

Abstract
Ultrafine particles (UFP) generated by combustion processes are often associated with adverse health effects. However, little is known about the inflammatory processes generated by UFP that may underlie their toxicological activity. Murine macrophages (J774.1 cells) and human peripheral blood mononuclear cells (PBMCs) were used to evaluate the molecular mechanism underlying the pro-inflammatory activity of UFP. The addition of soot particles to J774.1 cells induced a concentration-dependent release of IL-1α, IL-1β and IL-33 This effect was not associated with cell death and, in contrast to literature, was pronounced at very low concentrations (5–100 pg/ml). Similarly, UFP induced the release of IL-1α, IL-18 and IL-33 by PBMCs. However, this effect was solely observed in PBMCs obtained from smokers, as the PBMCs from non-smokers instead released higher levels of IL-10. The release of these cytokines after UFP exposure was caspase-1- and NLRP3 inflammasome-dependent in PBMCs from healthy smokers, whereas IL-1α release was calpain-dependent. These results show that UFP at very low concentrations are able to give rise to an inflammatory process that is responsible for IL-1α, IL-18 and IL-33 release, which is pronounced in PBMCs from smokers, confirming that these individuals are especially susceptible to inflammatory-based airway diseases once exposed to air pollution.
Epidemiological studies have widely demonstrated a direct link between air pollution and respiratory diseases. Diesel exhaust particles represent one of the major environmental insults responsible for adverse effects on the respiratory tract1,2,3. Combustion particles emitted by diesel engines consist of fine particles often referred to as soot4; in particular, sub-100 nm particles (ultrafine particles, UFP) are the most threatening, as they can localize into the low tract of the respiratory tree, leading to pulmonary diseases3. Several studies demonstrated that exposure to soot particles has remarkable effects on the immune system5,6,7,8. However, most of the studies are focused on allergic diseases9,10. In this regard, it was demonstrated that exposure to soot particles causes changes in lymphocyte homeostasis and immune responses in that it promotes autophagy in T cells with a Th2-like phenotype11. In recent years, dysfunctional autophagy has been linked to inflammatory pathways that promote oxidative stress and DNA damage and mutations, phenomena that can lead to cancer development12. Nevertheless, the exact mechanism underlying soot particle-induced immune cell dysfunction with ensuing inflammation, which can ultimately lead to toxicological effects on human health, is yet unknown. It was described that UFP are able to induce both epithelial and macrophagic cells to release reactive oxygen species (ROS), which are responsible for the induction of cell death via apoptosis and/or necrosis13,14,15. In this scenario, the role of mitochondria is pivotal in that alterations in the membrane depolarization-hyperpolarization equilibrium can promote the release of mitochondrial ROS (mtROS), which have recently been described as potential inducers of inflammatory pathways16. Shimada et al.17 demonstrated that the release of mtDNA together with mtROS can induce the activation of the inflammasome complex. The inflammasome is a multiprotein complex that is generated from the assembly of Nucleotide-binding oligomerization domain (Nod)-like receptors (NLRs) or HIN200 family receptors, such as absent in melanoma 2 (AIM2), which are able to bind to the adaptor apoptosis-associated speck-like protein containing a carboxy terminal CARD (ASC), which induces the autocleavage of caspase-1; this promotes the release of IL-1-like cytokines in their active form18. The activation of the inflammasome leads to the release of IL-1-like cytokines, which are responsible for early inflammation and can result in long-term, chronic inflammation18. In support of this, IL-1-like cytokines are highly detected in the serum of cancer patients. Here, UFP produced under controlled conditions in laboratory were characterized in terms of their ability to modify inflammatory cytokine production in human peripheral blood mononuclear cells (PBMCs). In contrast to the literature data, where the pro-inflammatory activity of soot particles is associated with concentrations on the order of μg/ml, combustion-generated soot particles were tested for the ability to induce IL-1-dependent inflammation at very low concentrations (pg/ml). Here, we found that combustion particle-treated PBMCs obtained from smoking, but not non-smoking, healthy volunteers were able to release IL-1α, IL-18 and IL-33 in a caspase-1-dependent manner. In contrast, PBMCs from non-smokers showed higher release of the immunosuppressive IL-10, suggesting host defence against the activity of IL-1-like cytokines. In contrast, although PBMCs from smokers still released IL-10 after the addition of soot particles, this effect was less pronounced than that in non-smokers, implying that both smoking and air pollution can prompt pulmonary inflammation in an IL-1-like manner.
Our data provide new perspectives for investigation into the role of inhaled combustion particles that, together with other pulmonary insults, can lead to the inflammation that may underlie lung pathologies such as asthma, lung fibrosis and cancer.
Results
Ultrafine soot particles induced a concentration-dependent release of IL-1-like cytokines in murine macrophages
Combustion-generated UFP have been widely demonstrated to be one of the main insults on human health3. To study the under-investigated potential pro-inflammatory activity of soot particles, we preliminarily considered an early step in the induction of inflammation, the release of IL-1-like cytokines, such as IL-1α, IL-1β, IL-18 and IL-33, in the J774.1 macrophagic/monocytic cell line. J774.1 cells were treated with the produced UFP in a concentration-dependent manner starting at 1 pg/ml and increasing up to 5 ng/ml. As shown in Fig. 1, soot particles were able to statistically increase the release of IL-1α (Fig. 1A), IL-1β (Fig. 1B) and IL-33 (Fig. 1C), but not IL-18 (data not shown), after 5 hours of treatment. However, it is worth noting that this effect was observed at concentrations much lower than what has been reported in the literature, which has described the pro-inflammatory and cytotoxic activities of diesel exhaust particles/air pollution-derived particles at concentrations of μg/ml7,19,20,21. Importantly, the bell-shaped release of IL-1α (Fig. 1A), IL-1β (Fig. 1B) and IL-33 (Fig. 1C) showed that the sub-maximal effect was reached at concentrations of 5, 10, 50 and 100 pg/ml rather than higher concentrations (500, 1000 and 5000 pg/ml). Notably, UFP treatment of cells stimulated the release of the cytokines at similar or higher levels than those observed for positive controls (LPS ± ATP).
Figure 1. The administration of soot particles for 5 hours induced the release of IL-1-like cytokines by murine macrophages.
