also distinctive for containing longer intergenic regions with elevated GC content and for being much richer in introns.
GC bias is common with both nuclear and mitochondrial genomes [38], and the GC contents of closely-related lineages can differ significantly [39]. The average intergenic GC content of C. gloeosporioides s.l. mitogenomes is around 10% higher than that of the genic regions. Although it remains to be determined whether such striking difference is prevalent in filamentous fungi, the lack of elevation in other Colletotrichum mitogenomes indicates a lineage specificity. The GC content elevation is prevalent among most intergenic regions, indicating that the elevation is not due to regional horizontal DNA transfer. Both neutral evolutionary events, such as biased mutation or biased gene conversion, and natural selection have been implicated to drive GC content evolution [40]. The striking GC content elevation observed in this study is specific to the intergenic regions where DNAs are under the least functional constraint, which would indicate neutral evolutionary forces. Mitochondrial DNA mutations, however, are AT biased due to increased likelihood of cytosine deamination or guanine oxidation [38]. Thus, if the GC content elevation is evolutionarily neutral, biased gene conversion would be a more likely mechanism. Gene conversion, however, relies on DNA mismatch repair system and DNA recombination events, the occurrence and frequency of which in C. gloeosporioides s.l. must be confirmed.
Regarding the natural selection hypothesis, genome GC content elevation has been implicated to confer an adaptation advantage toward high temperature, UV exposure or conditions where high DNA stability is required [41, 42]. If the intergenic DNAs are rich with regulatory elements, the intergenic GC content elevation observed here could in principle be relevant to the modulation of mitochondrial gene expressions, a mechanism proposed to explain the correlation between GC content and stress tolerance observed with monocot plants [43].
While the C. gloeosporioides s.l. mitogenome sequences are highly conserved, particularly at the coding regions, we have identified a 142-bp-long region in the cox3 ORF showing strong lineage-specific divergence. The short region contains 23 polymorphic sites, the frequency of which is over 300-fold higher than the rest of the coding regions. Moreover, up to 18 sites are non-synonymous, indicating functional relevance of the nucleotide changes. Most polymorphic sites are ZH03-specific singletons, demonstrating that this DNA region evolves differently in ZH03, likely involving recombination or horizontal transfer events. While its evolutionary origin and biological implications are unclear, the presence of such feature emphasizes the advantage of phylogenetic analysis based on entire mitogenome over partial mtDNA sequence.
Broad intron content variation exists among fungal mitogenomes, where both interspecific and intraspecific variations have been documented. Moreover, intron content variation has been a main source explaining the size variation commonly observed with closely-related mitogenomes [22, 44–47]. In this study, significant intron content variation exists both among and within C. gloeosporioides s.l. species, and most introns contain HEGs of either LAGLIDADG or GIY-YIG type. Such pattern suggests that horizontal intron cycling [48], a mechanism known to regulate intron gain and loss process, also plays a significant role in shaping the intron dynamics of C. gloeosporioides s.l.
Colletotrichum gloeosporioides was established in the 1950s, but understanding of its taxonomic position has shifted over time. The current broad concept (C. gloeosporioides s.l.) is defined by ITS phylogeny [1], and morphological phenotypes alone are unreliable for its identification from other lineages, such as C. boninense species complex [49] and C. cliviae [34]. Due to historical reason, many GenBank-deposited ‘C. gloeosporioides’ sequences are inaccurately annotated [6], which can be a problem for ITS-based identification. The development of a secondary C. gloeosporioides s.l. identification marker is thus advantageous. Introns at three positions (cob-1, cox1-4, and cox1-5) are conserved among all C. gloeosporioides s.l. mitogenomes; however, none of them was observed in mitogenomes outside of the C. gloeosporioides s.l. complex, indicating a potential utility for PCR-based detection. We further tested four primer pairs and identified P1/P2 targeting cob-1 as the one with the highest specificity. The P1/P2 primer pair identifies C. gloeosporioides s.l. based on amplicon size increase, instead of amplicon presence; such a strategy is more advantageous because: (1) all surveyed samples should produce amplification bands indicating successful PCR reaction, thus this is a good positive internal control of DNA template and PCR reagents; and (2) the strategy dramatically reduces the likelihood of non-specific primer binding and non-specific amplification, thus improving detection specificity and repeatability, which is a critical advantage in PCR-based detections. The P1/P2 primer-binding sites are highly conserved among very divergent species complexes (C. gloeosporioides s.l., C. actutatum s.l., C. orbiculare s.l., and C. graminicola s.l.), and are thus likely to be applicable across the whole Colletotrichum genus. Another advantage of the P1/P2 primer pair is that it detects mtDNA, which has a high copy number and is easily amplified. However, the P1/P2 effectiveness has been validated with only relatively few C. gloeosporioides s.l. and C. actutatum s.l. isolates, so it is critical to test its practical efficiency and specificity with more representative isolates in the future.
