

Abstract
Purpose
This study seeks to develop a liposomal formulation of diethylenetriamine NONOate (DN), a long acting nitric oxide (NO) donor, with a goal to replace inhaled NO (iNO) in the treatment of pulmonary arterial hypertension (PAH).
Methods
Liposomal formulations were prepared by a lipid film hydration method and modified with a cell penetrating peptide, CAR. The particles were characterized for size, polydispersity index (PDI), zeta potential, entrapment efficiency, storage and nebulization stability, and in-vitro release profiles. The cellular uptake and transport were assessed in rat alveolar macrophages (NR8383) and transforming growth factor β (TGF-β) activated rat pulmonary arterial smooth muscle cells (PASMCs). The fraction of the formulation that enters the systemic circulation, after intratracheal administration, was determined in an Isolated Perfused Rat Lung (IPRL) model. The safety of the formulations were assessed using an MTT assay and by measuring injury markers in the bronchoalveolar lavage (BAL) fluid; the pharmacological efficacy was evaluated by monitoring the changes in the mean pulmonary arterial (mPAP) and systemic pressure (mSAP) in a monocrotaline (MCT) induced-PAH rat model
Results
Liposome size, zeta potential, and entrapment efficiency were 171±4 nm, −37±3mV, and 46±5%, respectively. The liposomes released 70±5% of the drug in 8 h and were stable when stored at 4°C. CAR-conjugated-liposomes were taken up more efficiently by PASMCs than liposomes-without-CAR; the uptake of the formulations by rat alveolar macrophages was minimal. DN-liposomes did not increase lung weight, protein quantity, and levels of injury markers in the BAL fluid. Intratracheal CAR-liposomes reduced the entry of liposomes from the lung to blood; the formulations produced a 40% reduction in mPAP for 180 minutes.
Conclusion
This study establishes the proof-of-concept that peptide modified liposomal formulations of long-acting NO donor can be an alternative to short-acting iNO.
Keywords: Pulmonary arterial hypertension, nitric oxide donor, targeted drug delivery, CAR peptide, peptide link liposomes, inhalation delivery
1. INTRODUCTION
Nitric oxide, a potent endogenous vasodilator, helps maintain vascular tone(1). In the presence of inspiratory oxygen, endothelial nitric oxide synthase (eNOS) catalyzed reaction produces nitric oxide (NO) from L-arginine. Released NO then moves from endothelial to smooth muscle cells, activates guanylate cylclase (sGC) that converts GTP to cGMP. In the downstream, cGMP activates potassium channels but inactivates calcium channel and produces vasodilation (2). In PAH, a debilitating disease with high morbidity and mortality, NO production reduces because of endothelial cell proliferation and damage. The net result is disruption of cGMP mediated vasodilatation and subsequent vasoconstriction (3). To compensate the lack of NO supply in smooth muscles, gaseous NO has been used to treat newborns with persistent PAH and adults with acute and severe forms of pulmonary hypertension (4). Inhaled NO (iNO) increases oxygenicity, and reduces the intensity and duration of mechanical ventilation. NO also decreases lung barotrauma and oxygen toxicity, and thus helps improve patient outcomes (5).
However, iNO has a very limited use in PAH therapy; NO damages lung tissue by producing reactive oxygen species and altering surfactant functions. Like inhaled nitrogen dioxide and ozone, iNO damaged the lungs by altering the airway conductance, humoral immune responses, and host resistance (5). When used at higher concentrations, NO causes lung edema that may turn deadly in some patients (6). Excess NO in systemic circulation binds with hemoglobin and converts it to methohemoglobin, and thus prevents hemoglobin’s oxygen carrying capacity (7). Thus, iNO is contraindicated in patients with congenital or acquired methomoglobin reductase deficiency; also an abrupt cessation of iNO therapy in PAH patients often results in rebound PAH (8). iNO administration also poses a huge logistical hurdle. Because of its very short half-life (2–6 sec), NO is administered as a continuous inhalation using cumbersome delivery devices (9). The inhalation devices for NO are not portable and thus the drug can only be administered to hospitalized patients under the supervision of a trained healthcare worker (10, 11).
