

Abstract
Despite considerable research to uncover them, the anatomic and neuropathologic correlates of memory impairment in dementia with Lewy bodies (DLB) remain unclear. While some studies have implicated Lewy bodies in the neocortex, others have pointed to α-synuclein pathology in the hippocampus. We systematically examined hippocampal Lewy pathology and its distribution in hippocampal subfields in 95 clinically and neuropathologically characterized human cases of DLB, finding that α-synuclein pathology was highest in two hippocampal-related subregions: the CA2 subfield and the entorhinal cortex (EC). While the EC had numerous classic somatic Lewy bodies, CA2 contained mainly Lewy neurites in presumed axon terminals, suggesting the involvement of the EC → CA2 circuitry in the pathogenesis of DLB symptoms. Clinicopathological correlations with measures of verbal and visual memory supported a role for EC Lewy pathology, but not CA2, in causing these memory deficits. Lewy pathology in CA1—the main output region for CA2—correlated best with results from memory testing despite a milder pathology. This result indicates that CA1 may be more functionally relevant than CA2 in the context of memory impairment in DLB. These correlations remained significant after controlling for several factors, including concurrent Alzheimer's pathology (neuritic plaques and neurofibrillary tangles) and the interval between time of testing and time of death. Our data suggest that although hippocampal Lewy pathology in DLB is predominant in CA2 and EC, memory performance correlates most strongly with CA1 burden.
SIGNIFICANCE STATEMENT This study provides a detailed neuropathologic analysis of hippocampal Lewy pathology in human patients with autopsy-confirmed dementia with Lewy bodies. The approach—informed by regional molecular markers, concurrent Alzheimer's pathology analysis, and relevant clinical data—helps tease out the relative contribution of Lewy pathology to memory dysfunction in the disease. Levels of Lewy pathology were found to be highest in the hippocampal CA2 subregion and entorhinal cortex, implicating a potentially overlooked circuit in disease pathogenesis. However, correlation with memory performance was strongest with CA1. This unexpected finding suggests that Lewy pathology must reach a critical burden across hippocampal circuitry to contribute to memory dysfunction beyond that related to other factors, notably coexisting Alzheimer's disease tau pathology.
Keywords: α-synuclein, dementia with Lewy bodies, hippocampus, memory, spread, subregion
Introduction
Memory impairment is often a prominent feature of dementia with Lewy bodies (DLB), an age-related neurodegenerative disease that shares numerous features with Alzheimer's disease (AD) and Parkinson's disease (PD; Ballard et al., 1996; Salmon et al., 1996; Walker et al., 1997; Connor et al., 1998; Shimomura et al., 1998; Heyman et al., 1999; Calderon et al., 2001). These studies suggest early development of significant pathology in the hippocampus and surrounding cortical regions that are crucial for learning and memory (Squire, 1992). Lewy bodies (LBs) are intracellular protein aggregates containing α-synuclein and confirm the diagnosis of DLB at autopsy (Harding and Halliday, 2001). While brainstem LBs are thought to contribute to motor symptoms, the neural substrate for cognitive symptoms remains elusive (Colosimo et al., 2003; Parkkinen et al., 2005).
Few studies have systematically examined the relationship between Lewy pathology in the hippocampus and memory performance in DLB, choosing instead to study the dementia of late PD, a distinct clinical entity known as Parkinson's disease dementia, or PDD (Churchyard and Lees, 1997; Hall et al., 2014). More importantly, none have performed a detailed subregional analysis at the level of hippocampal subfields and related cortical regions. As the distinct functions (Wintzer et al., 2014) and molecular identities (Lein et al., 2004; Hawrylycz et al., 2012) of hippocampal subfields become clearer, specific localization of pathologies will help inform us of their role in symptomatology. Examining the distribution of pathology within hippocampal circuitry may also provide insight into disease pathogenesis, given the previous evidence that misfolded α-synuclein can propagate among interconnected regions (Luk et al., 2012).
The CA1 subfield of the hippocampus is an area important for memory function, as selective damage there can lead to amnesia (Zola-Morgan et al., 1986). In AD, CA1 is preferentially affected by plaque and tangle pathology, which may account for the severe amnesia that characterizes the disease; tangle pathology first emerges in the transentorhinal cortex, followed by entorhinal cortex (EC), a gateway region between the hippocampus and the rest of cortex (Hyman et al., 1984; Arnold et al., 1991; Braak and Braak, 1995). Lewy pathology is also found in CA1 and EC in DLB (Armstrong and Cairns, 2015), often coexisting with AD pathology (Hansen et al., 1993; Harding and Halliday, 2001; Horimoto et al., 2003; Tsuboi and Dickson, 2005). However, the relative contributions of Lewy and AD pathology in these regions to memory dysfunction remain unknown.
Although CA1 Lewy pathology may contribute to memory dysfunction, CA1 is less affected by Lewy pathology than other hippocampal subfields in either DLB (Dickson et al., 1994; Armstrong and Cairns, 2015) or sporadic PD (Braak et al., 2003; Bertrand et al., 2004; Armstrong et al., 2013). CA2/3 is usually cited as the predominant site of hippocampal Lewy pathology, due to difficulty in anatomically identifying the boundary between these subfields (Woodhams et al., 1993; Lein et al., 2005). Both regions have been implicated in various aspects of memory (e.g., social or episodic memory), notably in animal models (DeVito et al., 2009; Langston et al., 2010; Hitti and Siegelbaum, 2014); thus, given their heavy Lewy pathology burden, they could be involved in the prominent memory deficit in DLB (Hamilton et al., 2004).
In this study, we used molecular markers to help delineate specific hippocampal subfields and related parahippocampal regions in brains from patients with autopsy-confirmed DLB. We then evaluated the distribution and severity of Lewy pathology across these regions, accounting for the influence of concomitant AD pathology and time between memory testing and death. Based on previous reports, we hypothesized that Lewy pathology would be most prominent in CA2 or CA3 and in EC. We took advantage of this detailed analysis of hippocampal anatomy to address two questions. First, do subregions with heavy Lewy pathology burden contribute to memory impairment in DLB, or does CA1 need to be involved? Second, does lesion distribution within hippocampal subfields support a model of spreading pathology?
Materials and Methods
Research subjects.
Cases were selected from the brain bank of the Shiley-Marcos Alzheimer's Disease Research Center (ADRC) at the University of California, San Diego (UCSD). Subjects had been characterized using standardized and detailed clinical, neurological, and neuropsychological assessments during life as described previously (Salmon et al., 1996), and were followed longitudinally at the ADRC. UCSD ADRC procedures at autopsy are as follows: the brain is divided sagittally, and the left hemibrain is fixed in 10% buffered formalin, while the right hemibrain is sectioned coronally and then frozen at −70°C in sealed plastic bags. Routinely, tissue blocks from the right hemibrain of the midfrontal, inferior parietal, and superior temporal cortices, primary visual cortex in the occipital cortex, hippocampus, basal ganglia, substantia nigra, and cerebellum are removed and placed in 2% paraformaldehyde for subsequent thick sectioning by vibratome. Tissue blocks adjacent to the ones described above are stored at −70°C for subsequent immunoblot analysis for synaptic proteins and Aβ species (soluble and oligomers). Vibratome sections (40 μm thick) are stored in cryoprotective medium at −20°C for subsequent immunochemical studies. The formalin-fixed left hemibrain is serially sectioned in 1 cm slices, and tissue blocks from the regions described above are processed for histopathological examination by H&E and Thioflavin-S (Thio-S) to detect tau and β-amyloid deposits. TDP-43 staining and volumetric analyses are not routinely performed in these cases. LB pathology is evaluated using phosphorylated α-synuclein immunoreactivity. Pathological diagnoses of AD and DLB are made using National Institute on Aging-Alzheimer's Association (NIA-AA) guidelines (Montine et al., 2012) and guidelines proposed by the DLB Consortium (McKeith et al., 2005).
