

Abstract
The nucleotide excision repair protein complex ERCC1-XPF is required for incision of DNA upstream of DNA damage. Functional studies have provided insights into the binding of ERCC1-XPF to various DNA substrates. However, because no structure for the ERCC1-XPF-DNA complex has been determined, the mechanism of substrate recognition remains elusive. Here we biochemically characterize the substrate preferences of the helix-hairpin-helix (HhH) domains of XPF and ERCC-XPF and show that the binding to single-stranded DNA (ssDNA)/dsDNA junctions is dependent on joint binding to the DNA binding domain of ERCC1 and XPF. We reveal that the homodimeric XPF is able to bind various ssDNA sequences but with a clear preference for guanine-containing substrates. NMR titration experiments and in vitro DNA binding assays also show that, within the heterodimeric ERCC1-XPF complex, XPF specifically recognizes ssDNA. On the other hand, the HhH domain of ERCC1 preferentially binds dsDNA through the hairpin region. The two separate non-overlapping DNA binding domains in the ERCC1-XPF heterodimer jointly bind to an ssDNA/dsDNA substrate and, thereby, at least partially dictate the incision position during damage removal. Based on structural models, NMR titrations, DNA-binding studies, site-directed mutagenesis, charge distribution, and sequence conservation, we propose that the HhH domain of ERCC1 binds to dsDNA upstream of the damage, and XPF binds to the non-damaged strand within a repair bubble.
Keywords: DNA repair, DNA-protein interaction, NMR, nucleotide excision repair, structure-function, ERCC1-XPF, helix-hairpin-helix domain, XPF homodimer, single-stranded DNA binding
Introduction
To survive, cells require the ability to repair a plethora of DNA lesions. Therefore, cells contain several DNA repair mechanisms, including the versatile nucleotide excision repair (NER)2 pathway, a conserved DNA repair machinery that can remove a wide variety of DNA lesions (1, 2). Within a mammalian cell, 25–30 proteins are known to participate in two NER pathways: global genome and transcription coupled repair (3–5). Mutations in NER genes lead to impaired DNA repair. Presently, a dozen mutations in distinct NER genes have been identified in patients with eight overlapping phenotypes (6, 7). Most patients carrying a mutation in NER genes develop two distinct symptoms: sunlight-induced skin cancer and segmental progeria without cancer (8, 9).
ERCC1 and XPF form a stable heterodimeric complex that is essential for NER and functions as a structure-specific DNA endonuclease that is able to perform an incision 5′ to the DNA damage (10–13). Mutations in the ERCC1 and XPF genes can be linked to sunlight-induced skin abnormalities, late onset of skin cancers, neurodegeneration, and premature aging in both human patients and mice (7–9, 14). In the absence of ERCC1, only a marginal amount of XPF is present in fibroblasts and CHO cells (11, 13, 15–18). This suggests that the in vivo stability of full-length ERCC1-XPF depends on tight association between the two proteins. Consistent with this finding, XPF and ERCC1 knockout mice exhibit similar phenotypes (19–21). Furthermore, postnatal phenotypes of XPF and ERCC1 knockout mice suggest additional functions for ERCC1-XPF in double strand break repair (22), single strand annealing (23), interstrand cross-link repair (24, 25), telomere maintenance (26, 27), and gene-targeting events (28). All of these genome regulatory processes require binding of ERCC1-XPF at distinct DNA sequences, involving various protein complexes (29–31).
Biochemical and structural studies revealed that the helix-hairpin-helix (HhH) domain present in the C-terminal part of both proteins is essential for both ERCC1-XPF complex formation (11) and DNA binding (32–34). Structural studies by us and others showed that the HhH domain of the XPF protein serves as a scaffold for the correct folding of ERCC1, permitting formation of a stable heterodimer (34–36). This is further emphasized by the reduced stability of the ERCC1 (F231L)-XPF complex (37), a mutation that leads to severe DNA repair defects and death in early infancy (38–40).
A model for the binding of ERCC1 to a repair bubble was proposed before, where both the HhH domains of ERCC1 and XPF bind the ssDNA sequence (36). However, using NMR spectroscopy, we found that ERCC1 specifically recognizes dsDNA, probably through the hairpin sequences of the HhH domain of ERCC1 (34). Furthermore, the C-terminal ERCC1-XPF complex binds more tightly to ssDNA-dsDNA junctions, such as bubble and splayed arm substrates, than to either dsDNA or ssDNA alone (34). Previously, this led us to suggest that XPF might also contain an independent DNA-binding domain. We took advantage of an earlier observation that demonstrated that the isolated HhH domain of XPF is able to form a highly stable homodimer (41). Although XPF lacks one residue in the second hairpin motif, this domain adopts a canonical HhH domain structure (41). Using NMR, we showed that the homodimeric XPF HhH domain indeed binds ssDNA. Subsequently we determined the solution structure of XPF bound to ssDNA (42). We could show that besides nonspecific phosphate backbone contacts involving the second helix of the first HhH motif, a cavity is formed between the two motifs of the HhH domain, where a guanine base can be bound. These observations led us to propose that, in contrast to the model proposed by Tsodikov et al. (36), the ERCC1-XPF heterodimer recognizes DNA substrates involving the two individual DNA-binding surfaces present in ERCC1 and XPF that preferentially bind dsDNA and ssDNA, respectively (34).
Here we confirm and extend this model using in vitro DNA binding assays and NMR titration experiments, demonstrating the substrate preference of XPF and the ERCC1-XPF heterodimer for various DNA sequences. Based on these findings, we propose a model for the binding of the HhH domains of ERCC1-XPF heterodimers to DNA. In this model, the concerted binding of the HhH domains of ERCC1 and XPF to dsDNA and ssDNA, respectively, is essential for the correct positioning on the ssDNA/dsDNA junction.
Results
De Laat et al. (11) have shown that the C-terminal HhH domains of XPF and ERCC1 are indispensable for heterodimer formation and function. Similar to full-length ERCC1-XPF heterodimers, these HhH domains can together form stable complexes with various ss/ds junction-containing DNA, like bubble, hairpin, and splayed arm substrates (32–34). These findings suggest that structure-specific DNA binding by the ERCC1-XPF heterodimer is dependent on the HhH domain regions of both proteins. The ability of XPF to bind to ssDNA further supports this model (42). To elucidate the contribution of the HhH domain of XPF in ERCC1-XPF substrate preference, we first determine the binding preference of homodimeric XPF HhH domain to a variety of DNA substrates, as shown in supplemental Fig. S1.
We tested the binding of the XPF HhH domain homodimer to bubble10 (B10) because the ERCC1-XPF heterodimer can form a stable complex with this DNA sequence, as shown previously (34). Surprisingly, we noticed that the XPF homodimer binds to this substrate with even higher affinity (Fig. 1, A and B) than the ERCC1-XPF heterodimer (34). Quantification revealed an apparent KD of 0.5 ± 0.1 μm (Fig. 1B), which means more than 1 order of magnitude tighter binding than found earlier for the ERCC1-XPF heterodimer (34). It should be mentioned, however, that the ERCC1-XPF complex dissociates during electrophoresis, as is clear from the smear observed at the highest protein concentration, suggesting a faster on and off rate for the ERCC1-XPF complex for this substrate. If binding affinity would be determined based on the disappearance of free DNA, both complexes would bind with micromolar affinities. By performing binding experiments in lower-salt buffer (data not shown) or using agarose gels instead of polyacrylamide gel, dissociation is reduced significantly (Fig. 6). Also, under these conditions, the XPF HhH domain homodimer binds B10 DNA more tightly than ERCC1-XPF.
FIGURE 1.