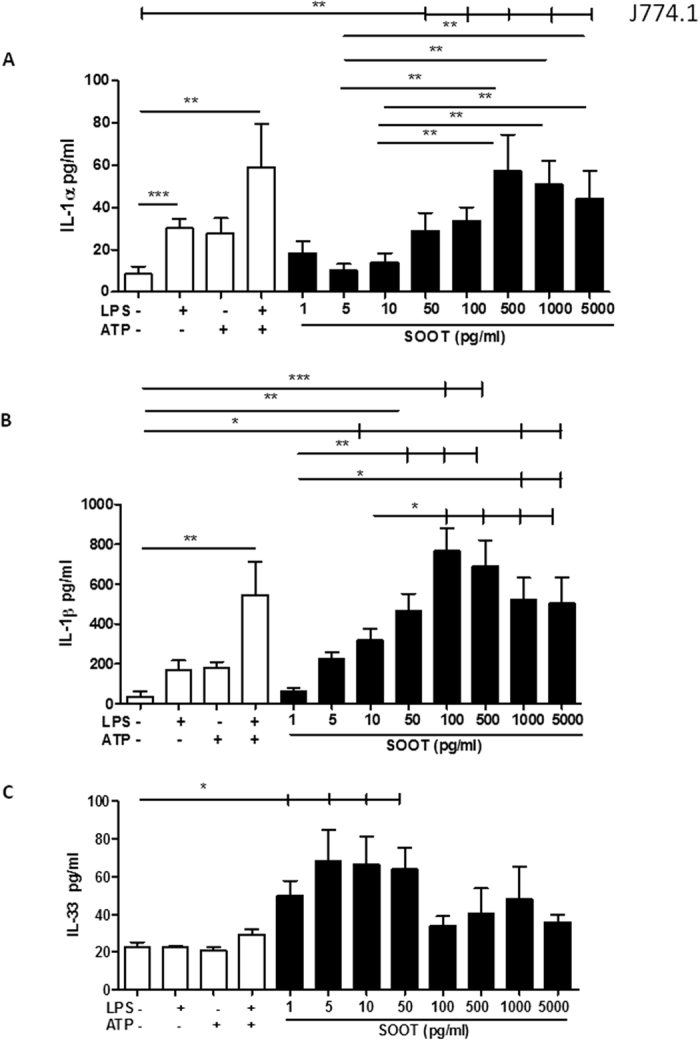
J774.1 cells (a murine macrophage cell line) were treated with soot particles in a concentration-dependent manner (1 pg/ml–5 ng/ml) for 5 hours. LPS (0.1 μg/ml) and/or ATP (0.5 mM) was used as a positive control. The addition of soot particles to macrophages induced the release of IL-1β (A), IL-1α (B) and IL-33 (C). Data are presented as the means ± SEM (n = 12). Statistically significant differences are denoted by *, ** and ***, indicating p < 0.05, p < 0.01 and p < 0.001, respectively, as determined by one-way ANOVA followed by Bonferroni’s multiple comparison post-test.
To understand the effect of UFP on other pro-inflammatory cytokines, the release of IL-6 and TNF-α was evaluated. As shown in Fig. 1 of the Supplementary Material, soot particles were not able to induce the release of IL-6 (Supplementary Figure 1A) and TNF-α (Supplementary Figure 1B) at our working concentrations. However, particles were able to induce IL-10 release (data not shown), implying that the phenotype of macrophages tended towards an M2 polarization.
A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test showed that J774.1 macrophagic/monocytic cells were viable after the addition of soot particles at both 5 hours (Supplementary Figure 2A) and 24 hours (Supplementary Figure 2B). To rule out any interference of soot particles to absorb at 550 nm, we also performed the MTT assay in a cell-free medium after the addition of the sole soot particles at the concentration of 1 pg/ml−5 ng/ml. Soot particles did not alter the absorbance of the MTT assay compared to the sole medium without particles (Supplementary 2C). To further confirm this observation, cells were treated with necrostatin, a well-known inhibitor of receptor-interacting protein (RIP) kinase involved in necrosis/cell death22. The addition of necrostatin did not change the levels of IL-1-like cytokines (data not shown), implying that soot particles do not induce necrosis. Moreover, soot particles were not able to induce the cleavage of caspase-3 (data not shown), a well-known enzyme involved in the apoptotic cascade.
These data imply that soot particles induce early inflammation in J774.1 cells via the release of IL-1-like cytokines without affecting cell viability.
Ultrafine soot particles induced a concentration-dependent release of IL-1-like cytokines from PBMCs from smokers
To verify if the observed effects on J774.1 cells were also valid for human primary cells, the effect of combustion-generated soot particles was evaluated in PBMCs isolated from healthy volunteers distinguished as non-smokers and smokers.
Interestingly, the addition of UFP to PBMCs obtained from healthy non-smokers did not induce the release of IL-1α (Fig. 2A, white bars), IL-33 (Fig. 2B, white bars) or IL-18 (Fig. 2C, white bars). In contrast, the addition of UFP (1 pg/ml–5 ng/ml) to PBMCs from healthy smokers significantly increased the release of all the cytokines (IL-1α, IL-33 and IL-18; Fig. 2A,B and C, respectively, dotted bars). Again, the release of these cytokines in response to treatment with soot particles was similar to or higher than the cytokine response of the positive control (smokers: LPS ± ATP, dotted bars). To understand the effect of soot particles on PBMCs from non-smokers, we analysed the release of an immunosuppressive cytokine, IL-1023, the level of which was significantly increased (Fig. 3A). Similarly, PBMCs obtained from smokers released IL-10 (Fig. 3B). However, it is important to note that the level of this cytokine was higher for PBMCs obtained from non-smokers (Fig. 3C, white bars) than PBMCs from smokers (Fig. 3C, dotted bars) after treatment with soot particles.
Figure 2. PBMCs obtained by healthy smokers were more susceptible to soot particle-induced IL-1-like cytokine release.
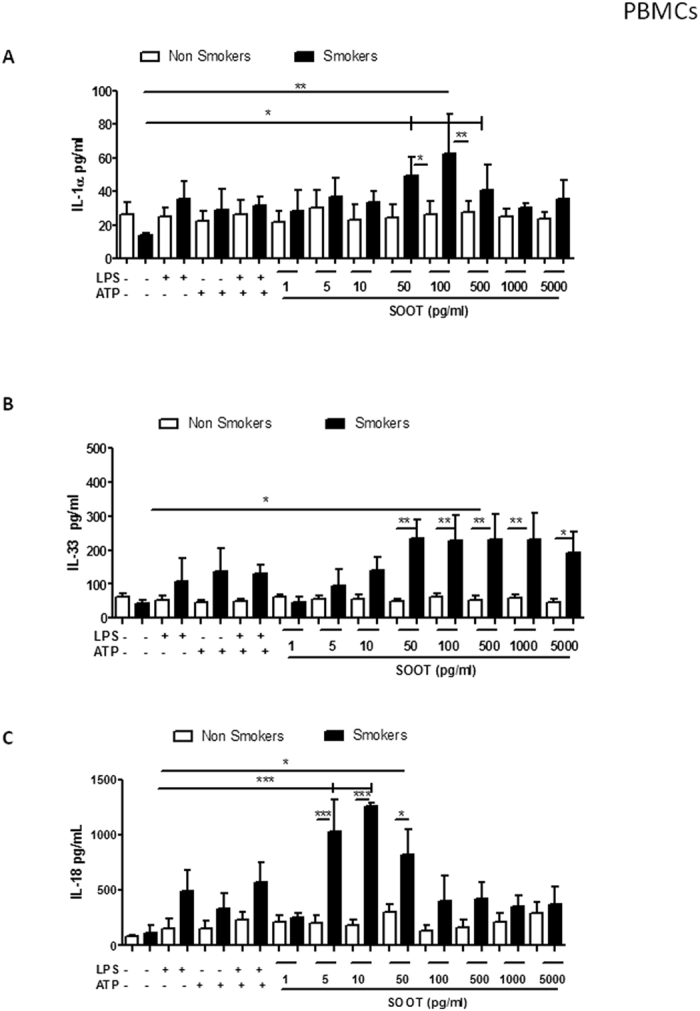
PBMCs were isolated from the blood of healthy non-smoking and smoking volunteers. The addition of soot particles (1 pg/ml–5 ng/ml) for 5 hours to PBMCs isolated from non-smokers did not induce the release of IL-1α (A) or IL-33 (C). However, soot particles slightly increased the release of IL-18 (50 pg/ml) (E). In sharp contrast, the addition of soot particles to PBMCs obtained from smokers significantly increased the release of IL-1α (B), IL-33 (D) and IL-18 (F). Data are presented as the means ± SEM (n = 5). Statistically significant differences are denoted by *, ** and ***, indicating p < 0.05, p < 0.01, p < 0.001 and p < 0.001, respectively, as determined by one-way ANOVA followed by Bonferroni’s multiple comparison post-test.