One hundred and seven cases (76 males, 31 females) were identified with neuropathologically confirmed DLB and available neuropsychological testing data (Table 1). Twelve of these cases were excluded for lack of hippocampal tissue, leaving a total of 95 cases to analyze for hippocampal Lewy pathology burden. Based on overall distribution of pathology, these cases were grouped into brainstem-predominant, limbic (transitional), or neocortical (diffuse) subtypes, as outlined in the most recent DLB Consortium report (McKeith et al., 2005). As for coexisting AD pathology, Braak stage for tau tangle distribution and Thal phase for amyloid plaques were also recorded. Vascular amyloid pathology was assessed semiquantitatively (0–3 scale, ranging from none to severe).
Table 1.
Subject demographics
| Male/female | Age | Death age | Education | MMSE | |
|---|---|---|---|---|---|
| All cases | 76/31 | 74.45 (6.8) | 80.36 (7.6) | 15.42 (3.0) | 24.08 (3.9) |
| Brainstem predominant | 5/0 | 71.00 (10.6) | 76.80 (12.7) | 16.40 (2.6) | 25.40 (5.1) |
| Limbic (transitional) | 18/9 | 76.78 (6.6) | 82.04 (7.2) | 15.59 (3.2) | 24.65 (4.0) |
| Neocortical (diffuse) | 42/21 | 74.27 (5.7) | 79.94 (6.9) | 15.43 (2.9) | 23.81 (3.8) |
The first row displays demographics for all study subjects (n = 107), some of which were excluded due to lack of stainable hippocampal tissue. Remaining subjects (n = 95) were classified into brainstem-predominant, limbic, and neocortical subtypes, with each row showing the corresponding demographics by DLB subtype. Means (SDs) are shown. MMSE, Mini Mental State Examination.
Immunohistochemistry.
Paraffin-embedded hippocampal blocks were sectioned using a microtome at the posterior level to visualize all subfields and were immunostained for phosphorylated α-synuclein using a mouse monoclonal antibody at 1:20,000 (BioLegend catalog #825701; RRID: AB_2564891) along with an H&E counterstain. Fluorescent immunohistochemistry was used for combined labeling of Lewy pathology and hippocampal subfield markers. Briefly, sections were deparaffinized using xylene and 100% ethanol. Endogenous peroxidase activity was blocked using 30% hydrogen peroxide in methanol, followed by antigen retrieval using a modified citrate buffer (Dako). Sections were permeabilized and blocked with TBS containing 0.25% Triton X-100 and 10% horse serum; primary antibodies for calbindin 28 kDa (rabbit; Swant catalog #C9638; RRID: AB_2314070), secretagogin (SCGN; rabbit; Thermo Fisher Scientific catalog # PA5-30392; RRID: AB_2547866), SLC30A3/zinc transporter 3 (ZnT3; rabbit; MBL International catalog #BMP094; RRID: AB_11126549), phospho-synuclein (P-syn)/81A (same as chromogenic; see above), and Synuclein-oligomeric (sYN-O2)/-fibrillar (F2) (mouse; kindly provided by O. El-Agnaf, Qatar Biomedical Research Institute) were used at 1:200, 1:200, 1:500, 1:20,000 and 1:500, respectively, and incubated for 72 h at 4°C. After rinsing three times with TBS, secondary antibodies, including anti-rabbit conjugated to Cy3 (donkey; Jackson ImmunoResearch catalog #711-165-152; RRID:AB_2307443) and anti-mouse conjugated to Alexa 488 (donkey; Jackson ImmunoResearch catalog #715-545-151; RRID:AB_2341099) were applied at 1:250 for 2 h at room temperature; after three TBS rinses, sections were coverslipped with PVA-DABCO (Sigma) mounting medium.
Assessment of pathology burden.
Lewy pathology burden—including both LBs and Lewy neurites (LNs)—was assessed semiquantitatively (0–4 ordinal scale) in EC, subiculum, CA1, CA2, CA3, CA4, and dentate gyrus (DG) based on NIA-AA criteria (Montine et al., 2012). Ratings were all done by one rater (D.H.A.) blinded to all case information and trained by a neuropathologist (S.R.). As in the DLB Consortium report, 0 corresponds to complete absence of Lewy pathology, 1 corresponds to sparse LBs/LNs, 2 corresponds to >1 LB/high power field (HPF; equivalent to 0.1 mm2) and sparse LNs, 3 corresponds to ≥4 LBs/HPF and scattered LNs, and 4 corresponds to numerous LBs/LNs (McKeith et al., 2005). We used the same anatomical definitions of hippocampal subregions as Armstrong and Cairns (2015), with the addition of mossy fiber (MF) staining using the marker calbindin to define the CA2/CA3 boundary, which is difficult to distinguish solely based on H&E staining. Plaque and tangle counts were obtained from each subregion through standard UCSD ADRC procedures using Thio-S staining, which also allowed for determination of Braak stage and Thal phase, as described above briefly and in previous studies (Salmon et al., 2015). Illustrative bright-field images were obtained on a Nikon Eclipse E800 upright microscope, and fluorescent images were obtained on a Zeiss LSM 780 side port laser-scanning confocal microscope.
Neuropsychological testing.
The California Verbal Learning Test (CVLT) and the Visual Reproduction test from the Wechsler Memory Scale (WMS) were administered to each patient. Patients were tested individually by a trained psychometrist in a quiet, well-illuminated room.
The CVLT is a standardized memory test that was developed to assess a variety of memory processes identified through cognitive psychological studies of normal memory (Delis et al., 1987). The CVLT assesses rate of learning, retention after short and long delay intervals, semantic encoding ability, recognition memory (i.e., discriminability), intrusion and perseverative errors, and response biases. Five presentation/free-recall trials for a list (List A) of 16 items (four items in each of four semantic categories) are administered followed by a single interference trial using a second, different list (List B) of 16 items. Immediately after the List B trial, free-recall and then cued-recall (using the names of the four categories) of the items on the initial list (List A) are elicited. Twenty minutes later, free-recall and cued-recall of the List A items are again elicited, followed by a yes/no recognition test consisting of the 16 List A items and 28 randomly interspersed distracter items.