Binding of the HhH domain of XPF to B10 substrate. A, electrophoretic mobility shift assay showing the binding of 0.1, 0.3, 1.0, 3.3, and 10 μm of XPF (homodimer concentration, top panel, left) or ERCC1-XPF (top panel, right) to 0.02 μm radiolabeled B10 substrate. Free DNA (F) and protein-DNA complex (C) are indicated. Bottom panel, SDS-PAGE of the protein samples used for the DNA binding experiments. B, quantification of a few representative DNA binding experiments (each experiment with different symbol type) of XPF and ERCC1-XPF (E/X) bound to the B10 substrate. The fraction bound is plotted as a function of the protein concentrations. Data were fitted as described before (34), and a simulated binding curve with the dissociation constants obtained was plotted. C, fraction of the complex formed on B10 bound by 1.25 μm XPF in the presence of 0.08, 0.12, 0.18, 0.26, 0.40, 0.59, 0.89, 1.33, and 2.0 μm non-labeled B10 oligonucleotide relative to the binding in the absence (0) of competitor. The inset shows the autoradiogram of the corresponding competition experiment. D, binding of the HhH domain of XPF to single/double-stranded DNA junctions. Shown are a representative binding experiment (top panel) and quantification of the competition experiments (bottom panel). Approximately 1 μm XPF in the absence (−) or presence of the indicated non-labeled probes (0.2 or 2 μm, supplemental Fig. S1) is bound to 0.02 μm B10 substrate. The relative competition efficiency is determined by quantification of the fraction bound in the presence of competitor. The ability to compete for B10 binding is compared with the competition obtained with non-labeled B10 substrate as shown in C. F, splayed arm. Using non-linear regression methods, this curve was fitted, and the competition obtained in the presence of the amount of heterologous probe was compared with the amount of B10 probe required to obtain the same inhibition of binding. For instance, a 10-fold relative competition efficiency means that 10 times more probe is required to obtain the same inhibition of binding by a given concentration of B10 competitor. If no competition is obtained at the highest amount of competitor, relative competition efficiency was estimated and is shown as being at least 500-fold less efficient. Mean ± S.D. of four independent experiments is presented.
FIGURE 6.

HhH domain surface residues affect binding of ERCC1-XPF to ss/dsDNA junctions. A, SDS-PAGE showing the ERCC1-XPF mutants used for binding experiments. B, binding of the indicated amounts of ERCC1 and XPF mutant proteins (micromolar) to B10 DNA, separated on an agarose gel. The ERCC1-XPF-DNA complex appears as a doublet, which might be due to the presence of two ss/ds junction binding sites in the B10 probe. F, free DNA; C, protein-DNA complex. C, quantification of the binding affinity of the indicated ERCC1 (red) and XPF (blue) mutants based on at least three independent experiments, calculated as mean apparent binding affinity ± S.D. relative to the binding found for wild-type ERCC1-XPF. D, binding curves for a few mutants obtained by plotting the simulated binding curve based on the calculated apparent dissociation constants, based on three independent binding experiments as shown by the indicated symbols.
The Homodimeric HhH Domain of XPF Binds Synergistically to ssDNA
The binding preferences of XPF for various probes were evaluated using competition experiments where an excess of the non-radiolabeled B10 oligonucleotide is added to the reaction mixture. As shown in Fig. 1C, binding of XPF to radiolabeled B10 is competed by a non-labeled oligonucleotide, as shown by the expected exponential dissociation curve. The affinity of XPF for various DNA substrates can be determined by comparing the ability of various probes to compete for the binding of XPF to the B10 substrate.
Using these competition assays, we find that XPF is unable to bind to 10-bp dsDNA or short ssDNA probes (10 or 20 nucleotides). On the other hand, probes containing single/double strand junctions or longer ssDNA (39 nt) or dsDNA (30 bp) fragments are found to have binding affinity for the XPF homodimer (Fig. 1D). Interestingly, for the bubble and hairpin substrates, the length of the ssDNA stretch influences DNA binding. Fig. 1D describes that both hairpin 20 (H20) and B10 are better substrates than probes containing either longer or shorter ssDNA stretches. Also, a splayed arm with two ssDNA sequences is a better XPF substrate than any DNA sequence containing one ssDNA strand (data not shown). Taken together, these data show that the XPF HhH domain homodimer binds to ssDNA. We argue that stable complex formation involves both DNA binding surfaces of the symmetric XPF dimer that can bind simultaneously to either one long ssDNA fragment or to a conformationally restricted DNA containing two ssDNA stretches. In contrast, weaker binding is observed for short ssDNA sequences and hairpin or bubble substrates with shorter ssDNA sequences. These short sequences may occupy only one binding site of the XPF homodimer. These data support the idea that the synergistic binding of an ssDNA fragment to the two DNA binding surfaces is required for high-affinity DNA binding by XPF homodimers.
Preference of the Homodimeric HhH Domain of XPF for G-rich ssDNA
To determine whether XPF possesses sequence preference in ssDNA-binding experiments, competition experiments were performed using 20-nt homopolymeric ssDNA substrates with a 39-nt ssDNA probe. XPF shows strongest binding for poly(dG) substrates, competing as effectively as the larger 39-nt ssDNA substrate (Fig. 2, A and B), whereas poly(dT) and poly(dC) compete 10-fold less effectively. It is remarkable that the purine poly(dG) binds well to XPF, whereas the poly(dA) binds at least 2 orders of magnitude less efficiently. Because XPF prefers binding to a poly(G) sequence, we hypothesized that XPF might recognize the telomeric hexanucleotide repeat sequence TTAGGG. However, competition experiments indicate that XPF does not bind specifically to telomere sequences (data not shown).
FIGURE 2.

The HhH domain of XPF binds preferentially to guanine-rich substrate. A, competition of XPF binding to ss39 in the absence (−) or presence of 0.02, 0.2, 2, and 20 μm dA20, dT20, dG20, and dC20. (competitor DNA). F, free DNA; C, protein-DNA complex. B, quantification of a representative competition experiment as described in A.
Two Independent DNA Binding Surfaces Contribute to Substrate Recognition by the Heterodimeric ERCC1-XPF Complex
Previously we found that the ERCC1-XPF heterodimer has preference for a ss/dsDNA junction-containing substrate (34). Combined with the finding that homodimeric XPF binds preferentially to single-stranded DNA (Fig. 2), this suggests that the binding preference of XPF might also contribute to substrate recognition in the heterodimeric complex. We therefore performed SPR experiments with various DNA substrates (Fig. 3A). Addition of DNA prevented the binding of the His-tagged ERCC1-XPF to the Ni2+-loaded NTA surface of the SPR chip. By fitting response values corresponding to bound ERCC1-XPF against the concentration of these DNA substrates, we find that ERCC1-XPF can bind to ssDNA and dsDNA with a KD of 0.8 and 2 μm, respectively. In agreement with earlier observations, the ss/dsDNA substrate has a 10-fold higher affinity for ERCC1-XPF than dsDNA of equal length. The higher affinity for splayed arm substrates underscores the importance of ss/ds junction-containing substrates for high-affinity binding by the ERCC1-XPF complex
FIGURE 3.

Two independent DNA binding surfaces contribute to substrate recognition by the heterodimeric ERCC1-XPF complex. A, SPR experiment showing the KDapp of the ERCC1-XPF HhH domain for the indicated DNA fragments. To calculate the KDapp, the response values at the end of loading (R60) were divided by the R60 value of the ERCC1-XPF HhH domain in the absence of DNA and plotted against the total concentration of the DNA and fitted considering a 1:1 binding model. B, SPR experiment showing the KDapp of the ERCC1-XPF HhH domain for dA10, dT10, dG10, and dC10. Experimental sensorgrams are shown in supplemental Fig. S4.