For recently divergent lineages, gene tree/species tree discordance is a critical factor confounding species inference. One approach to overcome this bias is to improve the number, representativeness, and independence of analyzed genetic markers. Taxonomic studies in C. gloeosporioides s.l. have relied almost entirely on nuclear markers, whereas we have diverged from this pattern by exploring the merit of a mitogenome tree. The length advantage of mitogenome alignment overcomes its drawback of low-level nucleotide diversity. In the present study, the final mitogenome alignment contains 2.6 times more phylogenetically informative sites than that of ApMAT. The alignment dataset produces a very solid tree in high resolution, based on which the relationships of very nascent lineages can be inferred. The merit of complete mitogenome sequences, however, lies not only in the generation of a gene tree in high resolution, but perhaps more importantly in the fact that it provides information about the lineage evolutionary histories based on a factor different from all prevailing markers in use. By comparing these independent markers, one can have a better chance of teasing out phylogenetic signals not related to species divergence, and delimitating nascent species with increased accuracy. The relationship between YT02 and SQ01 analyzed in this study is a striking illustration. Neither phylogeny based on ApMAT nor phylogeny based on the concatenation of ACT, TUB2, GADPH, CAL, CHS markers separate the two isolates. However, if GS and SOD2 markers were further included in the concatenation, the resulting tree would separate the two isolates into different C. siamense s.l. species. Without additional information, one would hesitate in deciding which phylogeny to put trust on. Yet, based on the information that SQ01 and YT02 have almost identical mitogenomes and the information that the mitogenome phylogeny is congruent with all other markers except GS and SOD2, one can confidently postulate that the strong lineage-specific sequence divergences of GS and SOD2 are due to evolutionary events independent of species divergence. Further examination of polymorphism sites did not identify evidence of selection, thereby indicating incomplete lineage sorting, one of the most common reasons causing species tree/gene tree incongruence in nascent species, as a plausible reason. Our study shows that by integrating mitogenome sequence information, we have a better chance of discovering and correcting phylogenetic incongruity among the prevailing nuclear markers, which is a very critical step for the accurate recognition of nascent species. Compared with the mitochondrial genome, the nuclear genome is much larger and contains far more genes. Phylogenomic approaches, particularly ones considering the genome-wide variation of gene tree topologies, will allow one to delimit species boundaries to a much higher degree of confidence and accuracy. Such analysis is within expectation as the cost of whole-genome sequencing keeps dropping.
When applying mtDNA markers for phylogenetic studies, it is important to keep in mind that mtDNA is a cytoplasmic factor, the inherence and population dynamics of which can be quite different from nuclear DNA. The genetic exchange of mtDNA is achieved via either vegetative hyphal cell fusion limited by vegetative compatibility (VC), or via maternal (perithecial parent) inheritance limited by female fertility. The exchange of nuclear DNA, on the other hand, occurs in a biparental direction and is limited by cross fertility. When outcrossing is common, mutations of both nuclear and mtDNA can spread in the population. When outcrossing is rare, the spread of nuclear DNA mutations is restricted, generating genetic isolation. The spread of mtDNA mutations, on the other hand, may still be unaffected if vegetative compatibility is not affected. In C. gloeosporioides s.l., the cross-fertility phenotype is highly variable among isolates and species, whereas the presence and number of VC groups in most species are unknown. Nuclear and mtDNA may also spread among closely-related species through interspecific hybridization or hyphal fusion events. The combined interaction effects of potential intraspecific genetic isolation and interspecific genetic exchange of nuclear as well as cytoplasmic markers are hard to predict, so it is difficult to conclude whether nuclear or mitochondrial markers are more advantageous for species inference in this context. Similarly, phylogenetic trees from the two sources may not necessarily be fully congruent, and both may deviate away from true species relationships, which means that one must stay cautious about relying fully on either one for species inference. On the other hand, conclusions derived from concordant phylogenetic information derived from two sources will be more reliable than from a single source. For nuclear as well as mitochondrial DNA markers, it is also important that an adequate number of isolates is sampled so as that the observed variations approximate species-level changes more closely.