The WMS Visual Reproduction test (Russell adaptation; Lezak, 1983) provides a measure of memory for geometric forms. On each of three trials, the subject must reproduce a complex geometric figure from memory immediately following a 10 s study period. Three increasingly complex stimuli containing from 4 to 10 components are presented on successive trials. As a measure of long-term retention, the subject is asked after 30 min of unrelated testing to again reproduce the figures from memory. Finally, the subject is asked to simply copy the stimulus figures to assess any visuoperceptual dysfunction that may be contaminating visual memory performance. The reproductions are scored for the number of correct components, out of 21, present from the original stimulus drawings. Thus, the maximum possible score for the immediate and delayed conditions is 21 points.
Statistical analysis.
Associations between ratings of Lewy pathology in various hippocampal subfields and scores on the CVLT and the Visual Reproduction test were examined using partial correlation analyses that controlled for the interval between time of testing and death. Four cases in which most of the hippocampal subfields were not visible were excluded from this analysis. Multiple hierarchical linear regression analyses were performed to examine the ability of CA1 Lewy pathology burden to predict various episodic memory scores (at their initial clinical evaluation) after adjusting for CA1 neuritic plaque and neurofibrillary tangle burden and for the interval between memory testing and death. The first model for CVLT predicted Trial 1–5 learning, the second model predicted short-delay free recall, the third model predicted long-delay free recall, and the fourth model predicted recognition discriminability. The first model for the Visual Reproduction test predicted immediate recall, and the second predicted delayed recall. Analyses were performed using SPSS Statistics software (version 21.0.0.0; IBM). Statistical significance was set at p < 0.05.
Results
Case characterization
Two-thirds of the DLB cases were of the neocortical subtype, indicating that diffuse pathology had reached the neocortex at the time of death. The remaining cases were mostly of the limbic, or transitional, variety, with only five cases with pathology restricted to the brainstem in our entire cohort. (Patients falling into this last category usually do not have dementia; therefore, we expect their cognitive symptoms to be a result of something other than Lewy pathology.) Neocortical and limbic cases displayed a 2:1 male predominance; there were no female brainstem-only cases (Table 1). In the overall group, mean age of testing was 74.45 years, and mean age of death was 80.36 years. The average test–death interval was 5.91 years. Average number of years of education was 15.42. The average score on the Mini Mental State Examination was 24.08, with a worsening trend from brainstem (25.40) to limbic (24.65) to neocortical (23.81) subtypes. In the analyzed cohort, average scores on the various CVLT measures were 22.6 (SD, 11.1) on Trial 1–5 learning, 2.7 (SD, 3.0) on short-delay free recall, 2.6 (SD, 3.3) on long-delay free recall, and 71.6 (SD, 17.1) on recognition discriminability. Average scores on the Visual Reproduction test were 5.6 (SD, 3.7) for immediate recall and 2.3 (SD, 3.3) for delayed recall. Neocortical cases performed significantly worse than the limbic cases only on the CVLT Trial 1–5 learning measure (t(83) = 2.47; p < 0.05) and long-delay free recall (t(83) = 2.00; p < 0.05).
Neuropathologic findings
All but three cases in our cohort had some level of neurofibrillary tangle pathology (Table 2). Of those three, two did not have any amyloid plaques, meaning that 93 out of 95 cases had some type of coexisting AD pathology in addition to Lewy pathology. Lack of plaques was more common overall (18 cases), especially among limbic cases (more than one-third without plaques), whereas the vast majority of cases had a moderate amount of amyloid pathology (Thal phase 3). Among the 18 cases without plaques, 16 had low Braak stage (II or less), which is often observed with age in cognitively normal adults (Crary et al., 2014); these could therefore be considered “pure DLB” cases. Braak stages V and VI were the most common among our cases, indicating severe tangle pathology. This finding was driven by the neocortical cases, since most limbic cases showed milder levels of tangles (Braak stage I–II).
Table 2.
Staging of concomitant Alzheimer's pathology
| All cases | Brainstem | Limbic | Neocortical | |
|---|---|---|---|---|
| Braak stage | ||||
| 0 | 3 | 1 | 1 | 1 |
| I–II | 30 | 2 | 12 | 16 |
| III–IV | 24 | 1 | 5 | 18 |
| V–VI | 38 | 1 | 9 | 28 |
| Thal phase | ||||
| 0 | 18 | 2 | 10 | 6 |
| 1 or 2 | 4 | 1 | 2 | 1 |
| 3 | 70 | 2 | 15 | 53 |
| 4 or 5 | 3 | 0 | 0 | 3 |
AD pathology including Braak neurofibrillary tangle stage and Thal amyloid phase are indicated for all cases and for each Lewy pathology subtype. While severe tau pathology was most commonly seen across all cases, severe amyloid pathology was seen only in three cases, all of which were neocortical. Alternatively, a lack of tau pathology was seen only in three cases, whereas no amyloid was found in 18 cases, most of which were limbic.
Lewy pathology was present in the hippocampus of all but eight cases (five of which were brainstem only, as mentioned above), with significantly higher overall burden in neocortical (mean total pathology across subfields, 7.76; SD, 3.72) compared to limbic cases (mean, 5.48; SD, 2.83; t(80) = −2.66; p < 0.01). On a subfield-by-subfield basis, neocortical cases had greater Lewy pathology than limbic cases in CA1, CA2, CA3, and EC. However, the pattern of severity across subfields did not differ between the two subtypes. As displayed in a representative case, Lewy pathology appeared most severe in CA2 (much more so than in CA3) and in EC (Fig. 1). Whereas CA2 pathology was mostly neuritic, consisting of LNs in putative axon terminals, EC pathology was mostly composed of LBs located in the soma of deep layer neurons.
Figure 1.

Visualization of hippocampal Lewy pathology in DLB. A–D, Phospho-synuclein staining in a representative case reveals mostly Lewy neurites in CA2 (A), Lewy bodies with surrounding Lewy grains in entorhinal cortex (D), and lighter, mixed staining in CA1 in between (B, C). Insets show 4× and 20× magnification (H&E counterstain).
Double labeling of phospho-synuclein along with the known DG marker calbindin allowed us to visualize the pathology in relation to the MFs projecting from DG to CA3 and terminating in CA2. Using this method, we confirmed that LNs localized to CA2, just distal to where the MFs tapered off, fitting both the classical definition of CA2 (Lorente de Nó, 1934) and the broader molecular definition (Lein et al., 2005). Thus, distal CA2 appears to be preferentially affected by Lewy pathology (Fig. 2A). Calbindin labels DG neurons in full, including their MF projections, but is also expressed at low levels in CA1 and CA2. Therefore, a more specific MF stain, ZnT3, was used to confirm the finding. Another α-synuclein antibody that recognizes early, oligomeric forms of the pathology (Vaikath et al., 2015) was used to confirm the specificity of the LN staining and indicated that the phospho-synuclein we observed encompassed the full extent of Lewy pathology. These two new antibodies showed that most of the Lewy pathology (Fig. 2B, green) was just distal to where the MFs (magenta) tapered off.
Figure 2.