Figure 3. The addition of soot particles induced PBMCs to release IL-10.
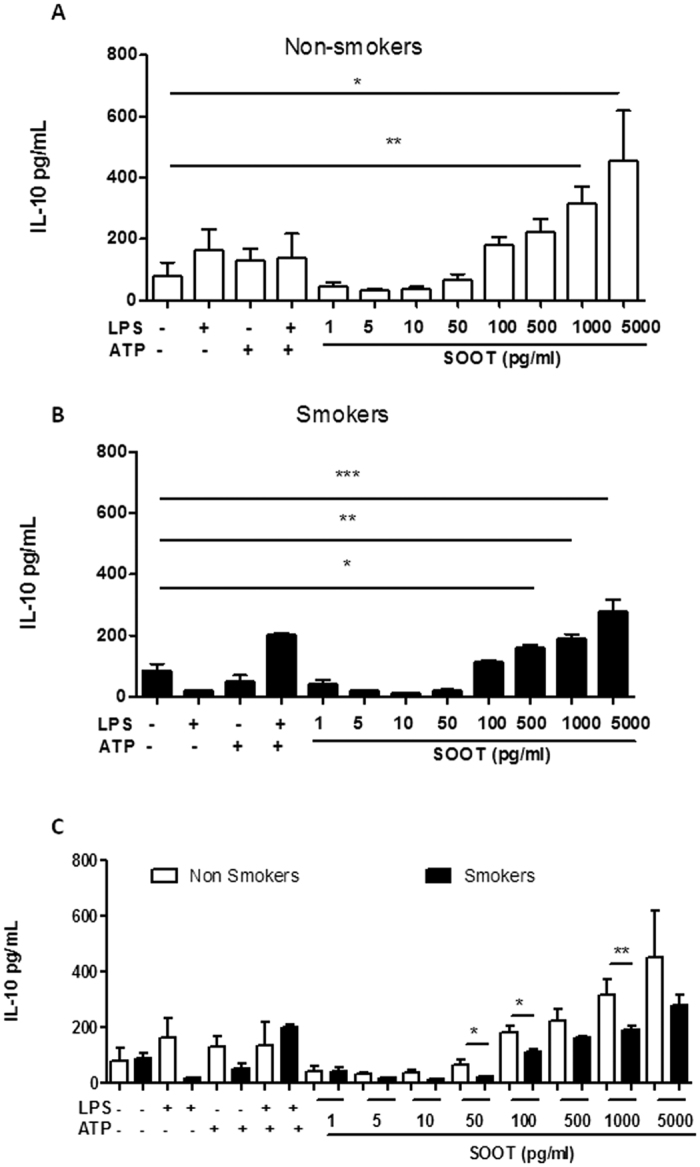
PBMCs were isolated from the blood of healthy non-smoking and smoking volunteers. The administration of soot particles (1 pg/ml–5 ng/ml) for 5 hours to PBMCs isolated from non-smokers significantly increased the release of IL-10 (A). In the same way, but to a lower extent, PBMCs isolated from smokers released IL-10 after soot particle addition (B). (C) Comparison of the levels of IL-10 between non-smokers (open white bars) and smokers (dotted bars). Data are presented as the means ± SEM (n = 5). Statistically significant differences are denoted by *, ** and ***, indicating p < 0.05, p < 0.01 and p < 0.001, respectively, as determined by one-way ANOVA followed by Bonferroni’s multiple comparison post-test.
The combined results imply that smokers exposed to UFP at very low concentrations are more susceptible to IL-1-dependent inflammation than non-smokers.
The release of IL-1-like cytokines after treatment of smoker-derived PBMCs with ultrafine soot particles was NLRP3/caspase-1-dependent
IL-1-like cytokine release is strictly dependent on the multimeric inflammasome complex18,22. To understand the molecular mechanism underlying the release of these cytokines after soot particle exposure, we tested the effect of well-known pharmacological inhibitors.