Conclusion
The C. gloeosporioides s.l. mitogenomes characterized in this study are highly conserved in DNA sequence, gene content, gene transcribing direction, and gene order. Many mitogenome features are also conserved at higher taxonomic levels. On the other hand, these mitogenomes collectively have higher intron content and non-homogenous GC content distribution which were not observed with any previously reported Colletotrichum mitogenomes. Despite high sequence similarity, considerable intron content polymorphism exists among C. gloeosporioides s.l. lineages, indicating its potential application as a genetic diversity marker. Phylogenetic analysis demonstrates the usefulness of mitogenome in complementing nuclear genes for C. gloeosporioides s.l. species delimitation. Taken together, the C. gloeosporioides s.l. mitogenomes revealed in this study are important resources for further studies into the biology, genetics, and evolution of this economically important fungal species complex.
Acknowledgements
We would like to thank the anonymous reviewers for their kind and helpful comments on the original manuscript.
Funding
This work was supported by the National Science Foundation of China (31601595), the General Financial Grant from the China Postdoctoral Science Foundation (2016 M592844), the Fundamental Research Funds of Northwest A&F University (Z109021610), and the China Agriculture Research System (CARS-28). The funders had no role in the design of the study or the collection, analysis or interpretation of the data or the writing of the manuscript.
Availability of data and materials
All sequences generated in this study are available in GenBank with the accession numbers listed in Additional file 1: Table S1 and Table S3.
Authors’ contributions
XL, RZ and GS conceived and designed the study. WL, WW, XT, QD, and TW isolated and characterized the fungal isolates. XL, BW and YM analyzed the sequence data. XL wrote the paper. XL, RZ, MLG, and GS revised the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- ACT
Actin
- BI
Bayesian inference
- CAL
Calmodulin
- CHS
Chitin synthase
- CTAB
Cetyl trimethylammonium bromide
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- GS
Glutamine synthetase
- HEGs
Homing endonuclease genes
- ML
Maximum likelihood
- mtDNA
Mitochondrial DNA
- SH test
Shimodaira-Hasegawa test
- SOD2
Manganese-superoxide dismutase
- TUB2
β-tubulin 2
Additional files
A list of isolated fungal strains with details about taxonomy, host, locality, and GenBank accessions of DNA markers. Table S2. A list of PCR primers. Table S3. General features of the Colletotrichum spp. mitogenomes. Table S4. Annotations of the mitogenome core genes (oxidative phosphorylation proteins, rRNAs, tRNAs). Table S5. Genic and intergenic regions GC contents (%) of C. gloeosporioides s.l. and C. actutatum s.l. mitogenomes. Table S6. Mitogenome codon usage frequencies. Table S7. A summary of the mitogenome intron features. (XLSX 55 kb)
SQ01 (C. siamense s.l.) mitogenome gene features and its sequence comparison with that of YT02 (C. siamense s.l.). (PDF 362 kb)
DNA sequence alignment of C. gloeosporioides s.l. mitogenomes at the short cox3 gene coding region (76–218 bp) where ZH03 (C. siamense s.l.) showed strong lineage-specific sequence divergence. (PDF 847 kb)
Primer design strategies for PCR-based identification of C. gloeosporioides s.l.. (PDF 394 kb)
PCR amplification results of primers tested for differentiating C. gloeosporioides s.l. from C. actutatum s.l.. (PDF 1260 kb)
Maximum likelihood phylogenetic trees based on concatenated mitochondrial proteins and concatenated coding DNAs. (PDF 322 kb)
Fifty percent majority rule consensus Bayesian trees based on different gene concatenation schemes. (PDF 475 kb)
DNA polymorphism sites in the GS (glutamine synthetase) and SOD2 (manganese-superoxide dismutase) alignments of C. siamense s.l. isolates. (PDF 464 kb)
Contributor Information
Xiaofei Liang, Email: xiaofeiliang@nwsuaf.edu.cn.