Using SPR, we further found similar binding preferences for guanine-rich DNA fragments to the ERCC1-XPF heterodimeric complex as for homodimeric XPF. The poly-dG10 fragment binds to the heterodimer with a KD of 2.5 ± 0.4 μm, whereas the poly-dT10 has a KD of 63 ± 14 μm. (Fig. 3B and supplemental Fig. S4). The binding affinity of dC10 and dA10 for the ERCC1-XPF heterodimer was about 2- and 4-fold lower than that of dT10. These findings demonstrate that the XPF ssDNA binding surface is relevant for high-affinity DNA binding in the ERCC1-XPF complex.
Determination of the ssDNA Binding Surface in XPF
Next we performed NMR titration experiments to determine the DNA binding surfaces in XPF using a 10-nt ssDNA sequence. Significant chemical shift changes for the 10-nt ssDNA sequences were only obtained under low-salt conditions (<50 mm NaCl), in agreement with the higher affinity observed in in vitro DNA binding studies under these conditions (data not shown). Note that, in agreement with the determined weak binding, especially for the ssDNA and dsDNA substrate, saturation could not be reached, as is clear from the significantly lower chemical shift perturbations for these probes in comparison with the splayed arm substrate.
We first compared the earlier determined amide chemical shift changes upon addition of ssDNA to homodimeric XPF (supplemental Fig. S2) with the CSPs for the ERCC1-XPF complex. We found that a similar surface of XPF in the heterodimeric complex is affected by addition of the 10-nt ssDNA sequence involving helix β and the following loop. Importantly, the DNA-binding surface of ERCC1 determined previously (supplemental Fig. S2) was not affected by addition of ssDNA (Fig. 4 and supplemental Fig. S3). In addition to amide proton chemical shift changes in the 15N-1H HSQC spectra of ERCC1-XPF, the 31P NMR spectrum of the ssDNA also reveals significant chemical shift changes upon addition of ERCC1-XPF, demonstrating complex formation (data not shown). The importance of the determined ssDNA binding surface of XPF for DNA binding by the ERCC1-XPF complex was further demonstrated by the 3- and 2-fold decreases in binding affinity upon mutation of His857 and Lys860 to alanine (see below). These experiments clearly demonstrate the ability of XPF to bind ssDNA in the heterodimeric ERCC1-XPF complex and show the importance of this ssDNA binding surface of XPF for substrate recognition.
FIGURE 4.

Separate DNA-binding surfaces in ERCC1 and XPF for dsDNA and ssDNA, respectively. A, representative 15N-1H HSQC spectra of affected residues of the ERCC1-XPF complex showing distinct chemical shift changes upon addition of various DNA sequences (supplemental Table S1). Free ERCC1-XPF spectra are shown in black, and spectra in the presence of a 4-fold excess of DNA are shown in red. For this experiment, the indicated DNA fragments (320 μm) were added to 80 μm ERCC1-XPF in a buffer containing 5 mm phosphate buffer and 100 mm NaCl. B, the determined mean CSP ± S.D. of three to five independent titration experiments (supplemental Fig. S3) is plotted on the surface of the ERCC1-XPF structure in two different views rotated by 200°. All residues (∼25) that were significantly affected (composite average chemical shift >0.2 ppm) were colored. The most affected residues are shown in red (>0.8 ppm), and the other residues are colored relatively to this maximum chemical shift in red shades. Missing or unambiguous residues are depicted in gray. The position of the most affected residues is labeled on the surface.
The HhH Domain of ERCC1 Specifically Recognizes dsDNA
The above results (Figs. 1–4) suggest that ERCC1 and XPF have complementary roles in, respectively, dsDNA and ssDNA recognition that could dictate the high selectivity of ERCC1-XPF in binding ss/dsDNA junction substrates. To test this hypothesis, we performed NMR titration experiments with dsDNA (10 or 20 bp) and with various splayed arm probes containing the 10-nt ssDNA sequence that was used to determine the XPF-ssDNA structure. The binding surfaces for these probes were determined by following the chemical shift changes upon addition of DNA under various salt conditions (supplemental Table 1). The results of these experiments are summarized in Fig. 4, showing a representative set of 15N-1H HSQC spectra for a few of the most affected residues (Fig. 4A). By calculating the average of three to five independent titration experiments using various DNA sequences (supplemental Fig. S3 and Table S1), the most affected residues were identified and plotted on the surface of the ERCC1-XPF structure (Fig. 4B).
Importantly, upon addition of dsDNA, chemical shift changes were observed on the ERCC1 surface, whereas the XPF surface remained mostly unaffected. Mainly residues located in the second hairpin of ERCC1 show pronounced shifts, whereas a few residues from the first hairpin and surrounding helices are somewhat affected (Fig. 4 and supplemental Fig. S3). The established dsDNA-binding surface of ERCC1 is similar to that found before (34) using hairpin DNA (supplemental Fig. S2). NMR studies using the splayed arm showed that, in addition to the dsDNA-binding surface of the ERCC1 protein, residues in XPF are also affected by the addition of this ssDNA-containing sequence. The most pronounced shifts in ERCC1 were found in the second hairpin regions, including Gly276 and Gly278, whereas the first hairpin region encompassing Lys243-Thr248 was affected to a lesser extent. For XPF, the regions 832–833 and 852–859 were mostly affected by the addition of splayed arm DNA. These experiments clearly establish that the hairpin regions of ERCC1 are involved in dsDNA binding, whereas the previously determined ssDNA binding surface of XPF is also involved in ssDNA binding in the heterodimeric complex.
To independently show that isolated ERCC1 can bind dsDNA, we took advantage of a recent observation revealing that the ERCC1-XPF complex dissociates during SPR experiments, leaving ERCC1 bound to the chip (37). Subsequent addition of 30-nt ssDNA (6.25 μm) to immobilized ERCC1 did not lead to an appreciable change in mass, whereas addition of the same concentration of the 30-nt dsDNA led to a significant increase in signal (Fig. 5). The on and off rates for ds30 binding to immobilized ERCC1 were determined to be 6 ± 3 × 103 m−1s−1 and 5.5 ± 0.1 × 10−2 s−1, respectively, giving a KD of 9 ± 3 μm. This relatively low binding affinity agrees well with the observed binding affinities of the ERCC1-XPF heterodimer for dsDNA in EMSA (34), NMR (Fig. 4), and SPR experiments (Fig. 3B). Taken together, these results indicate that the two independent DNA-binding surfaces present in ERCC1 and XPF together contribute to both substrate specificity and overall binding affinity of the complex.
FIGURE 5.
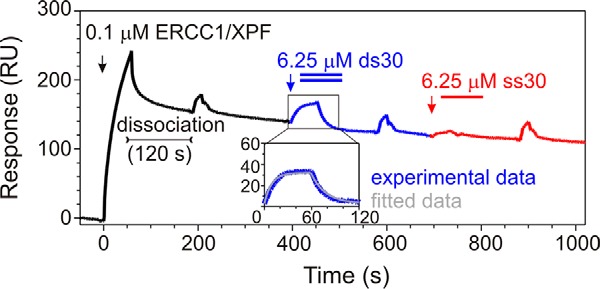
Substrate preference of ERCC1 for dsDNA established by SPR. Shown is the experimental curve (response units(RU)) of subsequential loading (black arrow) of 0.1 μm ERCC1-XPF complex to the Ni2+-loaded NTA SPR chip, followed by XPF dissociation, loading (blue arrow), and dissociation of ds30 probe and (red arrow) loading of the ss30 probe. From the ds30 experimental curve, the ds30 nonspecific binding to the chip surface was subtracted, fitted according to the Langmuir 1:1 binding model, and shown as an inset.