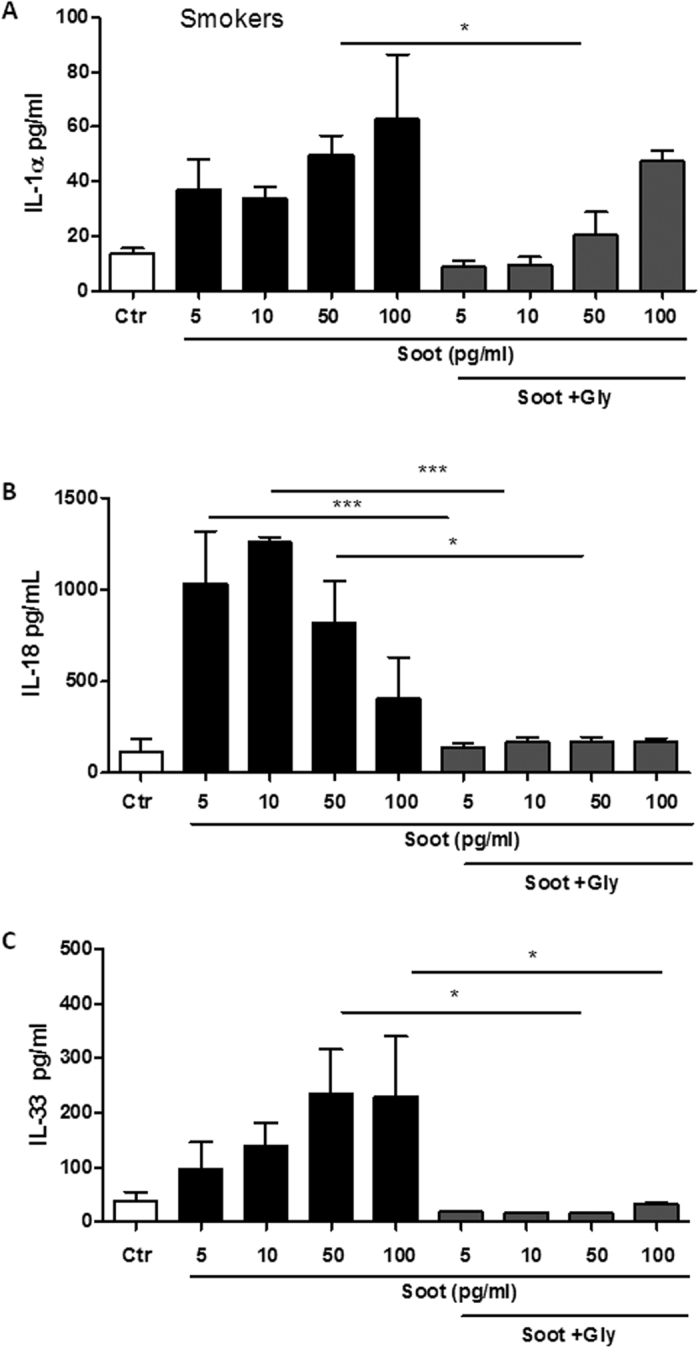
Cells were co-treated with soot particles and Y-Vad, a caspase-1 inhibitor. The administration of Y-Vad significantly decreased the release of all cytokines (IL-1α, IL-18 and IL-33 (Fig. 4A,B and C, respectively). However, higher concentrations of soot particles (100 pg/ml) still induced the release of IL-1α (Fig. 4A) after the addition of Y-Vad, implying that another mechanism was involved. Because IL-1α release is also dependent on the activation of calpain I/II22, we analysed whether this pathway was involved. Cells from smokers were treated with M66, a calpain I/II inhibitor22. The addition of M66 completely inhibited the release of IL-1α (Fig. 5) after treatment with soot particles, even at higher concentrations (50–100 pg/ml).
Figure 4. The release of IL-1-like cytokines by soot particle-treated PBMCs obtained from healthy smokers is caspase-1-dependent.
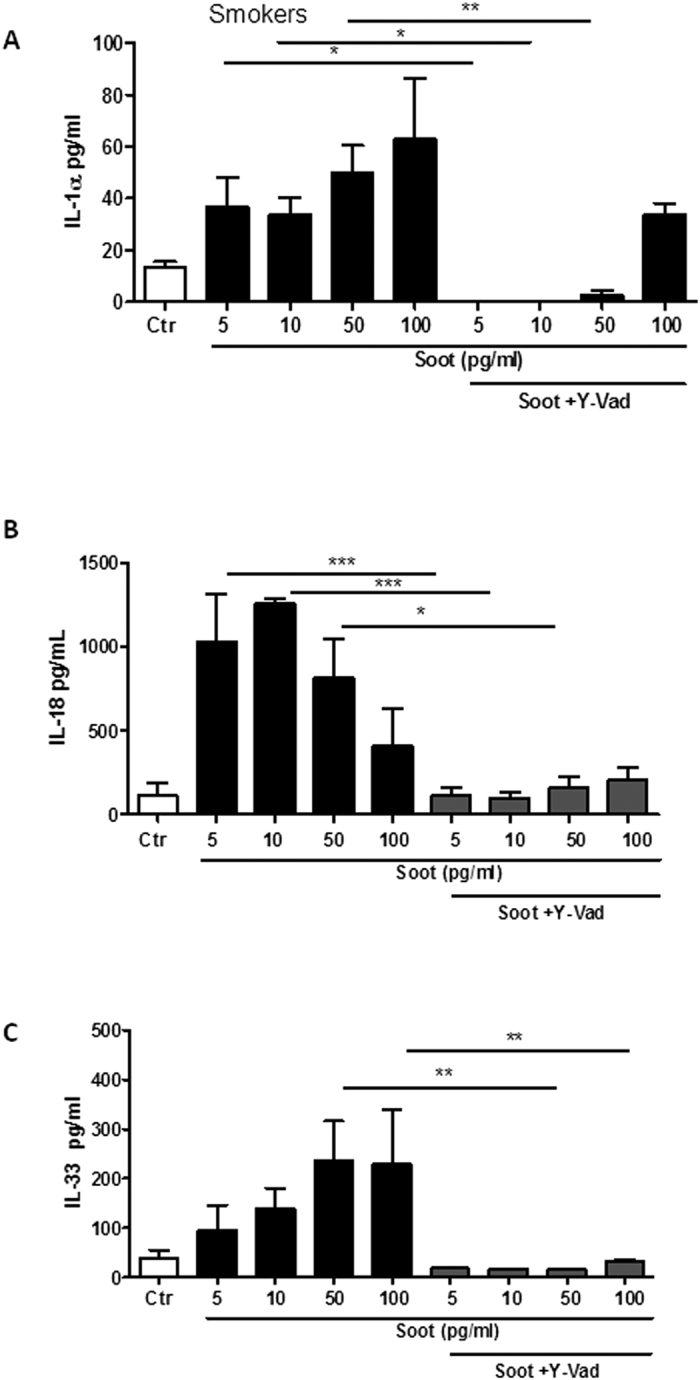
PBMCs obtained from smokers were treated with soot particles (1 pg/ml–5 ng/ml) in the presence or absence of Ac-YVad (Y-Vad: 1 μg/ml), a caspase-1 inhibitor. The inhibition of caspase-1 with Y-Vad significantly reduced the release of IL-1α (A), IL-18 (B) and IL-33 (C) from PBMCs treated for 5 hours. Data are presented as the means ± SEM (n = 5). Statistically significant differences are denoted by *, ** and ***, indicating p < 0.05, p < 0.01, and p < 0.001, respectively, as determined by one-way ANOVA followed by Bonferroni’s multiple comparison post-test.
Figure 5. NLRP3 is involved in the release of IL-1-like cytokines by soot particle-treated PBMCs obtained from healthy smokers.