Xianglin Tian, Email: 973552608@qq.com.
Wenkui Liu, Email: 493452583@qq.com.
Tingyu Wei, Email: 281650433@qq.com.
Wei Wang, Email: 575404665@qq.com.
Qiuyue Dong, Email: 1241597952@qq.com.
Bo Wang, Email: 253526973@qq.com.
Yanan Meng, Email: lingbo312@qq.com.
Rong Zhang, Email: rongzh@nwsuaf.edu.cn.
Mark L. Gleason, Email: mgleason@iastate.edu
Guangyu Sun, Email: sgy@nwsuaf.edu.cn.
References
- 1.Weir BS, Johnston PR, Damm U. The Colletotrichum gloeosporioides species complex. Stud Mycol. 2012;73(1):115–80. doi: 10.3114/sim0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyde KD, Cai L, Cannon PF, Crouch JA, Crous PW, Damm U, Goodwin PH, Chen H, Johnston PR, Jones EBG, et al. Colletotrichum – names in current use. Fungal Divers. 2009;39:147–82. [Google Scholar]
- 3.Yakoby N, Zhou R, Kobiler I, Dinoor A, Prusky D. Development of Colletotrichum gloeosporioides restriction enzyme-mediated integration mutants as biocontrol agents against Anthracnose disease in avocado fruits. Phytopathology. 2001;91(2):143–8. doi: 10.1094/PHYTO.2001.91.2.143. [DOI] [PubMed] [Google Scholar]
- 4.Goodwin PH. A molecular weed-mycoherbicide interaction: Colletotrichum gloeosporioides f. sp. malvae and round-leaved mallow, Malva pusilla. Can J of Plant Pathol. 2001;23(1):28–35.
- 5.Sutton BC. The genus Glomerella and its anamorph Colletotrichum. In: Bailey JA, Jeger MJ, editors. Colletotrichum: biology, pathogenicity, and control. Wallingford, UK: CAB International; 1992. p. 1–26.
- 6.Cai L, Hyde KD, Taylor PWJ, Weir B, Waller J, Abang MM, Zhang ZJ, Yang YL, Phoulivong S, Liu ZY, et al. A polyphasic approach for studying Colletotrichum. Fungal Divers. 2009;39:183–204. [Google Scholar]
- 7.Du M, Schardl CL, Nuckles EM, Vaillancourt LJ. Using mating-type gene sequences for improved phylogenetic resolution of Collectotrichum species complexes. Mycologia. 2005;97(3):641–58. doi: 10.3852/mycologia.97.3.641. [DOI] [PubMed] [Google Scholar]
- 8.Sharma G, Kumar N, Weir BS, Hyde KD, Shenoy BD. The ApMat marker can resolve Colletotrichum species: a case study with Mangifera indica. Fungal Divers. 2013;61(1):117–38.