To circumvent the use of heavy delivery equipment for NO, reduce PAH related hospitalizations, and minimize cost, we propose to develop a patient friendly and portable formulation of new generation long-acting NO donors. These NO generating substances are superior to gaseous NO in producing pulmonary selective vasodilation. Because of their long half-lives, NO donors may eliminate the requirement of continuous inhalation and reduce the incidences of PAH rebound. Of the various NO donors, diethylenetriamine NONOate (DN) has an NO-generating half-life of ~20 h (12). DN releases two moles of NO for each parent molecule at pH 7.4 at 37°C (13). Like gaseous NO, DN-based NO reduces smooth muscle cell proliferation and is safe for inhalational delivery (14). However, plain DN is unstable at room temperature, auto-releases NO, and thus cannot be administered in its native form (12). Despite being selective in reducing pulmonary pressure at a low concentration, DN, at higher concentration, produces pulmonary toxicity in acute respiratory distress syndrome (ARDS) and acute PAH patients (15).
For many years now, liposomes have been used as delivery chariots for anticancer drugs, thrombolytic agents, Si-RNA, DNA, gene and vaccines (16–22) a number of liposomal formulations are now commercially available (23). Liposomes reduce multi-organ toxicities by decreasing drug exposures to highly perfused organs and facilitate drug delivery to the site of action. To achieve targeted delivery, liposomes surface can be modified or engrafted with targeting ligands such as antibodies and peptides; such modifications empower the liposomes to accumulate preferentially at the disease site and maximize drug concentration at afflicted organs or cells (24–28). Based on the success of liposomes as drug carriers and to address the unmet need for a long-acting NO formulation, we hypothesize that encapsulation of DN in liposomes modified with a homing peptide improves stability, and direct DN to concentrate and release NO in PAH lesions of the pulmonary vasculature. To test this hypothesis, we loaded the drug in pegylated liposomes modified with a cyclic homing peptide (CAR) containing 9 amino acids (CARSKNKDC), which has a half-life of 27 h and binds with the heparan sulfate–a glucosamino glycan, an endogenous substance involved in inflammatory process (29); this peptide also preferentially accumulates on PAH lungs (30). We evaluated DN entrapped CAR conjugated liposomes for physical properties, stability, release profiles, cellular uptake, lung accumulation, safety and pharmacological efficacy in cellular, ex-vivo and in vivo models.
MATERIALS AND METHODS
2.1. Materials
Lipids were procured from Avanti Polar lipids Inc. (Alabaster, AL) and N-Succinimidyl1,3-(2-pyridyldithio) propionate (SPDP) from Molecular Biosciences (Boulder, CO). TGF-β was from PeproTech, (Rocky Hill, NJ) and diethylenetriamine NONOate (DN) from Cayman Chemical (Ann Arbor, MI). Other chemicals were procured from Fisher Scientific (Pittsburgh, PA) and Sigma Aldrich Inc. (St Louis, MO). All chemicals were of analytical grades and used without further purification. CAR peptide was obtained from Dr. Rouslahti’s lab at the University of California, Santa Barbara. Pulmonary arterial smooth muscle cells were a generous gift from Dr. Eva Nozik-Grayck (Denver, Colorado). Rat alveolar macrophage cells (NR8383) and cell media was purchased from ATCC (Manassas, VA).
2.2. Preparation of CAR conjugated liposomes of DN
DN liposomes were prepared by solvent evaporation and rehydration method with 50 μmol lipid composed of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol and 1,2 disteroyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) or 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) at a molar ratio of 70:30:5:5 (Table 1). Lipids were dissolved in 2 mL chloroform/methanol mixture in a round-bottom flask. A thin lipid film was developed using a Buchi R-114 Rotavapor (Buchi Laboratories AG, Postfach, Switzerland) under vacuum in a water bath at 45°C. Then 5 mg DN, dissolved in 10 mM NaOH (pH 8.0), was used to rehydrate the lipid film by agitation and sonication; the resulting vesicles were extruded through polycarbonate membrane (Avanti polar lipids, Inc. Alabaster, AL), first through the membrane of 400 nm pore and then through 200 pore, at room temperature. Drug free liposomes, with a volume-based diameter of ~1 μm, were prepared and used as a positive control in alveolar macrophage uptake studies; this liposomal preparation was extruded through a 1000 nm polycarbonate membrane.
Table 1.