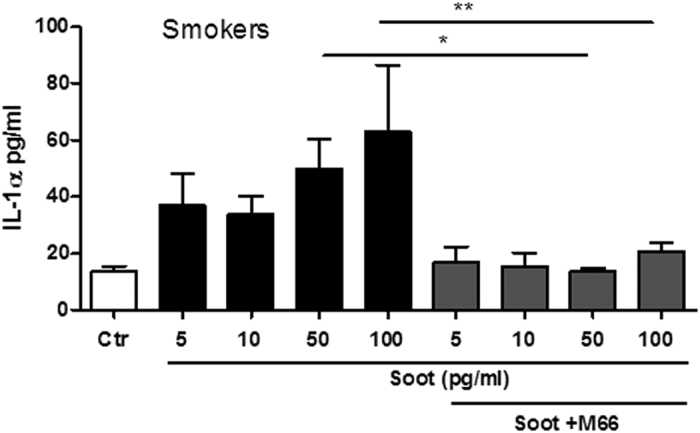
PBMCs obtained from smokers were treated with soot particles (1 pg/ml–5 ng/ml) in the presence or absence of glybenclamide (Gly: 1 μM), an NLRP3 inhibitor. The inhibition of NLRP3 with Gly significantly reduced the release of IL-1α (A), IL-18 (B) and IL-33 (C) from PBMCs treated for 5 hours. Data are presented as the means ± SEM (n = 5). Statistically significant differences are denoted by * and ***, indicating p < 0.05 and p < 0.001, respectively, as determined by one-way ANOVA followed by Bonferroni’s multiple comparison post-test.
Because the inflammasome complex comprises an intracellular receptor that assembles with ASC and caspase-1, the involvement of the NLRP3 inflammasome was examined. PBMCs from smokers were treated with soot particles in the presence of glybenclamide, a well-known inhibitor of the NLRP3 inflammasome22. The inhibition of NLRP3 significantly reduced the release of the cytokines IL-1α, IL-18 and IL-33, as reported in Fig. 6A–C. Similarly, IL-1α release was not completely reduced at the higher concentration of 100 pg/ml (Fig. 6A), confirming previous data (Fig. 4A) suggesting that other molecular mechanisms may be involved in IL-1α release (Fig. 5).
Figure 6. The release of IL-1-α from soot particle-treated PBMCs obtained from healthy smokers is calpain-dependent.
PBMCs obtained from smokers were treated with soot particles (1 pg/ml–5 ng/ml) in the presence or absence of M6690 (M66: 10 μM), a calpain I/II inhibitor. The inhibition of calpain I/II significantly reduced the release of IL-1α from PBMCs treated for 5 hours. Data are presented as the means ± SEM (n = 5). Statistically significant differences are denoted by * and **, indicating p < 0.05 and p < 0.01, respectively, as determined by one-way ANOVA followed by Bonferroni’s multiple comparison post-test.
These data support the hypothesis that the inflammasome plays a critical role in the inflammatory effect of soot particles on PBMCs from smokers.
Discussion
In this study, we found that human PBMCs derived from healthy smokers are more susceptible to ultrafine soot particle-induced IL-1-dependent inflammation. We found that treating PBMCs from smokers with soot particles induced the activation of the NLRP3 inflammasome, leading to caspase-1 activation and the ensuing release of IL-1α, IL-18 and IL-33, which are responsible for the pro-inflammatory activity of soot particles. The effect was observed at concentrations (50, 100 and 500 pg/ml) of soot particles much lower than those already reported in the literature (μg/ml)7,19,20,21. Instead, higher concentrations of soot particles led to the release of immunosuppressive IL-10 from PBMCs from both smokers and non-smokers, although IL-10 was higher from non-smokers. Moreover, in sharp contrast with previous studies24,25, the administration of soot particles did not lead to cell death.
IL-1α is an alarmin that under physiological conditions ‘instructs’ the adaptive immune system to trigger sterile inflammation18. Likewise, IL-1β and IL-18, according to the microenvironment, can promote T cell survival and the polarization of Th1, Th2 and Th17 cells and mediate leukocyte migration26. The results of this study showed that soot particles were able to induce the release of these cytokines at concentrations ranging from 5 pg/ml up to 100 pg/ml. In support of this, Bengalli et al.27 showed that treating a human monocyte cell line, THP-1 cells, with fine (10 nm) particulate matter caused the release of IL-1β in an NLRP3 inflammasome-dependent manner due to the recognition of particles by Toll-like receptors (TLRs) 2 and 4, which are first activated and stimulate the induction of a second signal, NLRP3 activation, that promotes the assembly of the inflammasome complex. Similarly, we found that soot particles can lead to IL-1α, IL-18 and IL-33 release via the activation of NLRP3 and caspase-1. However, the effect was observed only for PBMCs from smokers, while no effect could be observed for PBMCs from healthy non-smokers. Moreover, this effect was observed at concentrations much lower than what has been reported in the literature.
In addition, soot particles were able to release IL-33 from both macrophagic cells (J774.1) and PBMCs obtained from smokers. IL-33 is an alarmin of the IL-1 family that is crucial for the innate immune response, as it regulates immune cell infiltration and activation18.
The release of IL-1-like cytokines promotes the activation of NF-κB28, which is at the basis of inflammatory pathways. In support of this, several authors demonstrated that soot particles can lead to NF-κB activation in both macrophage/monocyte cell lines and a human epithelial cell line (A549)29,30.
According to the sterile inflammation theory, sterile, noninfectious insults, such as reactive oxygen species (ROS), oxidized and/or methylated DNA, high-mobility group box 1 (HMGB1), heat shock proteins, and ATP, all of which are generally identified as damage-associated molecular patterns (DAMPs), can induce chronic inflammation18. All these stimuli can behave as tumour promoters, as their underlying activity is the induction of chronic inflammation that, rather than providing a protective response to the loss of tissue homeostasis, can aberrantly facilitate tumour development. These insults, both endogenous and exogenous, are sensed by the inflammasome31. Our results demonstrate that UFP at very low concentrations can lead to NLRP3 inflammasome activation, which is responsible for the release of IL-1-like cytokines. However, it is very interesting that this phenomenon was observed only in PBMCs from smokers and not those from non-smokers, which instead showed immunosuppressive behaviour. Therefore, it is tempting to speculate that smokers, a high-risk population, exposed to ultrafine soot particles may be more susceptible to lung cancer than non-smokers. The inhalation of particulate matter together with smoking derivatives can lead to cell transformation, which is favoured by a pro-inflammatory scenario as in the case of IL-1-like cytokine release. In particular, because we observed that the release of pro-inflammatory cytokines occurs at low concentrations of UFP, it is likely that, in vivo, the presence of low amounts of soot particles in the respiratory tract together with smoking can induce adverse toxicological effects, such as cell transformation, in contrast to what has so far been found using high amounts (on the order of μg/ml) of soot particles. However, cell transformation occurs in epithelial cells, whereas our study was conducted on PBMCs, which are leukocytes. However, it is well-known that the immune system plays a pivotal role in tumour immune escape in that the immune cells are tolerogenic and therefore incapable of recognizing transformed cells as non-self32. In this context, immunosuppression overtakes the immunostimulatory activity of innate immune cells such as dendritic cells and macrophages, which in their tolerant phenotype can facilitate the immunosuppressive arm of the adaptive immune system22. In support of this, we found that higher concentrations of soot particles (500 pg/ml–5 ng/ml) were able to induce the release of IL-10 from macrophages and PBMCs. IL-10-producing macrophages are reported to be M2 polarized cells that, in the context of tumours, can facilitate tumour progression, confirming the hypothesis about the role of soot particle-induced IL-1-like cytokines in the tumour microenvironment33.