XPF Has a Preference for the Non-damaged Strand
Although the presence of two independent DNA-binding surfaces, which are probably occupied concurrently, permits positioning of the ERCC1-XPF heterodimer on ss/dsDNA junctions, it does not provide an explanation for the polarity of the cleavage. The binding preference of XPF could define this polarity by preferentially recognizing either the non-damaged (5′ overhang) or the damaged (3′ overhang) single strand.
We performed NMR titrations using a 10- or 20-bp stem substrate with the ssDNA sequence connected to either the 3′or 5′ of the stem. Both probes caused chemical shift perturbations in both ERCC1 and XPF upon binding of DNA irrespective of salt concentration or stem length. These changes mainly involve the two abovementioned ssDNA- and dsDNA-binding surfaces (Fig. 4). In agreement with the binding preference found in SPR experiments (Fig. 3B), the 5′ overhang splayed arm gives slightly more pronounced chemical shift changes than the 3′ overhang substrate. Although the overall CSPs were similar, the residues that were significantly affected are not identical, arguing that the two probes bind in a different way. In comparison with the splayed arm sequence, the DNA fragment containing the 5′ ssDNA sequence binds to the ssDNA- and the dsDNA-binding surfaces in a highly similar way. Also, residues outside of these main binding surfaces show similar CSPs (Fig. 4). This indicates that most CSPs for the splayed arm substrate come from binding to the 5′ ssDNA extension, suggesting that the non-damaged strand is the preferred substrate for XPF.
Mutation of the ERCC1 and XPF DNA Binding Interfaces Decreases the Binding Affinity
The full-length ERCC1-XPF complex processes ss/dsDNA junctions with high selectivity (32, 36, 43). Above, we described two DNA binding surfaces for XPF and ERCC1 proteins and explained the distinct roles of the XPF and ERCC1 helix-hairpin-helix domains in ss/dsDNA junction recognition. To validate this model, we mutated residues that could be in contact with DNA, as they show large chemical shift perturbations in DNA titrations (Fig. 4) or as they are located in between the two DNA binding domains that could therefore possibly affect the ability to bind the DNA substrate. Proteins were expressed and purified and concentrations normalized by SDS-PAGE. Despite the seemingly higher abundance of XPF, attributed to staining efficiency differences, no XPF HhH domain homodimer-DNA complex was detectable in the binding studies. The binding experiments were performed using an agarose gel instead of a polyacrylamide gel, which, in contrast to the results presented before (Fig. 1 and Ref. 34), yields a more stable complex that enables us to determine an apparent dissociation constant of ∼1 μm for wild-type ERCC1-XPF under these conditions. For the XPF mutants K860A and H857A, residues that are in direct contact with ssDNA (42), the binding to B10 was significantly reduced compared with wild-type ERCC1-XPF (Fig. 6). However, mutation of the hydrophobic residue I876A did not affect binding significantly. Mutations outside the ssDNA-binding surface (N834A, K860A, and D871A) cause only a small decrease in affinity, whereas a 2-fold decrease in binding was noted for the double mutant Q838A and D839A.
Mutation of the positively charged ERCC1 residues (K247E, R283E, and R284E) surrounding the hairpin residues that show the largest CSPs led to highly reduced affinities (Fig. 6). Mutation of the ERCC1 residues Glu261 and Gln262, both located outside of the dsDNA-binding surface, led to a much smaller 2-fold decrease in substrate binding.
Together, these experiments indicate the presence of two independent, functionally distinct DNA-binding surfaces in ERCC1 and XPF that both contribute to specificity and binding affinity. Mutation of residues that, in our model, are in contact with DNA significantly affected the ability of the ERCC1-XPF complex to bind ss/dsDNA substrates, underscoring the importance of these residues in substrate recognition.
Discussion
DNA damage removal requires correct positioning of the ERCC1-XPF complex with respect to the lesion. We determined the substrate preference for the HhH domains of ERCC1 and XPF. We show that XPF binds ssDNA in a non-sequence specific fashion but with a preference for substrates containing guanine-rich sequences (Figs. 2 and 3). NMR titrations revealed that XPF, irrespective whether present as a homo- or heterodimeric complex, binds ssDNA sequences using the same surface (Fig. 4). Importantly, ssDNA probes do not bind to the dsDNA-binding surface of HhH domains. By using various splayed arm probes, we show that the two nucleic acid binding surfaces of ERCC1 and XPF within the heterodimeric protein can both be bound concurrently using approximately the same interaction surfaces as for their preferred substrates (Fig. 4). Kinetic experiments and site-directed mutagenesis support the view that the two separate binding surfaces are required for both specificity and binding affinity.
ss/dsDNA Junction Recognition by the HhH Domains of ERCC1-XPF
Tsodikov et al. (36) suggest that both HhH domains of ERCC1 and XPF contain ssDNA binding surfaces and that each specifically binds to one of the two arms of the DNA substrate. Our NMR titration studies using hairpin 22 (34), ssDNA (42), dsDNA, and splayed arm substrates (Fig. 4 and supplemental Fig. S3) argue against such a model. We show that ssDNA is preferentially bound by XPF and not by ERCC1, whereas dsDNA substrates are specifically recognized by ERCC1 (Fig. 4). ss/dsDNA-containing probes make contact with both the XPF ssDNA-binding surface and the ERCC1 dsDNA-binding surface (Fig. 4). The importance of these separate DNA binding domains for recognition of ss/dsDNA sequences was confirmed by mutagenesis (Fig. 6).
The crystal structures of the Aeropyrum pernix XPF bound to DNA provide detailed insights into incision by XPF in archaea bacteria (44). Extrapolation of this structural information to eukaryotic repair factors is complicated, considering the distinct subunit composition and the different substrate specificities of both complexes (32, 44). Therefore, the question of how the eukaryotic ERCC1-XPF complex recognizes ss/dsDNA junctions remains to be answered. Our previous DNA-binding studies revealed that the HhH domains of ERCC1-XPF show a similar substrate specificity as the native complex, suggesting that the HhH domain region is required and sufficient for substrate recognition (34). Using site-directed mutagenesis (Fig. 6) and NMR spectroscopy (Fig. 4), we demonstrated that a dsDNA-binding site is located near the tip of the two hairpin structures in ERCC1. This conserved DNA-binding surface is similar to other HhH domain proteins (45, 46), including archaeal XPF (44). For the archaeal XPF homodimer, it was proposed that the HhH domains of the two protomers bind the two dsDNA sequences of a flap substrate (44). The structural homology suggests functional similarity, supporting the notion that ERCC1 recognizes dsDNA. Using the proposed model based on the A. pernix XPF (44), and knowing the polarity of the ERCC1-XPF heterodimer with respect to the damage, it can be expected that ERCC1 binds to the upstream dsDNA sequence, placing the catalytic domain of XPF in close proximity to cleave the damaged DNA strand (44, 47). This does not exclude that other regions of ERCC1-XPF or other repair proteins can further substantiate substrate specificity. Indeed, we and others noted that the central domain of ERCC1 also contains an ssDNA binding surface (47, 48). In addition, XPA and repair protein A (RPA), which bind to ssDNA and also interact with, respectively, ERCC1 (48) and XPF (49), can contribute to the correct positioning of the ERCC1-XPF complex near the damaged DNA. The presence of multiple weak DNA-binding surfaces within this DNA repair complex facilitates the correct positioning of the nuclease domain with respect to the damage and prevents inappropriate DNA binding and incision. Additional support for this model comes from a recent study by Su et al. (50) that shows that mutations of the individual DNA binding domains in full-length ERCC1 and XPF lead to a decrease in cleavage efficiency both in vitro and in vivo.