The composition and physicochemical properties of DN liposomes
| Formulation | Lipid Composition | Lipid molar ratio | Lipid quantity (μmol) | Mean size (nm) | PDI | Zeta potential (mV) | % Entrapment efficiency |
|---|---|---|---|---|---|---|---|
| F-1 | DPPC:CH:DSPE-PEG2000: DPPE | 70:30:5:5 | 50 | 171.1±4 | 0.006±0.002 | −36.97±3 | 46±5 |
| F-2 | DPPC:CH:DSPE-PEG2000: DSPE | 178.3±5.51 | 0.093±0.006 | −27.5±6.1 | 42.5±5.67 |
The liposomes were purified from un-entrapped drug by passing through a PD-10 column (Sephadex-25, GE Healthcare, Piscataway, NJ) with phosphate buffer saline (PBS, pH 7.4). Fluorescent liposomes, used in transport and IPRL studies, were prepared using rhodhamine labeled 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine (RDPE). To conjugate CAR peptide, the amine groups of phospholipid were activated by SPDP (N-Succinimidyl 3-[2-pyridyldithio]-propionate) chemistry (Fig. 1). Briefly, SPDP was dissolved in dimethylformamide (DMF), mixed with liposomes, and incubated for 30 min at room temperature. Excess SPDP was removed by ultra-centrifugation at 355,000xg for 4 h at 4°C using TL-100 Ultra-centrifuge (Beckman, USA). At this centrifugal force, liposomes settled on the tube, which was gently re-dispersed in PBS, and incubated with CAR-peptide, dissolved in PBS, for an hour at room temperature. To remove un-conjugated peptide, liposomes were further ultra-centrifuged as discussed above. Lastly, liposomes were re-suspended in PBS and stored at 4°C for additional assessment.
Fig. 1.

The steps involved in conjugation of CAR peptide on the liposomes surface.
2.3. Physicochemical characterization of DN CAR-liposomes
The size, polydispersity index (PDI), zeta potential, and entrapment efficiency of DN entrapped CAR liposomes were measured according to our published method (25). Particle size and zeta potential were measured in a Nano ZS90 Zetasizer (Malvern® Instruments Ltd., Worcestershire, UK). DN entrapment was quantified by lysing the liposomes with methanol using a validated UV spectroscopic method. Briefly, 20 μl liposomes was taken in an Eppendorf® tube, diluted with 1 mL methanol, and sonicated for 5 min. Following centrifugation for 15 min at 17000xg (Legend Micro 17R, Thermo Fisher Scientific, Waltham, MA), DN concentration was measured in a UV spectrophotometer (HP 8453A, Olis Inc. Bogart, GA) at 252 nm and the entrapment efficiency was determined using the following equation: L/T × 100 (Where L is the amount of DN in the liposomes and T is the total DN added to prepare the liposomes). To determine the extent of CAR conjugation on the liposomal surface, we assayed the amount of dithiopyridine (DTP) released upon interactions between SPDP-functionalized liposomes and CAR; the released DTP was read at 343 nm in a UV spectrophotometer. We calculated the amount of CAR, in mole, from the amount of DTP released, assuming one mole of DTP equals mole of CAR (31).
2.4. The stability of the formulations
The stability of the liposomal formulations, especially the thermo-stability, was studied by monitoring the size and drug amount in liposomes stored at 4°C, 25°C and 37°C for 21 days. Samples were collected once a week for three weeks, and the drug entrapment was assessed as indicated above and according to a published study (32). The robustness of liposomal aerosols was assessed by aerosolizing the liposomes with a MicroSprayer™ (PennCentury®, Inc., Philadelphia, PA). The size, zeta potential and entrapment efficiency were measured before and after aerosolization of liposomes (25).
2.5. In vitro release of DN
The in-vitro release profiles of DN was evaluated in a simulated lung fluid (SLF) according to our published report (25). We have used the Moss formula to prepare the SLF and maintained the pH at 7.0–7.2 (33). Ingredients were mixed with continuously stirred de-ionized water; no mucus was added into the SLF because <200 nm vesicles will land only on the alveolar space, an anatomical region that is devoid of mucus (34). Briefly, 500 μL of the liposomes were taken in a molecular cut-off cassette (Slide-A-Lyzer, MW 3500, 0.5–1 mL, Thermo Scientific, Waltham, MA) and placed in a beaker containing 50 mL SLF at 37°C. Samples were collected and replaced with 1 mL fresh PBS solution, and DN concentration was determined as described above. The total amount of DN (100%) in liposomes was assessed by disrupting the liposomes at time zero. The cumulative percent release was calculated using the following equation: % release = 100 X (Ft−F0)/(F100−F0), wherein Ft, F0 are the concentrations at time ‘t’ and time ‘0’, respectively. F100 represents 100% DN concentration from the liposomes.