Localization of Lewy neurite pathology using molecular markers. A, Lewy pathology, labeled by P-syn staining in green, localizes to CA2 mainly just beyond where mossy fibers from DG, labeled using calbindin in magenta, taper off (20× magnification composite). B, Confirmation using additional markers for mossy fibers and oligomeric α-synuclein: SLC30A3 (ZnT3) in magenta and Syn-O2 in green (20× magnification). C, Subcellular localization using CA2 marker SCGN in magenta and Syn-F2 in green (20× magnification composite, 20× maximum intensity projection for inset).
The observed pathology did not appear to be harbored by the CA2 neurons themselves, but rather by the axon terminals of the EC neurons that projected to CA2. Consistent with this hypothesis, the human CA2 marker SCGN showed that the Lewy pathology (Fig. 2C, green, labeled using a mature fibril-specific α-synuclein antibody that labels only fully formed aggregates) displayed very little colocalization with CA2 pyramidal neurons and their processes (Fig. 2C, magenta). A composite heat map representing the average pathology burden of each hippocampal subregion across all cases shows that Lewy pathology was highest in the CA2 subfield of the hippocampus proper and that EC was also highly affected (Fig. 3). CA2 Lewy pathology burden strongly correlated with EC burden (r = 0.772), CA1 burden with CA3 burden (r = 0.722), and CA3 burden with CA4 burden (r = 0.782). DG had the least amount of Lewy pathology: only ∼20% of cases had any DG Lewy pathology at all, and when it was present, only one or two LBs were found in the entire subfield.
Figure 3.
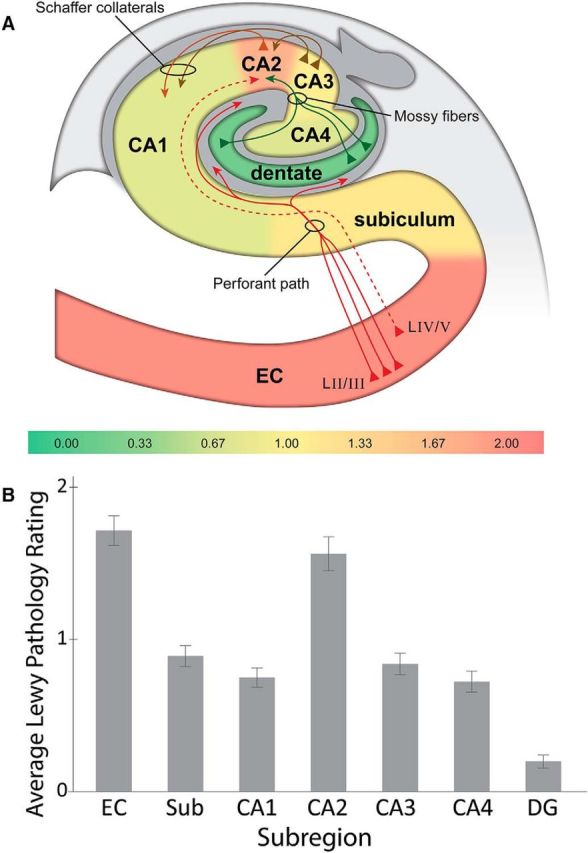
Distribution and severity of Lewy pathology show differential regional involvement. A, Heat map of Lewy pathology in hippocampal subfields from DLB cases (n = 95) shows increased burden in area CA2 and entorhinal cortex. The DG is relatively free of pathology by comparison. The color-coded scale is based on average pathology rating per subregion. Major known hippocampal projections are displayed as solid lines, whereas the suspected EC–CA2 circuit is shown as a dashed line (hippocampal diagram adapted with permission from Yang et al., 2008). B, Graphical representation of heat map data. Sub, Subiculum. Error bars indicate ± SE.
Correlations with neuropsychological testing
Partial correlations that accounted for the test–death interval revealed that increasing Lewy pathology burden in the CA1 subfield of the hippocampus was correlated with lower scores on CVLT Trial 1–5 learning (r = −0.249; p = 0.031) and on immediate (r = −0.249; p = 0.031) and delayed recall (r = −0.290; p = 0.012) on the Visual Reproduction test (Table 3, Fig. 4A,B). In addition, increasing Lewy pathology burden in the EC was correlated with lower scores on CVLT Trial 1–5 learning (r = −0.239; p = 0.023; Table 3, Fig. 4C). There were no significant correlations between memory performance and Lewy pathology burden in any other hippocampal subfields (Table 3). The relative selectivity of the relationship between CA1 Lewy pathology burden and memory should be viewed with caution since multiple comparisons increased the chance of false positive errors. We attempted to mitigate this problem to some degree by including only four key memory variables in the partial correlation analyses. We chose not to correct α for multiple comparisons because the analyses were exploratory, significant correlations consistently involved CA1 rather than occurring randomly, and the results of the partial correlations with CA1 were confirmed in subsequent regression analyses.
Table 3.
Clinicopathological correlations
| CVLT Trials 1–5 | CVLT long delay | Visual Reproduction immediate | Visual Reproduction delay | |
|---|---|---|---|---|
| Dentate | −0.074 | −0.042 | 0.064 | 0.001 |
| CA4 | −0.006 | 0.135 | −0.010 | −0.108 |
| CA3 | −0.063 | 0.077 | −0.092 | −0.141 |
| CA2 | −0.092 | 0.137 | 0.015 | 0.033 |
| CA1 | −0.249* | −0.161 | −0.249* | −0.290* |
| Subiculum | −0.119 | 0.001 | −0.011 | −0.058 |
| Entorhinal cortex | −0.258* | −0.035 | −0.071 | −0.135 |
Partial correlation coefficients (Pearson's r) showing the relationship between the degree of Lewy pathology in various hippocampal subfields and performance on verbal and nonverbal memory tests. The partial correlations control for the interval between testing and death.
*p < 0.05.
Figure 4.

Increasing Lewy pathology burden is associated with decreasing memory performance. A, Relationship between ratings of CA1 Lewy pathology and score on the CVLT Trial 1–5 learning measure (added interpolation line). B, C, Relationship between ratings of CA1 Lewy pathology and score on the Visual Reproduction test in immediate (B) and delay (C) conditions. D, Relationship between ratings of entorhinal cortex Lewy pathology and score on the CVLT Trial 1–5 learning measure. Error bars indicate ± SE.
A series of multiple regression analyses were performed to explore the relative contribution of Lewy pathology and AD pathology in CA1 to memory dysfunction. Lewy pathology ratings, neurofibrillary tangle counts, neuritic plaque counts, and the interval between time of testing and death were used to predict scores on various verbal and nonverbal memory measures (Table 4). We chose to use raw counts for tangles and plaques rather than Braak stage and Thal phase, as the latter reflect the overall distribution of the respective neuropathologic lesions more so than the burden in a specific subregion. Average tangle counts for CA1 in our cohort were 11.0/HPF (SD, 12.6) and average neuritic plaque counts were 7.7/HPF (SD, 7.8). These numbers are lower than those found in a comparable AD-only cohort from the UCSD ADRC (18.7 and 12.0, respectively), suggesting an additive effect of Lewy and AD pathologies.
Table 4.