The XPF HhH Domain Preferentially Binds to the Non-damaged Strand
To determine which ssDNA strand within the repair bubble is bound by XPF, we modeled the ssDNA sequence into the ERCC1-XPF heterodimer structure based on the previously determined solution structure of homodimeric XPF bound to ssDNA (42). The dsDNA is positioned based on homology with the archaeal XPF-DNA structure (44, 47). Assuming that the proposed models for dsDNA and ssDNA binding to ERCC1-XPF are correct, the gap between the dsDNA and the ssDNA can be filled by connecting the dsDNA fragment to either the 5′ or 3′ end of the ssDNA. As a result, in this model, XPF will bind to the damaged or the non-damaged strand, respectively.
If we assume that XPF binds the non-damaged strand (5′ extension), the 3′ end of the ssDNA would be connected to the dsDNA. Chemical shift changes for several of the residues in between these DNA binding interfaces were observed upon addition of a splayed arm substrate (e.g. Lys243, Met856). Also, the significant decrease in DNA binding by the Q838A/D839A mutant argues that this part of the protein is contributing to binding. Furthermore, the overall positive charge and higher sequence conservation combined with the substrate preference (Fig. 3) argue that XPF preferentially binds the non-damaged strand (5′ overhang) (Fig. 7, top panels).
FIGURE 7.

Model for binding of ERCC1-XPF HhH domains to an ss/dsDNA junction. The left panel shows a ribbon representation for ssDNA and dsDNA binding to, respectively, XPF (blue) and ERCC1 (red) based on structural models for archaeal XPF bound to dsDNA (PDB code 2BGW) (44) and human XPF bound to ssDNA (PDB code 2KN7) (42). In the top left panel, the 3′ end of ssDNA is connected with a yellow dashed line to the dsDNA (indicating a connection to the non-damaged strand). The left bottom panel is rotated 90°, and the dsDNA is connected with a green dashed line to the 5′ end of the ssDNA (indicating a connection to the damaged strand). The other panels show surface representations plotting charge distribution (Charge), sequence conservation (Conservation), effect of mutations on binding (Mutation), and the determined binding interface (Splayed arm 5′). The Charge panel is colored according to electrostatic surface potential calculated using the APBS software (67) (blue, positive; red, negative). The Conservation panel is colored according to sequence conservation calculated using the Consurf server (68) based on the complete multiple sequence alignment for eukaryotic repair proteins and plotted according to the default coloring: most conserved, red; least conserved, cyan. The Mutation panel is colored according to the contribution of a residue to the ability of the ERCC1-XPF heterodimeric HhH domain to bind to B10 DNA when mutated: most affected, red; least affected, light red. Residues that could be mutated without significant effect on binding are depicted in blue. The Splayed arm 5′ panel is colored according to chemical shift perturbation by a splayed arm substrate (as shown in Fig. 4): most affected, red. In all panels the relevant amino acids are depicted on the surface.
On the other hand, if the dsDNA connects to the 5′ end of the ssDNA (damaged strand), then the distance to the dsDNA would be substantially larger. Furthermore, the region between these two DNA binding surfaces is poorly conserved. Only small chemical shift perturbations are found upon addition of splayed arm substrate, and only limited effects on binding affinity by mutagenesis in this region were found. Combined with the overall negative charge for this side of the ERCC1-XPF molecule (Fig. 7, bottom panels), this would make the model where XPF would bind to a substrate with a 3′ ssDNA extension, being the damaged strand in our model, unlikely.
By combining the subtle CSP differences for the splayed arm substrate with the substrates containing one ssDNA strand (Fig. 4 and, supplemental Fig. S3), the effect of mutagenesis on the ability of ERCC1-XPF to bind DNA (Fig. 6), the charge, and the sequence conservation (Fig. 7), we propose that XPF recognizes the non-damaged strand. This agrees well with the previously reported binding and incision preference for full-length ERCC1-XPF in the presence of RPA (51).
Substrate Preference of XPF
In vitro DNA binding experiments demonstrated a clear preference of XPF for ssDNA substrates (Figs. 1–3). Most ssDNA sequences tested were suitable substrates for XPF irrespective whether XPF was present as homodimer or as heterodimer with ERCC1. However, guanine stretches were more effective in XPF recognition than thymidine or cytosine, whereas adenosine was a poor substrate for XPF (Figs. 2 and 3). Thus, DNA binding affinity and, thereby, the ability to repair damaged DNA may not be entirely sequence-independent. There are a few in vitro studies that also suggest that cleavage is not completely sequence-independent; for example, cleavage of an identical sequence where an acetylaminofluorene adduct positioned at three distinct guanines led to significant variation in incision efficiency (52). Similar differences were found for benzo[a]pyrenyl-guanine lesions placed at various positions (53, 54). In addition de Laat et al. (32) presented evidence for sequence specificity by showing that splayed arm substrates with distinct sequence composition around the junction were cleaved at distinct positions in the stem sequence, arguing that the incision position is somewhat dictated by the DNA sequence. Similar flanking sequence-dependent cleavage position variations were also found by Svoboda et al. (55). A more recent study by Bowles et al. (43) provides further support for differences in cleavage rate depending on the stem-loop sequence, although these studies show that the DEAH helicase-like domain is critically required for these effects. Interestingly, using excision repair sequencing, Hu et al. (56) succeeded in determining removed DNA sequences of excised DNA on a genome-wide scale. The position-dependent variation of the nucleotide sequences flanking the putative damaged pyrimidine dimers provides indirect support for context-dependent differences in cleavage efficiency; the relevance of this observation for DNA repair efficiency remains to be answered. This all argues that the incision position is weakly dictated by the DNA sequence. We propose that this is related to the observed preference of XPF for G-rich sequences, involving the recognition of a guanine base by XPF, as was found in the homodimer XPF-ssDNA structure (42).
Model Describing a Role for XPF in Sequence-dependent Incision
Following damage recognition, the ATP-dependent unwinding of DNA by transcription factor II H (TFIIH) (57) creates a DNA topology suitable for binding of RPA and XPA to the non-damaged and damaged DNA, respectively (58). Binding of these proteins further opens the damaged DNA and serves, through multiple interactions, as a platform for XPG and ERCC1-XPF, which subsequently perform the 3′ and 5′ incisions, respectively (12, 59, 60). This well orchestrated cleavage process (61–63) results in the removal of 24–32 nucleotides both in vitro and in vivo. The substantial variation in both cleavage position with respect to the damage and the length of the removed sequence (5, 64) suggests some heterogeneity in the cleavage mechanism, which is underscored by flanking sequence-dependent differences in cleavage efficiency in vitro (43, 52–55) and in vivo (56).
The noted preference of XPF for G-rich sequences (Figs. 2 and 3), which is consistent with the structure of the XPF ssDNA complex (42), may dictate the binding of the ERCC1-XPF complex. The presence of one or a few specific nucleotides within the accessible ssDNA sequence (of the non-damaged strand) can determine where cleavage will occur via the positioning of ERCC1-XPF on the DNA (32). We propose that both the DNA sequence-dependent differences in cleavage efficiency and the heterogeneity in the cleavage position by the ERCC1-XPF complex are the result of the deoxyguanosine preference of the ssDNA-binding domain of XPF.