2.6. Uptake of liposomes by rat pulmonary arterial smooth muscle (PASM) cells and rat alveolar macrophages
Both qualitative and quantitative methods were used to evaluate the uptake of the formulations by TGF-β activated PASM cells. For qualitative studies, fluorescent liposomes were first prepared using DPPC:CH:DSPE-PEG2000:DPPE:RD-PE (Rhodamie-PE). PASM cells were cultured in DMEM1/2 media (American type cell culture, Manassas, VA) in the presence of 10% FBS, penicillin/streptomycin, and glutamine. The cells were then seeded overnight on coverslips in a 12-well plate. Next day, cells were incubated in fresh media containing TGF-β and incubated for an additional 24 h (35). The TGF-β containing medium was then replaced with a fresh medium and the cells were incubated with fluorescent labeled CAR-liposomes for two hours. Following incubation with liposomes, the cells were washed thrice and fixed with 4% paraformaldehyde and incubated with 0.1% Triton™-X for 40 min. Then cells were further incubated with a blocking solution (goat serum and Tween®-20) for 2 h and with a monoclonal anti-β-actin primary antibody (Sigma-Aldrich, St. Louis, MO). Next day, cells were incubated with Alexa Fluor® 594 goat anti-mouse IgG (Invitrogen, Grand Island, NY) for 2 h at room temperature. Finally, cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). In each step, cells were washed thrice with PBS. Finally, the cover-slips containing the cells were fixed on a glass slide and examined under a uorescence microscope (IX-81, Olympus) (25).
The flow-cytometric study was performed to quantify the liposome uptake by PASM cells. Briefly, an aliquot of cells were attached on a Petri dish and the following day the medium was replaced with a TGF-β containing medium and incubated for 24 h. The TGF-β containing medium was removed and cells were incubated in a fresh medium containing fluorescent liposomes for 2 h. The cells were then washed with PBS, trypsinized for 3–5 min, centrifuged, re-suspended and washed with PBS to achieve a minimum of one million cells in each mL of PBS. The flow-cytometric analysis was performed in a BD FACSVerse™ (Qume Drive, San Jose, CA).
To assess the uptake by macrophages, we have used NR8383 alveolar macrophage cells. NR8383 cells were cultured in the DMEM: F12 (50:50) (American type cell culture, Manassas, VA) in the presence of 15% FBS, penicillin/streptomycin. The immunofluorescence and flow-cytometric analysis were performed upon incubation with different formulations as described above. CAR conjugated plain liposomes with a diameter of ~1 μm were used as a positive control.
2.7. Transport of liposomes across PASM Cells
The transport of plain liposomes and CAR-liposomes was studied in TGF-β activated PASM cells. For this study, PASM cells were cultured and seeded onto 12-well Transwell® plates (Corning Inc., NY). To evaluate the cell monolayer integrity, trans-epithelial electrical resistance (TEER) was measured and cells with a TEER of >300 ΩXcm2 were used. Of the 12 wells, six were incubated with the media containing TGF-β and the other six were incubated with media without TGF-β for 24 h. Next day, fluorescent (rhodamine B) labeled plain and CAR liposomes containing 0.025 μmol of rhodamine B were added to the apical side of each well and samples were collected from the basolateral side at different time points. The fluorescent intensity in samples was measured in a microplate reader at excitation/emission wavelengths of 557nm/571nm. The percentage of liposomes transported across the PASMC was calculated from the standard curve prepared with fluorescent liposomes.
2.8. Safety of the formulations
The safety of the formulation was assessed by an MTT assay and by quantifying injury markers in the bronchoalveolar lavage (BAL) fluid. The MTT assay was performed using bronchial epithelial cells (Calu-3) and PASM cells (25). Briefly, cells seeded in 96 wells plate were treated with saline, 0.1% sodium dodecyl sulfate, 10, 50 and 100 μM plain DN, and plain or CAR liposomes containing an equivalent concentration of DN. Following 24 h incubation with test and control samples, the media were removed and the cells were incubated with 0.5 mg/mL MTT for 4 h. The media containing MTT were then removed and 100 μL dimethyl sulfoxide (DMSO) was added to dissolve the crystalline precipitate. The absorbance was measured at 570 nm in a SynergyMX Microplate Reader (BioTek Instruments, Inc., Winooski, VT) and the percentage live cells was calculated using the following equation: % of viable cells = (A treated−AMTT)/(A control−AMTT) X 100.