- 9.Silva DN, Talhinhas P, Várzea V, Cai L, Paulo OS, Batista D. Application of the Apn2/MAT locus to improve the systematics of the Colletotrichum gloeosporioides complex: an example from coffee (Coffea spp.) hosts. Mycologia. 2012;104(2):396–409. doi: 10.3852/11-145. [DOI] [PubMed] [Google Scholar]
- 10.Liu F, Weir BS, Damm U, Crous PW, Wang Y, Liu B, Wang M, Zhang M, Cai L. Unravelling Colletotrichum species associated with Camellia: employing ApMat and GS loci to resolve species in the C. gloeosporioides complex. Persoonia. 2015;35:63–86. [DOI] [PMC free article] [PubMed]
- 11.Stewart JE, Timmer LW, Lawrence CB, Pryor BM, Peever TL. Discord between morphological and phylogenetic species boundaries: incomplete lineage sorting and recombination results in fuzzy species boundaries in an asexual fungal pathogen. BMC Evol Biol. 2014;14:38. doi: 10.1186/1471-2148-14-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kubatko LS, Degnan JH. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst Biol. 2007;56(1):17–24. doi: 10.1080/10635150601146041. [DOI] [PubMed] [Google Scholar]
- 13.Degnan JH, Rosenberg NA. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol. 2009;24(6):332–40. doi: 10.1016/j.tree.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Weisrock DW, Smith SD, Chan LM, Biebouw K, Kappeler PM, Yoder AD. Concatenation and concordance in the reconstruction of mouse lemur phylogeny: an empirical demonstration of the effect of allele sampling in phylogenetics. Mol Biol Evol. 2012;29(6):1615–30. doi: 10.1093/molbev/mss008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu F, Wang M, Damm U, Crous PW, Cai L. Species boundaries in plant pathogenic fungi: a Colletotrichum case study. BMC Evol Biol. 2016;16(1):1–14. doi: 10.1186/s12862-016-0649-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knowles LL. Estimating species trees: methods of phylogenetic analysis when there is incongruence across genes. Syst Biol. 2009;58(5):463–7. doi: 10.1093/sysbio/syp061. [DOI] [PubMed] [Google Scholar]
- 17.Sharma G, Pinnaka AK, Shenoy BD. Resolving the Colletotrichum siamense species complex using ApMat marker. Fungal Divers. 2015;71(1):247–64. doi: 10.1007/s13225-014-0312-7. [DOI] [Google Scholar]
- 18.Kang D, Hamasaki N. Maintenance of mitochondrial DNA integrity: repair and degradation. Curr Genet. 2002;41(5):311–22. doi: 10.1007/s00294-002-0312-0. [DOI] [PubMed] [Google Scholar]
- 19.Slack KE, Delsuc F, McLenachan PA, Arnason U, Penny D. Resolving the root of the avian mitogenomic tree by breaking up long branches. Mol Phylogenet Evol. 2007;42(1):1–13. doi: 10.1016/j.ympev.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Poortvliet M, Olsen JL, Croll DA, Bernardi G, Newton K, Kollias S, O’Sullivan J, Fernando D, Stevens G, Galván Magaña F, et al. A dated molecular phylogeny of manta and devil rays (Mobulidae) based on mitogenome and nuclear sequences. Mol Phylogenet Evol. 2015;83:72–85. doi: 10.1016/j.ympev.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Shen X-Y, Li T, Chen S, Fan L, Gao J, Hou C-L. Characterization and phylogenetic analysis of the mitochondrial genome of Shiraia bambusicola reveals special features in the order of Pleosporales. PLoS One. 2015;10(3):e0116466. doi: 10.1371/journal.pone.0116466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fourie G, van der Merwe NA, Wingfield BD, Bogale M, Tudzynski B, Wingfield MJ, Steenkamp ET. Evidence for inter-specific recombination among the mitochondrial genomes of Fusarium species in the Gibberella fujikuroi complex. BMC Genomics. 2013;14(1):1–14. doi: 10.1186/1471-2164-14-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tai TH, Tanksley SD. A rapid and inexpensive method for isolation of total DNA from dehydrated plant tissue. Plant Mol Biol Report. 1990;8(4):297–303. doi: 10.1007/BF02668766. [DOI] [Google Scholar]
- 24.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol İ. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117–23. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33(Suppl 2):W686–9. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6) doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism. Bioinformatics. 2009;25(11):1451–2. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 28.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaidya G, Lohman DJ, Meier R. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics. 2011;27(2):171–80. doi: 10.1111/j.1096-0031.2010.00329.x. [DOI] [PubMed] [Google Scholar]
- 30.Nylander JAA. MrModeltest v2. Program distributed by the author: Evolutionary Biology Centre, Uppsala University; 2004. [Google Scholar]