Multiple regression analyses
| β | t test | p | |
|---|---|---|---|
| CVLT Trials 1–5 | |||
| CA1 α-synuclein | −0.237 | −2.40 | 0.019 |
| CA1 plaques | −0.054 | −0.49 | 0.620 |
| CA1 tangles | −0.395 | −3.35 | 0.001 |
| Test–death interval | 0.537 | 4.97 | 0.000 |
| CVLT short delay | |||
| CA1 α-synuclein | −0.255 | −2.47 | 0.016 |
| CA1 plaques | 0.047 | 0.42 | 0.679 |
| CA1 tangles | −0.472 | −3.84 | 0.000 |
| Test–death interval | 0.409 | 3.63 | 0.001 |
| CVLT long delay | |||
| CA1 α-synuclein | −0.203 | −1.96 | 0.053 |
| CA1 plaques | 0.067 | 0.59 | 0.559 |
| CA1 tangles | −0.498 | −4.03 | 0.000 |
| Test–death interval | 0.422 | 3.73 | 0.000 |
| CVLT recognition | |||
| CA1 α-synuclein | −0.131 | −1.16 | 0.251 |
| CA1 plaques | −0.009 | −0.07 | 0.941 |
| CA1 tangles | −0.335 | −2.47 | 0.016 |
| Test–death interval | 0.279 | 2.25 | 0.028 |
| Visual reproduction: immediate | |||
| CA1 α-synuclein | −0.258 | −2.36 | 0.021 |
| CA1 plaques | 0.159 | 1.32 | 0.193 |
| CA1 tangles | −0.395 | −2.93 | 0.005 |
| Test–death interval | 0.476 | 3.86 | 0.000 |
| Visual reproduction: delay | |||
| CA1 α-synuclein | −0.304 | −2.81 | 0.006 |
| CA1 plaques | 0.105 | 0.88 | 0.383 |
| CA1 tangles | −0.481 | −3.62 | 0.001 |
| Test–death interval | 0.380 | 3.13 | 0.003 |
The degree of α-synuclein pathology in CA1 of the hippocampus makes a significant independent contribution to the memory deficit of patients with DLB, above and beyond the significant contribution made by the burden of CA1 AD tangle pathology. The same pattern of results was observed for the CVLT Trial 1–5 learning measure when total hippocampal α-synuclein burden (rather than just CA1) was used in the first model. All models were highly significant (p values < 0.001). Standardized β values and significance levels for inclusion in the various models are shown for each variable.
Overall regression models were significant for CVLT Trial 1–5 learning (R2 = 0.328; F = 8.77; p < 0.001), CVLT short-delay free recall (R2 = 0.270; F = 6.64; p < 0.001), CVLT long-delay free recall (R2 = 0.262; F = 6.38; p < 0.001), CVLT recognition discriminability (R2 = 0.127; F = 2.58; p < 0.05), Visual Reproduction test immediate recall (R2 = 0.233; F = 5.10; p < 0.001), and Visual Reproduction test delayed recall (R2 = 0.253; F = 5.68; p < 0.001). In each model, CA1 tangle burden accounted for a significant amount of variance in memory performance even after the interval between testing and death was taken into account (all p values < 0.02; Table 4). Furthermore, the burden of CA1 Lewy pathology explained a significant amount of variance in memory performance, beyond that explained by CA1 tangle pathology or the interval between testing and death, for CVLT Trial 1–5 learning (p = 0.019), CVLT short-delay free recall (p = 0.016), Visual Reproduction test immediate recall (p = 0.021), and Visual Reproduction test delayed recall (p = 0.006). A trend toward significance was observed for CVLT long-delay free recall (p = 0.053). There were no significant relationships between burden of neuritic plaque pathology in CA1 and performance on any of the memory measures.
Discussion
The present results provide clear evidence that the CA2 subregion of the hippocampus and the EC have a higher level of Lewy pathology than any other hippocampal region in patients with DLB, among both neocortical and limbic subtypes of pathology. The pathology was confirmed to be in CA2 rather than CA3 by virtue of being distal to MFs from the DG labeled using two distinct molecular markers (Kohara et al., 2014). Lewy pathology was also apparent in CA1, CA3, CA4, and the subiculum, although not to the same degree as in CA2 and EC. There was often little or no Lewy pathology in the DG. CA2 Lewy pathology burden was strongly correlated with EC Lewy pathology burden, but not with CA1 or CA3. However, CA1 and CA3 were highly correlated with each other, as were CA3 and CA4.
The predominance of Lewy pathology in CA2 and EC, in conjunction with a strong correlation between the degrees of Lewy pathology in both regions, implicates the EC–CA2 circuit in the pathogenesis of DLB. Although a direct connection between CA2 and EC has only been suspected in humans (Ding et al., 2010), it has been described in mice (Chevaleyre and Siegelbaum, 2010) and in nonhuman primates (Witter and Amaral, 1991), the latter of which specifically demonstrated a projection from deep EC to CA2, corresponding to where Lewy bodies and Lewy neurites were found in our study, respectively. CA2 in mice is reciprocally connected to the input layers of EC (layers II/III) via a single synapse (Rowland et al., 2013). In contrast, other hippocampal subregions receiving EC input do not project directly back to the region (e.g., DG) or only project back to the deeper output layers (IV/V or V/VI, depending on the nomenclature scheme) of EC (e.g., CA1). Our findings suggest that Lewy pathology begins in this potential circuit before spreading further to hippocampal subregions with less apparent but intercorrelated levels of Lewy pathology (e.g., CA1 and CA3), in line with the model of a propagating pathology (Luk et al., 2012). As the major output and input regions for CA2, respectively, CA1 and CA3 may be the next affected.
In our cohort, LBs in EC predominantly localized to the somata of layer IV/V neurons (rather than layer II/III, which project to the relatively spared DG), and LNs in CA2 were mainly distal to dentate MF input and of a punctate, presynaptic nature. Axonal aggregates in CA2 are likely to precede perikaryal LBs in EC, following the retrograde pattern of progression ascribed to Lewy pathology, with presynaptic aggregation of α-synuclein eventually leading to “dying-back” neurodegeneration (Schulz-Schaeffer, 2010; Scott et al., 2010; Kanazawa et al., 2012; Boassa et al., 2013). This sequence is consistent with Braak staging from sporadic PD, where the limbic system is affected earlier than the cortex (Braak et al., 2003). It also follows that neocortical cases had higher overall hippocampal Lewy pathology burden than limbic cases, in which the pathology had yet to reach cortical regions beyond the EC. The selective vulnerability of axon terminals in CA2 to Lewy pathology could be explained by the length and abundant branching of the projections and the high energy demands that engenders. This explanation has been given for the vulnerability of substantia nigra dopaminergic neurons to the same Lewy pathology in PD (Uchihara and Giasson, 2016). Other possibilities for the selective vulnerability of axon terminals in CA2 to Lewy pathology include the unique connectivity, electrophysiology and/or molecular identity of the neurons in the CA2 subfield (Caruana et al., 2012; Hawrylycz et al., 2012; Cui et al., 2013).