To analyze the injury markers in BAL (32), saline, lipopolysaccharide (LPS) (0.1 ng/ml), DN-liposomes with and without CAR-peptide (1 mg/mL) were administered intratracheally to anesthetized rats (n=3). Twenty four hours after the treatment, lungs were excised from anesthetized rats, weighed, lavaged with 5 mL ice cold PBS, BAL fluid was collected, centrifuged at 500xg for 10 min at 4°C, supernatant was collected and stored at −20°C for further analysis (36). The total protein content, lactate dehydrogenase (LDH) and alkaline phosphatase (ALP) activities were measured using commercial assay kits.
2.9. Liposome retention in Isolated Perfused Rat Lung (IPRL)
The fraction of liposomes that traverses the lung and enters the systemic circulation was evaluated in an IPRL model (Harvard Apparatus GmbH, Holliston, MA, USA) (37) using both healthy and PAH rat lungs. To excise lungs, the rats were anesthetized by ketamine and xylazine and the cardio-pulmonary area was surgically exposed. The trachea was intubated and the lungs were inflated by a positive air pressure. The diaphragm was then opened and 200 IU/Kg heparin in saline was administered to the right ventricle. A cannula was inserted in the pulmonary artery and tied using a ligature. A small incision was made in the left ventricle to insert another cannula that was pushed toward the right atrium to reach the pulmonary vein and tied. The lungs were then perfused with a physiological salt solution, composed of CaCl2, NaCl, KCl, MgSO4, NaH2PO4, Glucose, NaHCO3, Ficoll®-2.0, at pH 7.4 and 37°C. A 95% O2-5% CO2 gas was passed to the medium in the chamber. Finally, the lungs along with the heart were isolated from the chest cavity and placed in a humid artificial thoracic chamber under negative pressure at 37°C. The lungs were ventilated by humidified atmospheric air using a negative pressure ventilator; the tidal volume, ventilation frequency and perfusion rate were 1–3 mL, 60 cycles/min and 10mL/min, respectively. The lungs in the artificial thoracic chamber were stabilized after 5 min of perfusion and various treatments containing 0.05 μg/ml of rhodamine B or equivalent of rhodamine in liposomes were administered directly to the lung through the trachea using PenCentury™ Microsprayer®. Five sets of lungs (3 lungs in each set) were separated, three from healthy rats and two from PAH rats, to receive the five treatments: Three healthy lungs received (i) plain rhodamine B (RB), (ii) CAR-RB-liposomes, and (iii) RB-liposomes-with-no-CAR. Two sets of PAH lungs received (i) RB-liposomes-with-no-CAR, and (ii) CAR-RB-liposomes. The PAH lungs were collected from monocrotaline (MCT) induced PAH rats (38). The isolated lungs in the thoracic chamber were perfused with the above described physiological perfusate media and an aliquot of perfusate was collected periodically for 2 h. Each sample was replaced with an equivalent amount of fresh perfusate. The fluorescent intensity in the samples was measured in a fluorescent micro-plate reader. The percentage of liposomes retained in the lung was calculated from the standard curve of fluorescent intensity vs concentration of rhodamine-liposomes.
2.10. Hemodynamic studies in PAH rats
The efficacy of liposomal DN in reducing pulmonary arterial pressure was studied in a monocrotaline (MCT) induced PAH rodent model. Rats were subcutaneously injected with 50 mg/Kg MCT and allowed 4 weeks to develop PAH (39). The PAH rats were divided into four groups to receive the following treatments: (i) intravenous DN, (ii) intra-tracheal DN, (iii) intra-tracheal DN-liposomes-with-no-CAR, and (iv) DN-CAR-liposomes. To measure mean pulmonary arterial (mPAP) and mean systemic pressure (mSAP), PAH rats were anesthetized by intramuscular administration of ketamine and xylazine and the ventral neck area of the rats was shaved and cleaned using ethyl alcohol. To measure mPAP, a polyvinyl catheter (PV-1, Tygon®, Lima, OH) was inserted into the right jugular vein to reach the pulmonary artery via the right ventricle. For mSAP, a PE-50 catheter (BD Intramedic™, Sparks, MD) was inserted into the carotid artery up to 3 cm. The data were recorded in a PowerLab™ 16/30 system using LabChart Pro™ 7.0 software (AD Instruments, Inc., Colorado Springs, CO). Both mSAP and mPAP were measured using MEMSCAP™ SP844 physiological pressure transducers (MEMSCAP AS, Scoppum, Norway) and bridge amplifier (39). The baseline pressure was measured for 10 min, the rats were then treated with one of the above formulations, and mPAP and mSAP were recorded for 6 h. The percentage of reduction of mPAP and mSAP was calculated using the initial pressure as 100%. The targeting index (TI) of different treatment groups was calculated from ratio of area above the efficacy curve (AAC) for mPAP to AAC for mSAP using the following equation: TI= AACmPAP/AACmSAP.