Another issue to consider is the absence of cell death after treating both macrophages and PBMCs with soot particles. We found that neither J774.1 cells nor PBMCs underwent cell death after 5 and 24 hour of treatment with soot particles. These data are in contrast with literature data24,25. One explanation is that all the studies conducted so far have been performed with higher concentrations (μg/ml) of soot particles than our experimental conditions (1 pg/ml up to 5 ng/ml). In support of this, Pierdominici et al.11 demonstrated that T lymphocytes were not induced to undergo apoptosis or necrosis after soot particle treatment; rather, they found that the autophagy machinery was activated due to alterations in the mitochondrial membrane potential. Indeed, ROS are highly produced after the administration of soot particles. Several studies have reported that high concentrations of soot particles lead to the release of ROS, with subsequent cell death13,14,15. Similar to the findings of Pierdominici et al.11, we did not observe cell death after soot particle addition. The production of ROS by mitochondria is one of the signals that is able to induce NLRP3 inflammasome activation34. Therefore, although we still do not know whether soot particles activate the inflammasome in a direct or indirect manner, we can confirm that the activation of this complex leads to the release of not only IL-1β, as already reported, but also other alarmins, such as IL-1α, IL-18 and IL-33. Moreover, it is worth noting that IL-1α was induced not only after the activation of the inflammasome but also by the induction of the calpain machinery. Gross et al.35 demonstrated that the activation of the NLRP3, NLRC4 and absent in melanoma 2 (AIM2) inflammasomes can lead to the release of IL-1α, but not in a direct manner. In this scenario, the authors showed that IL-1α was not universally inflammasome dependent but rather was dependent on calcium-dependent calpain protease activity. Supporting this paper is the fact that IL-1α is not a substrate of caspase-1, although some danger molecules can regulate the inflammasome-dependent processing and release of IL-1α in mouse bone-marrow-derived macrophages. In this context, we found that calpain is involved in soot particle-induced IL-1α release by PBMCs obtained from healthy smokers, in contrast to the results for non-smokers. However, based on our data, it is most likely that the addition of IL-1α was primarily dependent on the activation of calpain I/II, which is calcium-dependent. It is worthy to note that UFP were reported to induce intracellular calcium transients36. Moreover, in our previous study, we found that plasmacytoid dendritic cells (pDCs) derived from the cancerous lesions of patients with lung cancer were able to release IL-1α, rendering pDCs tolerogenic in that they highly populated tumour lesions, thus implying their role in tumour proliferation22. Similarly, PBMCs from smokers, a population well known to be at high risk of lung cancer, released higher levels of IL-1α and IL-10 than PBMCs from non-smokers, implying that the release of both types of cytokines may favour cell transformation and thus carcinogenesis. However, the levels of IL-1α were not as high as that for the other cytokines (IL-33 and IL-18). This issue could be due to the limits of the ELISA kit, or it could imply that even low concentrations have biological impacts. In support of this, Cohen et al. found that UV-irradiated cells released approximately 200 pg/ml of IL-1α, which in this paper is considered to be an alarmin that signals genotoxic stress37. Similarly, in our previous study22, we found that approximately 150–200 pg/ml of IL-1α were released from cancerous cells after stimulation. In contrast, healthy cells had a basal release of IL-1α at approximately 20 pg/ml, while the release from lung cancer cells was higher (~34 pg/ml), suggesting that even low (detectable) levels can be involved in lung pathogenesis. Based on this result, we believe that the statically significant increase in IL-1α from PBMCs post soot particle treatment can interfere with host immune responses. It is to note though, that treatment of cells with recombinant IL-1α needed higher concentrations (approximately μg/ml) of this cytokine to observe a biological effect in in vitro studies.
In conclusion, our study highlights the molecular mechanism by which very small nanoparticles induce the release of more IL-1α, IL-18 and IL-33 in smoking individuals than non-smokers, who instead showed higher release of the immunosuppressive cytokine IL-10, implying host defence against the pro-inflammatory activity of IL-1-like cytokines. In contrast, although PBMCs from smokers released IL-10 after the addition of soot particles at high concentrations (500 pg/ml–5 ng/ml), the levels of IL-10 were lower than those in non-smokers, implying that both smoking and air pollution can induce pulmonary inflammation in an IL-1-like manner.
The findings of the present study demonstrate the molecular mechanism that underlies the pronounced susceptibility of smokers to inflammatory-based airway diseases once exposed to air pollution. Human PBMCs derived from healthy smokers are more susceptible to ultrafine soot particle-induced IL-1-dependent inflammation via activation of the NLRP3 inflammasome, which leads to caspase-1 activation and the ensuing release of IL-1α, IL-18 and IL-33.
Our data provide new perspectives for the investigation of the role of inhaled combustion ultrafine particles that together with other pulmonary insults can lead to inflammation that may underlie allergic diseases, lung fibrosis and lung cancer. Taken together, these data provide new insight into pulmonary inflammation that can be induced by very small nanoparticles after exposure to air pollution.
Materials and Methods
Human Samples
We used blood from healthy volunteers (smokers and non-smokers) recruited at the “Monaldi-Azienda Ospedaliera (AORN)-Ospedale dei Colli” Hospital in Naples, Italy, after their approval according to the Review Board of the hospital and the patients’ informed consent. In addition, all experimental protocols were, as stated above, approved by the Review Board of “Monaldi-AORN-Ospedale dei Colli” Hospital (approval number 1254/2014). In addition all experimental protocols were performed in accordance with the guidelines and regulations provided and accepted by the Review Board of the “Monaldi-AORN-Ospedale dei Colli” Hospital. The healthy volunteers were 50 ± 10 years of age. All subjects had no history of allergic diseases or chronic respiratory conditions. Blood was collected and used within 24 hours.
Isolation of human PBMCs
Mononuclear cells were isolated according to Ficoll’s protocol. Briefly, blood (5 ml) was mixed with cell medium (5 ml) supplemented with sole antibiotics and Ficoll medium (Life Sciences, Italy). Then, it was centrifuged at 1125 g for 20 minutes. The PBMC layer was collected, diluted with cell medium and centrifuged at 753 g to remove the remaining Ficoll solution. Platelets were separated from the PBMCs by centrifugation at 149 g for 10 minutes. PBMCs were collected in cell medium and plated.
Cell culture
Macrophagic cells, J774.1 cells, and PBMCs were cultured in Roswell Park Memorial Institute (RPMI) 1640 (Cambrex Biosciences, Microtech, Naples, Italy) supplemented with 10% heat-inactivated foetal bovine serum (FBS), 100 units/ml penicillin, 100 units/ml streptomycin and 2 mM L-glutamine (Cambrex Biosciences, Microtech, Naples, Italy). Cells were seeded and treated with LPS (0.1 μg/ml, Alexis, Vincibiochem, Italy), ATP (0.5 mM, Sigma Aldrich, Italy), soot particles (1 pg/ml–5 ng/ml), Ac-YVad (Y-Vad: 1 μg/ml), or M6690 (M66: 10 μM). Cells were treated for 5 or 24 hours, as reported in the Results section.