A previous study suggested that Lewy pathology in the hippocampus is harbored in local interneurons, at least in the case of PDD (Flores-Cuadrado et al., 2016). However, no such colocalization was observed in our study (data not shown), and only very little was found in another study focusing on DLB (Bernstein et al., 2011). Early work examining neuronal subtypes containing LBs in temporal cortex revealed that the majority localized to pyramidal cells and not interneurons positive for parvalbumin (Wakabayashi et al., 1995). More recent work in mice confirmed that native, nonaggregated α-synuclein was detected at excitatory synapses, but not in inhibitory interneurons in the hippocampus; due to this intrinsic difference in endogenous α-synuclein levels, hippocampal inhibitory interneurons were less likely to form inclusions in culture when triggered by exogenous α-synuclein fibrils (Taguchi et al., 2014). This discrepancy may suggest distinct substrates underlying the dementia of PD and that of DLB.
Although the CA1 Lewy pathology burden was less than in CA2, only the former correlated with measures of learning and memory. This correlation was observed even after accounting for the significant impact of CA1 tangle pathology. These results indicate that the effects of Lewy pathology on learning and memory become most apparent at a later stage of pathology since all cases (except for one) with CA1 pathology also had pathology in CA2, but not vice versa, suggesting that CA2 pathology is a prerequisite for CA1 pathology. The finding that CA1 Lewy pathology burden had a stronger correlation with learning and memory than CA2 burden, despite less Lewy pathology in CA1, is consistent with results of previous MRI studies showing that CA1 volume was most predictive of memory scores in patients with DLB despite relative preservation of the hippocampus (CA1 in particular) in DLB compared to AD (Mak et al., 2016; Delli Pizzi et al., 2016). CA2 Lewy pathology may contribute to cognitive symptoms that are not detected by standard memory testing. Previous studies in rodents have advanced a role for CA2 in social memory (Hitti and Siegelbaum, 2014; Stevenson and Caldwell, 2014; e.g., memory necessary for interactions with conspecifics) or temporal aspects of memory (e.g., separating similar memories acquired at different times; Mankin et al., 2015). Given the predominance of CA2 Lewy pathology we observed, future studies should examine these aspects of memory in patients with DLB.
Our study has several limitations. One is the relatively long delay between time of neuropsychological testing and death, approaching six years. Despite controlling for this variable interval, it results in a degree of postmortem pathology not fully reflective of pathology burden at the time of testing, which is most likely worse given the progressive nature of the disease. Another limitation is that the pathogenic form of aggregated α-synuclein is unknown. While we focused on the version hyperphosphorylated at serine-129, previous reports suggest that oligomeric forms of the protein are most toxic (Roberts et al., 2015). We attempted to address this issue by cross-validating some of our findings using conformer-specific antibodies for α-synuclein, including one that preferentially recognizes oligomers. While the best method to assess Lewy pathology burden remains unclear, our rigorous anatomical and molecular approach has yielded important insights.
Eight of our cases lacked hippocampal Lewy pathology, yet still had memory dysfunction. It is conceivable that Lewy pathology was present in these cases but overlooked from our sampling method (one single hippocampal section from the posterior left hemisphere). For instance, the amygdala and fornix, two prominent regions involved in memory consolidation but not specifically examined in this study, may have harbored Lewy pathology. The majority of these cases did have significant AD pathology in the hippocampus, however, and that along with significant atrophy may have caused their memory impairment. As for the couple of cases also lacking AD pathology, unusual alternate disease processes may be to blame; for instance, one case had concomitant Fahr's syndrome, causing calcifications in the basal ganglia and cortex that have been associated with dementia.
While CA1 tangles were pervasive in our cohort and strongly correlated with both verbal and visual memory dysfunction, Lewy pathology in CA1 contributed to memory performance above and beyond the effects of tau pathology. This finding suggests a convergence of pathologies with a possible additive effect on cognitive dysfunction. The relationship may even be causal: it has been suggested that phospho-synuclein promotes the phosphorylation of tau (Iseki et al., 2002; Guo et al., 2013). Our findings therefore support the view that many cases of dementia are not due to a single type of neuropathologic lesion, but rather a mix of several. Additional studies will be necessary to elucidate the precise sequence of progression of Lewy pathology in DLB and how it relates to cognitive decline. However, the results lend credence to a model whereby impairment of learning and memory becomes most apparent once Lewy pathology has reached CA1 and EC, potentially via CA2.
This study demonstrates a specific and selective distribution of Lewy pathology in the hippocampus of confirmed patients with DLB. Although CA2 and EC are most affected, memory impairment corresponds closest to pathology in the CA1 subregion that is thought to be downstream of this main pathology. This finding suggests that hippocampal Lewy pathology must spread beyond its initial site to cause memory dysfunction. A better understanding of the mechanisms governing aggregate formation and spread along interconnected hippocampal circuits could lead to new avenues for treating the disease.
Footnotes
This work was supported by the NIH/NIA Neuroplasticity of Aging Predoctoral Training Grant 2T32 AG00216-23, the Shiley-Marcos Alzheimer's Disease Research Center (NIA Grant P50 AG05131), the Helmsley Trust, the JPB Foundation, the Lookout Foundation, the Mathers Foundation, and NIH grant AG18440. We thank the participants and staff of the Shiley-Marcos Alzheimer's Disease Research Center at the University of California, San Diego. We also thank the Center for Advanced Laboratory Medicine at the University of California, San Diego, particularly Dr. Don Pizzo, for help with processing the patient brain specimens; Jamie Simon at the Salk Institute for Biological Studies for assistance with the hippocampal heat map; Dr. Omar El-Agnaf from the Qatar Biomedical Research Institute for providing us with the conformer-specific antibodies for α-synuclein pathology; and Mary Lynn Gage for help with editing this manuscript.
The authors declare no competing financial interests.