We have performed all animal studies according to the NIH Guidelines for the Care and Use of Laboratory Animals and the protocol for animal use (AM-02004) was approved by Texas Tech University Health Sciences Center (TTUHSC) Animal Care and Use Committee. Rats were housed at TTUHSC Amarillo animal facility.
2.10. Data analysis
The data are presented as mean±SD and differences between various treatment groups was analyzed by one-way ANOVA followed by Tukey’s post-hoc test (GraphPad Prism, version 5.0, GraphPad Software, San Diego, CA). Statistical significance was considered at p<0.05.
3. RESULTS AND DISCUSSION
3.1. Liposomes were colloidally stable
We determined the mean hydrodynamic diameter (z-average size), polydispersity index (PDI) and zeta potential of our liposomal preparation. Similar to our previously published studies (25), liposomes’ size were <200 nm; they were monodispersed, and had the dispersity indices (PDI) between 0.006 and 0.093. The formulations were colloidally stable; the zeta potential and drug entrapment were −27.5 to −36.97 mV and 42.5–46%, respectively. The colloidal properties and drug entrapment of the two formulations were very similar (Table 1). The physical properties of the liposomes did not change when the product was purified and separated from unconjugated CAR by ultra-centrifugation. The purified liposomes, containing no unconjugated CAR, were gently re-dispersed in PBS; the size, drug entrapment, and the PDI of the re-dispersed liposomes were the same as those of original liposomes. The final preparations contained 4.76 μmol CAR in each μmol of lipids, which was indirectly determined from the release of dithiopyridine (DTP).
3.2. Liposomal DN was stable upon refrigeration and aerosolization
DN is a thermo-labile drug, the recommended storage temperature for plain DN is −80°C. The drug has a half-life of 56 hr., at room temperature, which decreases to 22 hr. when stored at 37°C (40). The encapsulation of DN in PLGA particles could not even save the drug from degradation: DN, loaded in PLGA particles and stored at room temperature, decomposes to 50–90% of initial concentration in five to 30 days (40). We have evaluated the degradation pattern of DN, encapsulated in liposome, at three storage temperatures: 4°C, 25°C and 37°C. The drug was stable in liposomes stored at 4°C; but 80–90% of the liposomal drug underwent degradation when stored at 25°C and 37°C. The drug in F2 formulation degraded more than the drug in F-1 (Figs. 2A and 2B) because the later formulation contained DSPE. F-1 formulation was more stable because the lipid, DPPE, has a slightly higher transition temperature, which may have reduced the drug degradation. We have then examined whether the formulations form any aggregates upon refrigeration (Fig. 2C): The liposomes showed no change in size or formed no aggregates, suggesting that the formulations stable were upon refrigeration (Fig. 2C). DN releases ~100% NO at pH 4 or below; thus we maintained the liposomes’ vesicular pH at 8.0, (40). Elevated pH in combination with refrigeration may have contributed to the enhanced stability of DN in the liposomes.
Fig. 2.

The degradation patterns of DN in Formulation F1 containing DPPC:CH:DSPE-PEG2000:DPPE (A), and Formulation F2 containing PPC:CH:DSPE-PEG2000:DSPE (B). Both formulations were stored at 4°C, 25°C and 37°C. The changes in size (C) of the liposomes upon storage at 4°C for two weeks.
We chose to use DPPE based liposomes (F1 formulation) in the nebulization stability study because of its high drug entrapment. The aerosolization of DPPE-based liposomes, with PenCentury MicroSprayer® (Table 2), did not induce any changes in the size, PDI, and the entrapment efficiency of the formulations. Thus, the storage and nebulization stability of F1 formulation were optimal for using this preparation as an inhaled drug formulation.
Table 2.