Soot samples
Soot particles were collected at the exhaust of an atmospheric pressure acetylene/air premixed flame operated in fuel-rich conditions (equivalence ratios greater than 3). The flame was ignited on a McKenna burner, and it was stabilized on the burner by a metal plate located approximately 3 cm from the burner outlet. The soot particles deposited on the metal plate after some hours of flame operation were removed by mechanical ablation and collected as a powder. The obtained sample was washed with dichloromethane to remove the organic fraction condensed on the particles, dried and used for spectroscopic, elemental and morphologic analyses. Ultraviolet-visible (UV-vis) and infrared spectroscopy and Raman spectroscopy were used for structural analysis of the carbon-network constituting the soot particles38. Soot particles appeared as a network of aromatic structures with few peripheral H-atoms. The size of the aromatic network was relatively small, as the dimension of the polyaromatic subunits retrieved from Raman spectroscopy was smaller than 2 nm. Elemental analysis confirmed the low presence of H atoms in the soot particles, as the amount of C was approximately 96%, in mass, of the total material, and H was approximately 2.5–2.8%, with the rest being trace compounds. Atomic force microscopy of the material deposited directly from the flame on mica disks and before any further sample treatment showed that the soot particles were made up of spherical particles with sizes on the order of 20–30 nm and larger, chain-like, soot agglomerates with sizes up to a few hundred nanometers39,40.
Sample preparation
Soot particles were dispersed in bidistilled water to obtain a suspension with a concentration of 5 ppm (5 μg/ml). This constituted the pristine sample from which different concentrations could be obtained. The suspension was sonicated to allow particle disaggregation and prevent further agglomeration. The UV-vis absorption spectrum for this suspension was measured, and the spectral extinction coefficient was retrieved. The particle concentration in the suspension was then obtained from the spectral extinction coefficient using the Lambert-Beer law, known as the spectral absorption cross section of soot from the literature41,42,43. The mass concentration was determined assuming a soot density of 1.8 g/cm3. After 24 hours, we observed a deposit of soot particles and a decrease in the concentration of soot in UV-vis absorption measurements performed at intervals of 6 hours. To obtain a stable suspension, we decided to add 10% dimethyl sulfoxide (DMSO) to the suspension and, after sonication, we did not observe any deposits after 72 hours. UV-vis absorption confirmed that the concentration of soot remained constant after 72 hours as well. Dynamic light scattering was thus used to determine the size distribution of the particles effectively suspended in water/DMSO solution. The sizes of the suspended particles ranged from 20 nm up to approximately 300 nm, with mean sizes ranging from 80 to 120 nm.
Cytokine measurements
Interleuchin (IL) -1α, IL-1β, IL-33, IL-18, IL-10, tumor necrosis factor (TNF) -α and IL-6 were measured in cell-free supernatants using commercially available enzyme-linked immunosorbent assay kits (ELISAs) (eBioscience, CA, USA).
MTT assay
To assess cell viability, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to medium-free cells post treatment. DMSO was used to dissolve the purple formazan crystals. The formazan concentration was determined by measuring the optical density. The data are presented as absorbance (550 nm) vs treatment.
Statistical Analysis
Data are reported as the mean ± standard error media (SEM). Statistical differences were assessed with one-way Analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison post-test, and p values less than 0.05 were considered significant.
Additional Information
How to cite this article: De Falco, G. et al. Human peripheral blood mononuclear cells (PBMCs) from smokers release higher levels of IL-1-like cytokines after exposure to combustion-generated ultrafine particles. Sci. Rep. 7, 43016; doi: 10.1038/srep43016 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was supported by funds from the University of Salerno for basic research (FARB), European Funds for regional development (FSER)-Regional programme (POR) Campania FESR 2007–2013- RETE DELLE BIOTECNOLOGIE IN CAMPANIA - Progetto “MOdelli in Vivo di Patologie umanE - MOVIE”, and POR-FSE 2007–2013 Research in Experimental Medicine (CREME), awarded to AP. RS was supported by a fellowship from the Italian Ministry for Research (MIUR) Protocol N. 62198. MT was supported by a fellowship from the PhD programme in Drug Discovery and Development, University of Salerno.
Footnotes
The authors declare no competing financial interests.
Author Contributions G.D.F., M.T., M.S., M.C., A.D.A., R.P.A., P.A., and R.S. designed the experimental protocol. G.D.F. and M.T. performed the experiments. M.C. and M.S. prepared the soot particle samples. A.D.A., R.P.A., P.A., and R.S. interpreted the data and wrote the manuscript. All authors read and approved the final manuscript.
References
- Lotti M., Olivato I. & Bergamo L. Inflammation and short-term cardiopulmonary effects of particulate matter. Nanotoxicology 3(1), 27–32 (2009). [Google Scholar]
- Oberdörster G., Stone V. & Donaldson K. Toxicology of nanoparticles: A historical perspective. Nanotoxicology 1(1), 2–25 (2007). [Google Scholar]
- Terzano C., Di Stefano F., Conti V., Graziani E. & Petroianni A. Air pollution ultrafine particles: toxicity beyond the lung. Eur Rev Med Pharmacol Sci. 14(10), 809–821 (2010). [PubMed] [Google Scholar]
- D’Anna A. Combustion-formed Nanoparticles-an invited topical review. Proc. Combust. Inst. 32, 593–613 (2009). [Google Scholar]
- Beamer C. A. et al. IL-33 mediates multi-walled carbon nanotube (MWCNT)-induced airway hyper-reactivity via the mobilization of innate helper cells in the lung. Nanotoxicology 7(6), 1070–81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Zhang Q., Kaplan B. L., Baker G. L. & Kaminski N. E. Induced T cell cytokine production is enhanced by engineered nanoparticles. Nanotoxicology 8 Suppl 1, 11–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provoost S. et al. Diesel exhaust particles stimulate adaptive immunity by acting on pulmonary dendritic cells. J. Immunol. 184(1), 426–432 (2010). [DOI] [PubMed] [Google Scholar]
- Shaw C. A. et al. Protein corona formation in bronchoalveolar fluid enhances diesel exhaust nanoparticle uptake and pro-inflammatory responses in macrophages. Nanotoxicology 10(7), 981–91 (2016). [DOI] [PubMed] [Google Scholar]
- Acciani T. H., Brandt E. B., Khurana Hershey G. K. & Le Cras T. D. Diesel exhaust particle exposure increases severity of allergic asthma in young mice. Clin. Exp. Allergy. 43(12), 1406–1418 (2013). [DOI] [PubMed] [Google Scholar]