References
- Armstrong RA, Cairns NJ (2015) Comparative quantitative study of “signature” pathological lesions in the hippocampus and adjacent gyri of 12 neurodegenerative disorders. J Neural Transm 122:1355–1367. 10.1007/s00702-015-1402-8 [DOI] [PubMed] [Google Scholar]
- Armstrong RA, Kotzbauer PT, Perlmutter JS, Campbell MC, Hurth KM, Schmidt RE, Cairns NJ (2013) A quantitative study of α-synuclein pathology in fifteen cases of dementia associated with Parkinson disease. J Neural Transm 121:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW (1991) The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex 1:103–116. 10.1093/cercor/1.1.103 [DOI] [PubMed] [Google Scholar]
- Ballard C, Patel A, Oyebode F, Wilcock G (1996) Cognitive decline in patients with Alzheimer's disease, vascular dementia and senile dementia of Lewy body type. Age Ageing 205:209–213. [DOI] [PubMed] [Google Scholar]
- Bernstein HG, Johnson M, Perry RH, Lebeau FE, Dobrowolny H, Bogerts B, Perry EK (2011) Partial loss of parvalbumin-containing hippocampal interneurons in dementia with Lewy bodies. Neuropathology 31:1–10. 10.1111/j.1440-1789.2010.01117.x [DOI] [PubMed] [Google Scholar]
- Bertrand E, Lechowicz W, Szpak GM, Lewandowska E, Dymecki J, Wierzba-Bobrowicz T (2004) Limbic neuropathology in idiopathic Parkinson's disease with concomitant dementia. Folia Neuropathol 42:141–150. [PubMed] [Google Scholar]
- Boassa D, Berlanga ML, Yang MA, Terada M, Hu J, Bushong EA, Hwang M, Masliah E, George JM, Ellisman MH (2013) Mapping the subcellular distribution of α-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: implications for Parkinson's disease pathogenesis. J Neurosci 33:2605–2615. 10.1523/JNEUROSCI.2898-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E (1995) Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging 16:271–278; discussion 278–284. 10.1016/0197-4580(95)00021-6 [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. 10.1016/S0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- Calderon J, Perry RJ, Erzinclioglu SW, Berrios GE, Dening TR, Hodges JR (2001) Perception, attention, and working memory are disproportionately impaired in dementia with Lewy bodies compared with Alzheimer's disease. J Neurol Neurosurg Psychiatry 70:157–164. 10.1136/jnnp.70.2.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruana DA, Alexander GM, Dudek SM (2012) New insights into the regulation of synaptic plasticity from an unexpected place: hippocampal area CA2. Learn Mem 19:391–400. 10.1101/lm.025304.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Siegelbaum SA (2010) Strong CA2 pyramidal neuron synapses define a powerful disynaptic cortico-hippocampal loop. Neuron 66:560–572. 10.1016/j.neuron.2010.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchyard A, Lees AJ (1997) The relationship between dementia and direct involvement of the hippocampus and amygdala in Parkinson's disease. Neurology 49:1570–1576. 10.1212/WNL.49.6.1570 [DOI] [PubMed] [Google Scholar]
- Colosimo C, Hughes AJ, Kilford L, Lees AJ (2003) Lewy body cortical involvement may not always predict dementia in Parkinson's disease. J Neurol Neurosurg Psychiatry 74:852–856. 10.1136/jnnp.74.7.852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor DJ, Salmon DP, Sandy TJ, Galasko D, Hansen LA, Thal LJ (1998) Cognitive profiles of autopsy-confirmed Lewy body variant vs pure Alzheimer disease. Arch Neurol 55:994–1000. 10.1001/archneur.55.7.994 [DOI] [PubMed] [Google Scholar]
- Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Gearing M, Grinberg LT, Hof PR, Hyman BT, Jellinger K, Jicha GA, Kovacs GG, Knopman DS, et al. (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128:755–766. 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Gerfen CR, Young WS 3rd (2013) Hypothalamic and other connections with dorsal CA2 area of the mouse hippocampus. J Comp Neurol 521:1844–1866. 10.1002/cne.23263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delis DC, Kramer JH, Kaplan E, Ober BA (1987) California Verbal Learning Test. San Antonio, TX: Psychological Corp. [Google Scholar]
- Delli Pizzi S, Franciotti R, Bubbico G, Thomas A, Onofrj M, Bonanni L (2016) Atrophy of hippocampal subfields and adjacent extrahippocampal structures in dementia with Lewy bodies and Alzheimer's disease. Neurobiol Aging 40:103–109. 10.1016/j.neurobiolaging.2016.01.010 [DOI] [PubMed] [Google Scholar]
- DeVito LM, Konigsberg R, Lykken C, Sauvage M, Young WS 3rd, Eichenbaum H (2009) Vasopressin 1b receptor knock-out impairs memory for temporal order. J Neurosci 29:2676–2683. 10.1523/JNEUROSCI.5488-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Schmidt ML, Lee VM, Zhao ML, Yen SH, Trojanowski JQ (1994) Immunoreactivity profile of hippocampal CA2/3 neurites in diffuse Lewy body disease. Acta Neuropathol 87:269–276. 10.1007/BF00296742 [DOI] [PubMed] [Google Scholar]
- Ding SL, Haber SN, Van Hoesen GW (2010) Stratum radiatum of CA2 is an additional target of the perforant path in humans and monkeys. Neuroreport 21:245–249. 10.1097/WNR.0b013e328333d690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Cuadrado A, Ubeda-Bañon I, Saiz-Sanchez D, de la Rosa-Prieto C, Martinez-Marcos A (2016) Hippocampal α-synuclein and interneurons in Parkinson's disease: data from human and mouse models. Mov Disord 31:979–988. 10.1002/mds.26586 [DOI] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VMY (2013) Distinct α-Synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. 10.1016/j.cell.2013.05.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall H, Reyes S, Landeck N, Bye C, Leanza G, Double K, Thompson L, Halliday G, Kirik D (2014) Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson's disease. Brain 139:2493–2508. [DOI] [PubMed] [Google Scholar]
- Hamilton JM, Salmon DP, Galasko D, Delis DC, Hansen LA, Masliah E, Thomas RG, Thal LJ (2004) A comparison of episodic memory deficits in neuropathologically-confirmed dementia with Lewy bodies and Alzheimer's disease. J Int Neuropsychol Soc 10:689–697. [DOI] [PubMed] [Google Scholar]
- Hansen LA, Masliah E, Galasko D, Terry RD (1993) Plaque-only Alzheimer disease is usually the Lewy body variant, and vice versa. J Neuropathol Exp Neurol 52:648–654. 10.1097/00005072-199311000-00012 [DOI] [PubMed] [Google Scholar]
- Harding AJ, Halliday GM (2001) Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol 102:355–363. [DOI] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, Abajian C, Beckmann CF, Bernard A, Bertagnolli D, Boe AF, Cartagena PM, Chakravarty MM, Chapin M, Chong J, Dalley RA, et al. (2012) An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489:391–399. 10.1038/nature11405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyman A, Fillenbaum GG, Gearing M, Mirra SS, Welsh-Bohmer KA, Peterson B, Pieper C (1999) Comparison of Lewy body variant of Alzheimer's disease with pure Alzheimer's disease: Consortium to Establish a Registry for Alzheimer's Disease, Part XIX. Neurology 52:1839–1844. 10.1212/WNL.52.9.1839 [DOI] [PubMed] [Google Scholar]
- Hitti FL, Siegelbaum SA (2014) The hippocampal CA2 region is essential for social memory. Nature 508:88–92. 10.1038/nature13028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horimoto Y, Matsumoto M, Nakazawa H, Yuasa H, Morishita M, Akatsu H, Ikari H, Yamamoto T, Kosaka K (2003) Cognitive conditions of pathologically confirmed dementia with Lewy bodies and Parkinson's disease with dementia. J Neurol Sci 216:105–108. 10.1016/S0022-510X(03)00220-X [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL (1984) Alzheimer's disease: cell-specific pathology isolates the hippocampal formation. Science 225:1168–1170. 10.1126/science.6474172 [DOI] [PubMed] [Google Scholar]