The changes in size, zeta potential, and drug entrapment efficiency after nebulization of the liposomal preparations.
| Nebulization | Size (nm) | PDI | Zeta Potential (mV) | Entrapment efficiency (%) |
|---|---|---|---|---|
| No | 171.1±4 | 0.006±0.002 | −37±3 | 46±5 |
| Yes | 170±4.58 | 0.0084±0.025 | −34.96 ± 2.84 | 45.5±4.58 |
3.3. The liposomal formulations release drug in a continuous fashion
In SLF, both formulations released DN for 8 h: Formulations F-1 and F-2 released 70% and 63% of the drug, respectively (Fig. 3). Intact DN was released from the liposomes because the in vitro release study was performed at pH ~7.2; DN decomposes to NO at lower pH (40). The slight difference in the drug release from two formulations can be explained by the variation in lipid composition. Drug release and liposome membrane permeability depends on the lipid composition of the liposomal formulations. The presence of cholesterol and acid acyl chain length of lipids elevates the phase transition temperature of the liposomes; lipids with higher phase transition temperature slow drug release by modulating gel-liquid transition; drug release decreases at gel state but increases upon gel to liquid transition when lipid temperature nears phase transition temperature (41, 42). The liposomes contained ~ 5% DSPE or DPPE; DSPE has a higher phase transition temperature (Tm 74°C) that could increase the phase transition temperature of the liposomes. This may be the reason why DPPE-based liposomes (Tm 63°C) released more drug than DSPE-based preparations. However, the presence of <5 mol% DSPE-PEG2000 may have offset the variations caused by the lipids of different transition temperature. Thus, the drug loading, release patterns and stability of both liposomal formulations were very similar. We chose to use DPPE based liposomes in cellular, ex vivo and in vivo studies, described below, because this liposome preparation contained a greater amount of drug.
Fig. 3.

The in vitro release profiles of plain DN and DN-encapsulated CAR-liposomes in a simulated lung fluid at 37°C. The data represent mean ± SD, n=3.
3.4. CAR-DN-liposomes were preferentially taken up by PASM cells but not by alveolar macrophages
The uptake of CAR-DN-liposomes by two cells was evaluated in both normal and TGF-β activated PASM cells. TGF-β activated PASM cells internalized more CAR-DN-liposomes than normal PASM cells (Fig. 4). The enhanced internalization by TGF-β activated PASM cells is possibly associated with a better binding of CAR with cell surface heparan sulfate, expressed by PASM cells after TGF-β treatment. TGF-β increases the expression of different proteoglycans including heparan sulfate (HS), a cell surface ligand for CAR (35, 43–45), by increasing the transcriptional activity for HS or influencing the factors that suppresses HS transcription (45).
Fig. 4.

The fluorescence microscopic images of the uptake of CAR-conjugated liposomes by pulmonary arterial smooth muscle cells (A) and TGF-β activated pulmonary arterial smooth muscle cells (B). Green = cellular structure; blue = cell nucleus stained with DAPI; Red = rhodamine B labeled liposomes; the right most panel is the overlay. Scale bar = 5 μm.
The CAR-mediated enhanced uptake of liposomes was further confirmed by a flow-cytometric analysis of cellular upatke (Fig. 5). The internalization of CAR-modified-DN-liposomes by TGF-β-activated PASMCs was twofold greater than that of DN-liposomes-with-no-CAR. The uptake by normal cells was similar for both CAR and no-CAR liposomes, suggesting that the higher uptake of CAR-liposome is selective to activated smooth muscle cells. Thus, both microscopic and flow cytometry data demonstrate that CAR-decorated-liposomes specifically bind with TGF-β treated proliferating cells that express heparan sulfate (35, 46).
Fig. 5.

The flow cytometric data on the uptake of DN-plain-liposomes and DN-CAR-liposomes by normal and TGF-β treated pulmonary arterial smooth muscle cells (PASMC): The histogram (A), and the bar diagram (B) representing the cellular uptake. The formulations were incubated in normal and TGF-β treated PASMC for two hours. The data represent mean ± SD, n=3.