- Brandt E. B. et al. Exposure to allergen and diesel exhaust particles potentiates secondary allergen-specific memory responses, promoting asthma susceptibility. J. Allergy Clin. Immunol. 136(2), 295–303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierdominici et al. Diesel exhaust particle exposure in vitro impacts T lymphocyte phenotype and function. Part Fibre Toxicol. 11, 74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poillet-Perez L., Despouy G., Delage-Mourroux R. & Boyer-Guittaut M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biology 4, 184–192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerloff K., Albrecht C., Boots A. W., Förster I. & Schins R. P. F. Cytotoxicity and oxidative DNA damage by nanoparticles in human intestinal Caco-2 cells. Nanotoxicology 3(4), 355–364 (2009). [Google Scholar]
- Tseng C. Y., Wang J. S. & Chao M. W. Causation by Diesel Exhaust Particles of Endothelial Dysfunctions in Cytotoxicity, Pro-inflammation, Permeability, and Apoptosis Induced by ROS Generation. Cardiovasc. Toxicol., doi: 10.1007/s12012-016-9364-0 (2016). [DOI] [PubMed] [Google Scholar]
- Vesterdal L. K. et al. Pulmonary exposure to particles from diesel exhaust, urban dust or single-walled carbon nanotubes andoxidatively damaged DNA and vascular function in apoE(−/−) mice. Nanotoxicology 8(1), 61–71 (2014). [DOI] [PubMed] [Google Scholar]
- Rimessi A., Previati M., Nigro F., Wieckowski M. R. & Pinton P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. The International Journal of Biochemistry & Cell Biology S1357–2725(16), 30155–8, doi: 10.1016/j.biocel.2016.06.015 (2016). [DOI] [PubMed] [Google Scholar]
- Shimada K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36(3), 401–414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlizzi M., Casolaro V., Pinto A. & Sorrentino R. Inflammasome: cancer’s friend or foe? Pharmacol. Ther. 143(1), 24–33 (2014). [DOI] [PubMed] [Google Scholar]
- Girtsman T. A., Beamer C. A., Wu N., Buford M. & Holian A. IL-1R signalling is critical for regulation of multi-walled carbon nanotubes-induced acute lung inflammation in C57Bl/6 mice. Nanotoxicology 8(1), 17–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabolli V. et al. The alarmin IL-1α is a master cytokine in acute lung inflammation induced by silica micro- and nanoparticles. Part Fibre Toxicol. 11, 69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totlandsdal A. I., Cassee F. R., Schwarze P., Refsnes M. & Låg M. Diesel exhaust particles induce CYP1A1 and pro-inflammatory responses via differential pathways in human bronchial epithelial cells. Part Fibre Toxicol. 7, 41 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrentino R. et al. A. Human lung cancer-derived immunosuppressive plasmacytoid dendritic cells release IL-1α in an AIM2 inflammasome-dependent manner. Am. J. Pathol. 185(11), 3115–3124 (2015). [DOI] [PubMed] [Google Scholar]
- Mittal S. K., Cho K. J., Ishido S. & Roche P. A. Interleukin 10 (IL-10)-mediated Immunosuppression: March-i induction regulates antigen presentation by macrophages but not dendritic cells. J. Biol. Chem. 290(45), 27158–27167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo M. et al. Diesel exhaust particle-induced cell death of human leukemic promyelocytic cells HL-60 and their variant cells HL-NR6. Biol. Pharm. Bull. 24(4), 357–363 (2001). [DOI] [PubMed] [Google Scholar]
- Mazzarella G. et al. Effects of diesel exhaust particles on human lung epithelial cells: an in vitro study. Respir Med. 101(6), 1155–1162 (2007). [DOI] [PubMed] [Google Scholar]
- Joosten L. A., Netea M. G. & Dinarello C. A. Interleukin-1β in innate inflammation, autophagy and immunity. Semin. Immunol. 25(6), 416–424 (2013). [DOI] [PubMed] [Google Scholar]
- Bengalli R. et al. Release of IL-1 β triggered by Milan summer PM10: molecular pathways involved in the cytokine release. Biomed Res Int. 2013, 158093, doi: 10.1155/2013/158093 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoesel B. & Schmid J. A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer. 12, 86 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton D. B. et al. Primary alveolar macrophages exposed to diesel particulate matter increase RAGE expression and activate RAGE signaling. Cell Tissue Res. 358(1), 229–238 (2014). [DOI] [PubMed] [Google Scholar]
- Nam H. Y. et al. Diesel exhaust particles increase IL-1β-induced human β-defensin expression via NF-κB-mediated pathway in human lung epithelial cells. Particle and Fibre Toxicology 3, 9 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O., Thomas C. J., Guarda G. & Tschopp J. The inflammasome: an integrated view. Immunol. Rev. 243(1), 136–151 (2011). [DOI] [PubMed] [Google Scholar]
- Zitvogel L., Tesniere A. & Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat. Rev. Immunol. 6(10), 715–727 (2006). [DOI] [PubMed] [Google Scholar]
- Solinas G., Germano G., Mantovani A. & Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 86(5), 1065–1073 (2009). [DOI] [PubMed] [Google Scholar]
- Heid M. E. et al. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 191(10), 5230–5238 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O. et al. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36(3), 388–400 (2012). [DOI] [PubMed] [Google Scholar]
- Moller W., Brown D. M., Kreyling W. G. & Stone V. Ultrafine particles cause cytoskeletal dysfunctions in macrophages: role of intracellular calcium. Particle and Fibre Toxicology 2, 7 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I. et al. IL-1α is a DNA damage sensor linking genotoxic stress signaling to sterile inflammation and innate immunity. Sci Rep. 5, 14756, doi: 10.1038/srep14756 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C., D’Anna A., Ciajolo A. & Sirignano M. Analysis of the chemical features of particles generated from ethylene and ethylene/2,5 dimethyl furan flames. Combust. Flame. 167, 268–273 (2016). [Google Scholar]
- Barone A. C., D’Alessio A. & D’Anna A. Morphological Characterization of the Early Process of Soot Formation by Atomic Force Microscopy. Combust. Flame. 132(1/2), 181–187 (2003). [Google Scholar]
- De Falco G., Commodo M., Minutolo P. & D’Anna A. Flame-Formed Carbon Nanoparticles: Morphology, Interaction Forces and Hamaker Constant from AFM. Aerosol Science and Technology 49(5), 281–289 (2015). [Google Scholar]
- Dalzell W. H. & Sarofim A. F. Optical Constants of Soot and their Application to Heat-Flux Calculations. J. Heat Transfer 91(1), 100–104 (1969). [Google Scholar]
- Cecere D. et al. Evidence and Characterization of Nanoparticles produced in non-Sooting Premixed Flames. Combust. Sci. Technol. 174(11–12), 377–398 (2002). [Google Scholar]
- D’Anna A., Rolando A., Allouis C., Minutolo P. & D’Alessio A. Nano-Organic Carbon and Soot Particle Measurements in a Laminar Ethylene Diffusion Flame. Proc. Combust Inst. 30(1), 1449–1456 (2005). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.