Experimental Procedures
Protein Expression and Purification
The HhH domains of the ERCC1-XPF heterodimer were expressed and purified as described before (34). Homodimeric XPF HhH domain expression and purification have also been described before (41). The ERCC1-XPF mutants were prepared using the QuikChange protocol (Stratagene) and expressed as the wild-type ERCC1-XPF complex (34). Because of the absence of tryptophan or tyrosine residues, XPF homodimer protein concentration was based on SDS-PAGE, which leads to relatively large errors, in part because of differences in Coomassie staining efficiency. Therefore, whenever applicable, we normalized proteins based on SDS-PAGE and used UV absorbance of the heterodimeric complex to quantify.
Electrophoretic Mobility Shift Assay
EMSA experiments were performed as described before (34, 65) using the radiolabeled bubble 10 probe, ss39 or Holliday junction as substrate in a buffer containing 10 mm Tris (pH 7.5), 100 mm NaCl, 10% glycerol, 1 mm DTT, and BSA (final concentration, 20 μg/ml). All oligonucleotides were purchased from Operon or Eurogentec and annealed by incubating the two mixed strands (final concentration, 50 μm) for 5 min at 95 °C, followed by a cooling step for 1 h in a solution containing 10 mm Tris (pH 8.0) and 100 mm NaCl. For the competition experiments, the indicated amount of competitor (supplemental Fig. S1) and the radiolabeled gel-purified probe were mixed in a tube, and, subsequently, the protein-containing solution (∼1 μm) was added to this mixture. After incubation for 30 min on ice, samples were loaded on a 0.5× Tris borate-EDTA-buffered 5% acrylamide gel, and electrophoresis was carried out for 2.5 h at 160 V at room temperature. Analysis and quantification were performed as described before (65). Alternatively, complexes were separated on a 0.5× Tris borate-EDTA-buffered 3% agarose gel for 2 h at 80 V at 4 °C.
Surface Plasmon Resonance Measurements
SPR experiments were performed in 10 mm Hepes (pH 7.5), 50 mm NaCl, and 0.005% (w/v) Tween 20 (SPR buffer) at 10 μl/min at 12 °C using a Biacore® X system (Biacore AB) (66). The ERCC1-XPF HhH domain was dialyzed to the SPR buffer using Zeba Desalt spin columns (Thermo Scientific). Low-binding tubes and tips were used to prevent loss of the sample during the incubations and dilutions. Before each experiment, 5 μl of 0.3 m Ni2+ was loaded on flow cell 2 of the NTA sensor chip (Biacore AB), and flow cell 1 was used as a reference surface. 50 nm ERCC1-XPF HhH domain was incubated in SPR buffer on ice for 20 min in the absence or presence of different oligonucleotides (concentration ranging from 0.01–100 μm). Then it was injected over the NTA sensor chip, followed by association for 60 s and dissociation for 120 s. Flow cell 1 (without Ni2+) baseline curves were subtracted from the flow cell 2 experimental curves using Biaevaluation 3.2 software. Between consecutive injections, the chip was regenerated with 10 μl of 0.25 m EDTA in 3.5 m guanidium (pH 8). All experiments were performed at least in duplicate.
Because the addition of DNA prevented the binding of the His-tagged ERCC1-XPF HhH domain on the Ni-NTA surface, the relative amount of (DNA-free) protein (F) was determined as a response value at the end of loading at 60 s (R60) divided by the R60 value of the ERCC1-XPF HhH domain in the absence of DNA. To calculate the apparent dissociation constant (KDapp) for binding of each oligonucleotide to the ERCC1-XPF HhH domain, the relative amount of the DNA-free protein (F) was fitted against the total oligonucleotide concentration and according to a simple 1:1 model of interaction using GraphPad Prism: (1-F) = [DNA]/(KDapp + [DNA]), where F represents the relative amount of unbound ERRC1-XPF and KDapp the apparent equilibrium binding constant. For ERCC1 DNA binding studies, 6.25 μm ssDNA or dsDNA was loaded on the immobilized ERCC1 after dissociation of XPF from the ERCC1-XPF complex by extensive washing with binding buffer.
NMR Experiments
NMR titrations were followed by recording 15N-1H HSQC spectra of ERCC1-XPF by adding small volumes of a concentrated solution of commercially purchased DNA oligonucleotides (Eurogentec or Operon). The 15N-labeled ERCC1-XPF protein and unlabeled DNA were dissolved in the same buffer containing 5–50 mm sodium phosphate buffer and ∼10–100 mm NaCl (pH 7.0). All NMR data were collected at 22 °C on a Bruker DRX600 spectrometer equipped with a z gradient triple resonance cryoprobe or a Bruker Avance 900 spectrometer equipped with a 5-mm z gradient triple resonance probe. A set of 15N-1H HSQC spectra was acquired with successive addition of ssDNA, dsDNA, and splayed arm DNA substrates to 40–100 μm 15N-labeled ERCC1-XPF. The NMR data were processed and analyzed as described before (34). To compare the chemical shift changes on the DNA backbone of a 10-nt ssDNA fragment (42) upon addition of protein, 1H-decoupled 1D 31P spectra of the free and bound ssDNA were acquired on a Bruker DRX500 spectrometer equipped with a QXI probe.
Author Contributions
D. D., M. F., and G. E. F. conducted most of the NMR experiments. L. K. performed and analyzed the SPR analysis, and most DNA binding studies were performed by G. E. F. R. B., R. K., and G. E. F. conceived the idea for the studies, and D. D. and M. F. wrote the paper with G. E. F.
Supplementary Material
Acknowledgments
We thank Annet Vliegenthart, Jolinde Soesbergen, Mehmet Demirci, and Donal van Uunen for various contributions during the project.
This work was supported by The Netherlands Foundation for Chemical Research, the Center for Biomedical Genetics, European Commission Project 031220/Spine2-complexes and Project 261863/Bio-NMR, the Slovenian Infrastructural Center for Analysis of Molecular Interaction, and the Slovenian Research Agency (Z1–4071). Funding for the open access charge was provided by Utrecht University. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S4 and Table S1.
- NER
- nucleotide excision repair
- HhH
- helix-hairpin-helix
- ssDNA
- single-stranded DNA
- Ni-NTA
- nickel-nitrilotriacetic acid
- CSP
- chemical shift perturbation
- SPR
- surface plasmon resonance
- HSQC
- heteronuclear single-quantum coherence.