- 31.Ronquist F, Teslenko M, Mark P, Ayres DL, Darling A, Höhna S. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–5. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–52. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 34.Yang YL, Liu ZY, Cai L, Hyde KD, Yu ZN, McKenzie EHC. Colletotrichum anthracnose of Amaryllidaceae. Fungal Divers. 2009;39:123–46. [Google Scholar]
- 35.Wikee S, Cai L, Pairin N, McKenzie EHC, Su YY, Chukeatirote E. Colletotrichum species from Jasmine (Jasminum sambac) Fungal Divers. 2011;46(1):171–82. doi: 10.1007/s13225-010-0049-x. [DOI] [Google Scholar]
- 36.Grum-Grzhimaylo AA, Debets AJ, van Diepeningen AD, Georgieva ML, Bilanenko EN. Sodiomyces alkalinus, a new holomorphic alkaliphilic ascomycete within the Plectosphaerellaceae. Persoonia. 2013;31:147–58. doi: 10.3767/003158513X673080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Réblová M, Gams W, Seifert KA. Monilochaetes and allied genera of the Glomerellales, and a reconsideration of families in the Microascales. Stud Mycol. 2011;68:163–91. doi: 10.3114/sim.2011.68.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith D. Updating our view of organelle genome nucleotide landscape. Front Genet. 2012;3:175. doi: 10.3389/fgene.2012.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barth D, Berendonk TU. The mitochondrial genome sequence of the ciliate Paramecium caudatum reveals a shift in nucleotide composition and codon usage within the genus Paramecium. BMC Genomics. 2011;12(1):1–11. doi: 10.1186/1471-2164-12-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith DR, Lee RW. Mitochondrial genome of the colorless green alga Polytomella capuana: a linear molecule with an unprecedented GC Content. Mol Biol Evol. 2008;25(3):487–96. doi: 10.1093/molbev/msm245. [DOI] [PubMed] [Google Scholar]
- 41.Nishio Y, Nakamura Y, Kawarabayasi Y, Usuda Y, Kimura E, Sugimoto S, Matsui K, Yamagishi A, Kikuchi H, Ikeo K, et al. Comparative complete genome sequence analysis of the amino acid replacements responsible for the thermostability of Corynebacterium efficiens. Genome Res. 2003;13(7):1572–9. doi: 10.1101/gr.1285603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Musto H, Naya H, Zavala A, Romero H, Alvarez-Valin F, Bernardi G. Genomic GC level, optimal growth temperature, and genome size in prokaryotes. Biochem Biophys Res Commun. 2006;347(1):1–3. doi: 10.1016/j.bbrc.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 43.Šmarda P, Bureš P, Horová L, Leitch IJ, Mucina L, Pacini E, Tichý L, Grulich V, Rotreklová O. Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc Natl Acad Sci U S A. 2014;111(39):E4096–102. doi: 10.1073/pnas.1321152111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Reedy RM, Malireddy R, Dillman CB, Kennell JC. Comparative analysis of Fusarium mitochondrial genomes reveals a highly variable region that encodes an exceptionally large open reading frame. Fungal Genet Biol. 2012;49(1):2–14. doi: 10.1016/j.fgb.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Zhang S, Zhang G, Liu X, Wang C, Xu J. Comparison of mitochondrial genomes provides insights into intron dynamics and evolution in the caterpillar fungus Cordyceps militaris. Fungal Genet Biol. 2015;77:95–107. doi: 10.1016/j.fgb.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 46.Formey D, Molès M, Haouy A, Savelli B, Bouchez O, Bécard G, Roux C. Comparative analysis of mitochondrial genomes of Rhizophagus irregularis – syn. Glomus irregulare – reveals a polymorphism induced by variability generating elements. New Phytol. 2012;196(4):1217–27. doi: 10.1111/j.1469-8137.2012.04283.x. [DOI] [PubMed] [Google Scholar]
- 47.Torriani SFF, Penselin D, Knogge W, Felder M, Taudien S, Platzer M, McDonald BA, Brunner PC. Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet Biol. 2014;62:34–42. doi: 10.1016/j.fgb.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 48.Joardar V, Abrams NF, Hostetler J, Paukstelis PJ, Pakala S, Pakala SB, Zafar N, Abolude OO, Payne G, Andrianopoulos A, et al. Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genomics. 2012;13(1):1–12. doi: 10.1186/1471-2164-13-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Damm U, Cannon PF, Woudenberg JHC, Johnston PR, Weir BS, Tan YP, Shivas RG, Crous PW. The Colletotrichum boninense species complex. Stud Mycol. 2012;73(1):1–36. doi: 10.3114/sim0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All sequences generated in this study are available in GenBank with the accession numbers listed in Additional file 1: Table S1 and Table S3.