- Iseki E, Takayama N, Marui W, Uéda K, Kosaka K (2002) Relationship in the formation process between neurofibrillary tangles and Lewy bodies in the hippocampus of dementia with Lewy bodies brains. J Neurol Sci 195:85–91. 10.1016/S0022-510X(01)00689-X [DOI] [PubMed] [Google Scholar]
- Kanazawa T, Adachi E, Orimo S, Nakamura A, Mizusawa H, Uchihara T (2012) Pale neurites, premature α-synuclein aggregates with centripetal extension from axon collaterals. Brain Pathol 22:67–78. 10.1111/j.1750-3639.2011.00509.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K, Pignatelli M, Rivest AJ, Jung HY, Kitamura T, Suh J, Frank D, Kajikawa K, Mise N, Obata Y, Wickersham IR, Tonegawa S (2014) Cell type–specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nat Neurosci 17:269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston RF, Stevenson CH, Wilson CL, Saunders I, Wood ER (2010) The role of hippocampal subregions in memory for stimulus associations. Behav Brain Res 215:275–291. 10.1016/j.bbr.2010.07.006 [DOI] [PubMed] [Google Scholar]
- Lein ES, Zhao X, Gage FH (2004) Defining a molecular atlas of the hippocampus using DNA microarrays and high-throughput in situ hybridization. J Neurosci 24:3879–3889. 10.1523/JNEUROSCI.4710-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Callaway EM, Albright TD, Gage FH (2005) Redefining the boundaries of the hippocampal CA2 subfield in the mouse using gene expression and 3-dimensional reconstruction. J Comp Neurol 485:1–10. 10.1002/cne.20426 [DOI] [PubMed] [Google Scholar]
- Lezak MD. (1983) Neuropsychological assessment. New York: Oxford UP. [Google Scholar]
- Lorente de Nó R. (1934) Studies on the structure of the cerebral cortex. II. Continuation of the study of the ammonic system. J Psychol Neurol 46:113–177. [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak E, Su L, Williams GB, Watson R, Firbank M, Blamire A, O'Brien J (2016) Differential atrophy of hippocampal subfields: a comparative study of dementia with Lewy bodies and Alzheimer disease. Am J Geriatr Psychiatry 24:136–143. [DOI] [PubMed] [Google Scholar]
- Mankin EA, Diehl GW, Sparks FT, Leutgeb S, Leutgeb JK (2015) Hippocampal CA2 activity patterns change over time to a larger extent than between spatial contexts. Neuron 85:190–201. 10.1016/j.neuron.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S,. et al. (2005) Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 65:1863–1872. 10.1212/01.wnl.0000187889.17253.b1 [DOI] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, National Institute on Aging, Alzheimer's Association (2012) National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123:1–11. 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkkinen L, Kauppinen T, Pirttilä T, Autere JM, Alafuzoff I (2005) Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol 57:82–91. 10.1002/ana.20321 [DOI] [PubMed] [Google Scholar]
- Roberts RF, Wade-Martins R, Alegre-Abarrategui J (2015) Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson's disease brain. Brain 138:1642–1657. 10.1093/brain/awv040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland DC, Weible AP, Wickersham IR, Wu H, Mayford M, Witter MP, Kentros CG (2013) Transgenically targeted rabies virus demonstrates a major monosynaptic projection from hippocampal area CA2 to medial entorhinal layer II neurons. J Neurosci 33:14889–14898. 10.1523/JNEUROSCI.1046-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon DP, Galasko D, Hansen LA, Masliah E, Butters N, Thal LJ, Katzman R (1996) Neuropsychological deficits associated with diffuse Lewy body disease. Brain Cogn 165:148–165. [DOI] [PubMed] [Google Scholar]
- Salmon DP, Heindel WC, Hamilton JM, Filoteo JV, Cidambi V, Hansen LA, Masliah E, Galasko D (2015) Recognition memory span in autopsy-confirmed dementia with Lewy bodies and Alzheimer's disease. Neuropsychologia 75:548–555. 10.1016/j.neuropsychologia.2015.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ. (2010) The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol 120:131–143. 10.1007/s00401-010-0711-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S (2010) A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci 30:8083–8095. 10.1523/JNEUROSCI.1091-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura T, Mori E, Yamashita H, Imamura T, Hirono N, Hashimoto M, Tanimukai S, Kazui H, Hanihara T (1998) Cognitive loss in dementia with Lewy bodies and Alzheimer disease. Arch Neurol 55:1547–1552. 10.1001/archneur.55.12.1547 [DOI] [PubMed] [Google Scholar]
- Squire LR. (1992) Nondeclarative memory: multiple brain systems supporting learning. J Cogn Neurosci 4:232–243. 10.1162/jocn.1992.4.3.232 [DOI] [PubMed] [Google Scholar]
- Stevenson EL, Caldwell HK (2014) Lesions to the CA2 region of the hippocampus impair social memory in mice. Eur J Neurosci 9:3294–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Watanabe Y, Tsujimura A, Tatebe H, Miyata S, Tokuda T, Mizuno T, Tanaka M (2014) Differential expression of alpha-synuclein in hippocampal neurons. PLoS One 9:e89327. 10.1371/journal.pone.0089327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi Y, Dickson DW (2005) Dementia with Lewy bodies and Parkinson's disease with dementia: are they different? Parkinsonism Relat Disord 11:47–51. [DOI] [PubMed] [Google Scholar]
- Uchihara T, Giasson BI (2016) Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol 131:49–73. 10.1007/s00401-015-1485-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaikath NN, Majbour NK, Paleologou KE, Ardah MT, van Dam E, van de Berg WD, Forrest SL, Parkkinen L, Gai WP, Hattori N, Takanashi M, Lee SJ, Mann DM, Imai Y, Halliday GM, Li JY, El-Agnaf OM (2015) Generation and characterization of novel conformation-specific monoclonal antibodies for α-synuclein pathology. Neurobiol Dis 79:81–99. 10.1016/j.nbd.2015.04.009 [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Hansen LA, Masliah E (1995) Cortical Lewy body-containing neurons are pyramidal cells: laser confocal imaging of double-immunolabeled sections with anti-ubiquitin and SMI32. Acta Neuropathol 89:404–408. 10.1007/BF00307643 [DOI] [PubMed] [Google Scholar]
- Walker Z, Allen RL, Shergill S, Katona CL (1997) Neuropsychological performance in Lewy body dementia and Alzheimer's disease. Br J Psychiatry 170:156–158. 10.1192/bjp.170.2.156 [DOI] [PubMed] [Google Scholar]
- Wintzer ME, Boehringer R, Polygalov D, McHugh TJ (2014) The hippocampal CA2 ensemble is sensitive to contextual change. J Neurosci 34:3056–3066. 10.1523/JNEUROSCI.2563-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, Amaral DG (1991) Entorhinal cortex of the monkey: v. projections to the dentate gyrus, hippocampus, and subicular complex. J Comp Neurol 307:439–459. [DOI] [PubMed] [Google Scholar]
- Woodhams P, Celio MR, Ulfig N, Witter MP (1993) Morphological and functional correlates of borders in the entorhinal cortex and hippocampus. Hippocampus 3:303–311. [PubMed] [Google Scholar]
- Yang Y, Kim S, Kim JH (2008) Ischemic evidence of transient global amnesia: location of the lesion in the hippocampus. J Clin Neurol 4:59–66. 10.3988/jcn.2008.4.2.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zola-Morgan S, Squire LR, Amaral DG (1986) Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci 6:2950–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]