References
- 1. Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem. 65, 43–81 [DOI] [PubMed] [Google Scholar]
- 2. Cleaver J. E., and Crowley E. (2002) UV damage, DNA repair and skin carcinogenesis. Front. Biosci. 7, d1024–d1043 [DOI] [PubMed] [Google Scholar]
- 3. Friedberg E. C. (2003) DNA damage and repair. Nature 421, 436–440 [DOI] [PubMed] [Google Scholar]
- 4. Hanawalt P. C. (2002) Subpathways of nucleotide excision repair and their regulation. Oncogene 21, 8949–8956 [DOI] [PubMed] [Google Scholar]
- 5. Araujo S. J., and Wood R. D. (2000) Protein complexes in nucleotide excision repair. Mutat. Res. DNA Repair 435, 23–33 [DOI] [PubMed] [Google Scholar]
- 6. Cleaver J. E., Thompson L. H., Richardson A. S., and States J. C. (1999) A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum. Mutat. 14, 9–22 [DOI] [PubMed] [Google Scholar]
- 7. Cleaver J. E. (2005) Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat. Rev. Cancer 5, 564–573 [DOI] [PubMed] [Google Scholar]
- 8. Cleaver J. E., and Revet I. (2008) Clinical implications of the basic defects in Cockayne syndrome and xeroderma pigmentosum and the DNA lesions responsible for cancer, neurodegeneration and aging. Mech. Ageing Dev. 129, 492–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Niedernhofer L. J., Garinis G. A., Raams A., Lalai A. S., Robinson A. R., Appeldoorn E., Odijk H., Oostendorp R., Ahmad A., Van Leeuwen W., Theil A. F., Vermeulen W., van der Horst G. T., Meinecke P., Kleijer W. J., et al. (2006) A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444, 1038–1043 [DOI] [PubMed] [Google Scholar]
- 10. Bessho T., Sancar A., Thompson L. H., and Thelen M. P. (1997) Reconstitution of human excision nuclease with recombinant XPF-ERCC1 complex. J. Biol. Chem. 272, 3833–3837 [DOI] [PubMed] [Google Scholar]
- 11. de Laat W. L., Sijbers A. M., Odijk H., Jaspers N. G., and Hoeijmakers J. H. (1998) Mapping of interaction domains between human repair proteins ERCC1 and XPF. Nucleic Acids Res. 26, 4146–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsunaga T., Mu D., Park C. H., Reardon J. T., and Sancar A. (1995) Human DNA-repair excision nuclease: analysis of the roles of the subunits involved in dual incisions by using anti-Xpg and anti-Ercc1 antibodies. J. Biol. Chem. 270, 20862–20869 [DOI] [PubMed] [Google Scholar]
- 13. Sijbers A. M., de Laat W. L., Ariza R. R., Biggerstaff M., Wei Y. F., Moggs J. G., Carter K. C., Shell B. K., Evans E., de Jong M. C., Rademakers S., de Rooij J., Jaspers N. G., Hoeijmakers J. H., and Wood R. D. (1996) Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 86, 811–822 [DOI] [PubMed] [Google Scholar]
- 14. Nishigori C., Ishizaki K., Takebe H., Imamura S., and Hayakawa M. (1986) A case of xeroderma pigmentosum group F with late onset of clinical symptoms. Arch. Dermatol. 122, 510–511 [PubMed] [Google Scholar]
- 15. Gaillard P. H., and Wood R. D. (2001) Activity of individual ERCC1 and XPF subunits in DNA nucleotide excision repair. Nucleic Acids Res. 29, 872–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sijbers A. M., van der Spek P. J., Odijk H., van den Berg J., van Duin M., Westerveld A., Jaspers N. G., Bootsma D., and Hoeijmakers J. H. (1996) Mutational analysis of the human nucleotide excision repair gene ERCC1. Nucleic Acids Res. 24, 3370–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hayashi T., Takao M., Tanaka K., and Yasui A. (1998) ERCC1 mutations in UV-sensitive Chinese hamster ovary (CHO) cell lines. Mutat. Res. DNA Repair 407, 269–276 [DOI] [PubMed] [Google Scholar]
- 18. Yagi T., Wood R. D., and Takebe H. (1997) A low content of ERCC1 and a 120 kDa protein is a frequent feature of group F xeroderma pigmentosum fibroblast cells. Mutagenesis 12, 41–44 [DOI] [PubMed] [Google Scholar]
- 19. Tian M., Shinkura R., Shinkura N., and Alt F. W. (2004) Growth retardation, early death, and DNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol. Cell Biol. 24, 1200–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weeda G., Donker I., de Wit J., Morreau H., Janssens R., Vissers C. J., Nigg A., van Steeg H., Bootsma D., and Hoeijmakers J. H. (1997) Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol. 7, 427–439 [DOI] [PubMed] [Google Scholar]
- 21. McWhir J., Selfridge J., Harrison D. J., Squires S., and Melton D. W. (1993) Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat. Genet. 5, 217–224 [DOI] [PubMed] [Google Scholar]
- 22. Ahmad A., Robinson A. R., Duensing A., van Drunen E., Beverloo H. B., Weisberg D. B., Hasty P., Hoeijmakers J. H., and Niedernhofer L. J. (2008) ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell Biol. 28, 5082–5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Al-Minawi A. Z., Saleh-Gohari N., and Helleday T. (2008) The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res. 36, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuraoka I., Kobertz W. R., Ariza R. R., Biggerstaff M., Essigmann J. M., and Wood R. D. (2000) Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J. Biol. Chem. 275, 26632–26636 [DOI] [PubMed] [Google Scholar]
- 25. Fisher L. A., Bessho M., and Bessho T. (2008) Processing of a psoralen DNA interstrand cross-link by XPF-ERCC1 complex in vitro. J. Biol. Chem. 283, 1275–1281 [DOI] [PubMed] [Google Scholar]
- 26. Zhu X. D., Niedernhofer L., Kuster B., Mann M., Hoeijmakers J. H., and de Lange T. (2003) ERCC1/XPF removes the 3′ overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol. Cell 12, 1489–1498 [DOI] [PubMed] [Google Scholar]
- 27. Muñoz P., Blanco R., Flores J. M., and Blasco M. A. (2005) XPF nuclease-dependent telomere loss and increased DNA damage in mice overexpressing TRF2 result in premature aging and cancer. Nat. Genet. 37, 1063–1071 [DOI] [PubMed] [Google Scholar]
- 28. Niedernhofer L. J., Essers J., Weeda G., Beverloo B., de Wit J., Muijtjens M., Odijk H., Hoeijmakers J. H., and Kanaar R. (2001) The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J. 20, 6540–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fekairi S., Scaglione S., Chahwan C., Taylor E. R., Tissier A., Coulon S., Dong M. Q., Ruse C., Yates J. R. 3rd, Russell P., Fuchs R. P., McGowan C. H., and Gaillard P. H. (2009) Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138, 78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Svendsen J. M., Smogorzewska A., Sowa M. E., O'Connell B. C., Gygi S. P., Elledge S. J., and Harper J. W. (2009) Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138, 63–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Muñoz I. M., Hain K., Déclais A. C., Gardiner M., Toh G. W., Sanchez-Pulido L., Heuckmann J. M., Toth R., Macartney T., Eppink B., Kanaar R., Ponting C. P., Lilley D. M., and Rouse J. (2009) Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 35, 116–127 [DOI] [PubMed] [Google Scholar]
- 32. de Laat W. L., Appeldoorn E., Jaspers N. G., and Hoeijmakers J. H. (1998) DNA structural elements required for ERCC1-XPF endonuclease activity. J. Biol. Chem. 273, 7835–7842 [DOI] [PubMed] [Google Scholar]
- 33. Park C. H., Bessho T., Matsunaga T., and Sancar A. (1995) Purification and characterization of the XPF-ERCC1 complex of human DNA repair excision nuclease. J. Biol. Chem. 270, 22657–22660 [DOI] [PubMed] [Google Scholar]
- 34. Tripsianes K., Folkers G., Ab E., Das D., Odijk H., Jaspers N. G., Hoeijmakers J. H., Kaptein R., and Boelens R. (2005) The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair. Structure 13, 1849–1858 [DOI] [PubMed] [Google Scholar]
- 35. Choi Y. J., Ryu K. S., Ko Y. M., Chae Y. K., Pelton J. G., Wemmer D. E., and Choi B. S. (2005) Biophysical characterization of the interaction domains and mapping of the contact residues in the XPF-ERCC1 complex. J. Biol. Chem. 280, 28644–28652 [DOI] [PubMed] [Google Scholar]
- 36. Tsodikov O. V., Enzlin J. H., Schärer O. D., and Ellenberger T. (2005) Crystal structure and DNA binding functions of ERCC1, a subunit of the DNA structure-specific endonuclease XPF-ERCC1. Proc. Natl. Acad. Sci. U.S.A. 102, 11236–11241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Faridounnia M., Wienk H., Kovačič L., Folkers G. E., Jaspers N. G., Kaptein R., Hoeijmakers J. H., and Boelens R. (2015) The Cerebro-oculo-facio-skeletal syndrome point mutation F231L in the ERCC1 DNA repair protein causes dissociation of the ERCC1-XPF complex. J. Biol. Chem. 290, 20541–20555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bogliolo M., Schuster B., Stoepker C., Derkunt B., Su Y., Raams A., Trujillo J. P., Minguillón J., Ramírez M. J., Pujol R., Casado J. A., Baños R., Rio P., Knies K., Zúñiga S., et al. (2013) Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 92, 800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jaspers N. G., Raams A., Silengo M. C., Wijgers N., Niedernhofer L. J., Robinson A. R., Giglia-Mari G., Hoogstraten D., Kleijer W. J., Hoeijmakers J. H., and Vermeulen W. (2007) First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am. J. Hum. Genet. 80, 457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kashiyama K., Nakazawa Y., Pilz D. T., Guo C., Shimada M., Sasaki K., Fawcett H., Wing J. F., Lewin S. O., Carr L., Li T. S., Yoshiura K., Utani A., Hirano A., Yamashita S., et al. (2013) Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 92, 807–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Das D., Tripsianes K., Jaspers N. G., Hoeijmakers J. H., Kaptein R., Boelens R., and Folkers G. E. (2008) The HhH domain of the human DNA repair protein XPF forms stable homodimers. Proteins 70, 1551–1563 [DOI] [PubMed] [Google Scholar]
- 42. Das D., Folkers G. E., van Dijk M., Jaspers N. G., Hoeijmakers J. H., Kaptein R., and Boelens R. (2012) The structure of the XPF-ssDNA complex underscores the distinct roles of the XPF and ERCC1 helix-hairpin-helix domains in ss/ds DNA recognition. Structure 20, 667–675 [DOI] [PubMed] [Google Scholar]
- 43. Bowles M., Lally J., Fadden A. J., Mouilleron S., Hammonds T., and McDonald N. Q. (2012) Fluorescence-based incision assay for human XPF-ERCC1 activity identifies important elements of DNA junction recognition. Nucleic Acids Res. 40, e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Newman M., Murray-Rust J., Lally J., Rudolf J., Fadden A., Knowles P. P., White M. F., and McDonald N. Q. (2005) Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J. 24, 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shao X., and Grishin N. V. (2000) Common fold in helix-hairpin-helix proteins. Nucleic Acids Res. 28, 2643–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nishino T., Komori K., Ishino Y., and Morikawa K. (2005) Structural and functional analyses of an archaeal XPF/Rad1/Mus81 nuclease: asymmetric DNA binding and cleavage mechanisms. Structure 13, 1183–1192 [DOI] [PubMed] [Google Scholar]
- 47. Tripsianes K., Folkers G. E., Zheng C., Das D., Grinstead J. S., Kaptein R., and Boelens R. (2007) Analysis of the XPA and ssDNA-binding surfaces on the central domain of human ERCC1 reveals evidence for subfunctionalization. Nucleic Acids Res. 35, 5789–5798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsodikov O. V., Ivanov D., Orelli B., Staresincic L., Shoshani I., Oberman R., Schärer O. D., Wagner G., and Ellenberger T. (2007) Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 26, 4768–4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fisher L. A., Bessho M., Wakasugi M., Matsunaga T., and Bessho T. (2011) Role of interaction of XPF with RPA in nucleotide excision repair. J. Mol. Biol. 413, 337–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Su Y., Orelli B., Madireddy A., Niedernhofer L. J., and Schärer O. D. (2012) Multiple DNA binding domains mediate the function of the ERCC1-XPF protein in nucleotide excision repair. J. Biol. Chem. 287, 21846–21855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. de Laat W. L., Appeldoorn E., Sugasawa K., Weterings E., Jaspers N. G., and Hoeijmakers J. H. (1998) DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev. 12, 2598–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mu D., Bertrand-Burggraf E., Huang J. C., Fuchs R. P., Sancar A., and Fuchs B. P. (1994) Human and E.coli excinucleases are affected differently by the sequence context of acetylaminofluorene-guanine adduct. Nucleic Acids Res. 22, 4869–4871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cai Y., Patel D. J., Geacintov N. E., and Broyde S. (2007) Dynamics of a benzo[a]pyrene-derived guanine DNA lesion in TGT and CGC sequence contexts: enhanced mobility in TGT explains conformational heterogeneity, flexible bending, and greater susceptibility to nucleotide excision repair. J. Mol. Biol. 374, 292–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cai Y., Patel D. J., Geacintov N. E., and Broyde S. (2009) Differential nucleotide excision repair susceptibility of bulky DNA adducts in different sequence contexts: hierarchies of recognition signals. J. Mol. Biol. 385, 30–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Svoboda D. L., Taylor J. S., Hearst J. E., and Sancar A. (1993) DNA repair by eukaryotic nucleotide excision nuclease: removal of thymine dimer and psoralen monoadduct by HeLa cell-free-extract and of thymine dimer by Xenopus laevis oocytes. J. Biol. Chem. 268, 1931–1936 [PubMed] [Google Scholar]
- 56. Hu J., Adar S., Selby C. P., Lieb J. D., and Sancar A. (2015) Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 29, 948–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Coin F., Oksenych V., and Egly J. M. (2007) Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol. Cell 26, 245–256 [DOI] [PubMed] [Google Scholar]
- 58. Missura M., Buterin T., Hindges R., Hübscher U., Kaspárková J., Brabec V., and Naegeli H. (2001) Double-check probing of DNA bending and unwinding by XPA-RPA: an architectural function in DNA repair. EMBO J. 20, 3554–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. O'Donovan A., Davies A. A., Moggs J. G., West S. C., and Wood R. D. (1994) Xpg endonuclease makes the 3′ incision in human DNA nucleotide excision repair. Nature 371, 432–435 [DOI] [PubMed] [Google Scholar]
- 60. Wakasugi M., Reardon J. T., and Sancar A. (1997) The non-catalytic function of XPG protein human nucleotide excision repair. J. Biol. Chem. 272, 16030–16034 [DOI] [PubMed] [Google Scholar]
- 61. Mocquet V., Lainé J. P., Riedl T., Yajin Z., Lee M. Y., and Egly J. M. (2008) Sequential recruitment of the repair factors during NER: the role of XPG in initiating the resynthesis step. EMBO J. 27, 155–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wakasugi M., and Sancar A. (1999) Order of assembly of human DNA repair excision nuclease. J. Biol. Chem. 274, 18759–18768 [DOI] [PubMed] [Google Scholar]
- 63. Riedl T., Hanaoka F., and Egly J. M. (2003) The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 22, 5293–5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. de Laat W. L., Jaspers N. G., and Hoeijmakers J. H. (1999) Molecular mechanism of nucleotide excision repair. Genes Dev. 13, 768–785 [DOI] [PubMed] [Google Scholar]
- 65. de Jong R. N., Truffault V., Diercks T., Ab E., Daniels M. A., Kaptein R., and Folkers G. E. (2008) Structure and DNA binding of the human Rtf1 Plus3 domain. Structure 16, 149–159 [DOI] [PubMed] [Google Scholar]
- 66. Nieba L., Nieba-Axmann S. E., Persson A., Hämäläinen M., Edebratt F., Hansson A., Lidholm J., Magnusson K., Karlsson A. F., and Plückthun A. (1997) BIACORE analysis of histidine-tagged proteins using a chelating NTA sensor chip. Anal. Biochem. 252, 217–228 [DOI] [PubMed] [Google Scholar]
- 67. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ashkenazy H., Erez E., Martz E., Pupko T., and Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.