The alveolar macrophages can clear or engulf 250 nm sized inhaled particles (47); particles that are smaller than 250 nm can evade macrophage uptake (47); the escaped particles can reside longer in their place of landing, the lungs (48, 49). To test whether the particles can escape microphage uptake, we measured the internalization of the formulations by rat alveolar macrophages. Similar to published data, NR8383 rat alveolar macrophages could not internalize CAR modified-liposomes (Fig. 6); the size (<250 nm) and PEG chain of liposomes surface have possibly reduced the macrophage uptake (50). However, NR8383 rat alveolar macrophages internalized a greater amount of liposomes that were ~1000 nm in size. The macrophage uptake was further evaluated by flow cytometry using liposomes of ~1000 nm as a positive control (Fig. 7). The uptake of both F-1 and F-2 was significantly lower than the positive control. Thus, both microscopic and flow cytrometric data showed that the uptake of liposomes by rat alveolar macrophages depends on the size of liposomes: macrophages engulf vesicles of 1000 nm more efficiently than the vesicles that are smaller than 250 nm. Little or no internalization by macrophages, but enhanced uptake by activated PASM cells, suggest that CAR-modified-liposomes can reside in the lungs for an extended time and elicit superior therapeutic efficacy by releasing NO in PASM cells.
Fig. 6.

The fluorescence microscopic images of the uptake of DN CAR-liposomes by NR8383 alveolar macrophage after incubation for an hour. Green = FAM-labeled liposome; blue = cell nucleus stained with DAPI; red = beta-actin. Scale bar = 10 μM.
Fig. 7.

The flow cytometric data on the uptake of DN CAR-liposomes by NR8383 rat alveolar macrophage after incubation for an hour: The histogram (A) and the bar-diagram (B) showing the extent of uptake by NR8383 alveolar macrophages upon incubation with different formulations. PC was used as the positive control. The data represent mean ± SD, n=3.
3.5. CAR-liposomes can move across the pulmonary arterial cells
Since the target site of DN is the pulmonary vasculature, we evaluated whether our liposomal formulations can traverse the smooth muscle cell membrane. Liposomes-with-no-CAR could traverse neither TGF-β-activated nor normal cells; but CAR-modified-liposomes was capable of navigating across TGF-β treated smooth muscle cells. The transport of CAR-liposomes across the TGF-β activated cells was 20% greater (Fig. 8) than that across the un-activated cells. The fluorescent labeled liposomes with a size of <200 nm, and the flexibility of the liposome structure, are believed to be a good candidate for lipid-mediated free transport or lipid-mediated endocytosis across the cell monolayer (51). The enhanced uptake of CAR conjugated liposomes were because of CAR’s propensity to bind with cell surface HS, produced due to TGF-β treatment; TGF-β enhances the expression of cell surface heparan sulfate, a ligand for CAR peptide; CAR peptide also possesses the cell-penetrating property (52). The propensity of CAR to bind heparan sulfate couple with the peptide’s cell-penetrating property may have facilitated the transport of CAR-modified-liposomes across the smooth muscle cells. Thus, the proposed formulation can cross the smooth muscle cells and produce vasodilatory action in the pulmonary vasculature.
Fig. 8.

The percentage of DN plain liposomes and DN CAR liposomes transported across the normal and TGF-β activated PASMCs. Both plain and CAR-liposomes were prepared with DPPC:CH:DSPE-PEG2000:DPPE. The data represent mean ± SD, n=3.
3.6. CAR-liposomes accumulated over Isolated perfused rat lung (IPRL)
The central assumption for inhalation delivery of liposomal DN formulation is that the drug resides over the pulmonary vasculature and reduces the systemic exposure. A useful tool to test this hypothesis is an IPRL model that allows quantitation of the drug, in intact form, which leaves the pulmonary circulation. The accumulation of CAR-liposomes over PAH lungs was greater than those of plain and CAR-liposomes over the healthy lungs (Figs. 9A and 9B): In case of PAH lungs, accumulation was 95% and that for healthy lungs was 85% after two h of perfusion. However, when we used rhodamine B alone as a control, only 33% of the dye was in the lungs after the perfusion (Fig. 9C), suggesting that the fluorescent dye remained conjugated with the liposomes during the perfusion. We used both healthy and PAH rat lungs to study whether the disease state, PAH, influences the extent of accumulation of CAR-liposomes in the lungs. The buildup of CAR-liposomes in PAH lungs was greater than in healthy lungs, but that of liposomes-without-CAR was identical in both healthy and PAH lungs. The greater retention in PAH lungs suggest that CAR-liposomes preferentially concentrate over PAH lesions. Because the formulations concentrated on the respiratory epithelium, one might argue that CAR-liposomes, which are expected to elicit therapeutic effect on the pulmonary vasculature, lack targeting specificity. However, we posit that formulations that accumulate over the respiratory epithelium can also navigate through the epithelium, enter the vasculature, and produce therapeutic effects.
Fig. 9.
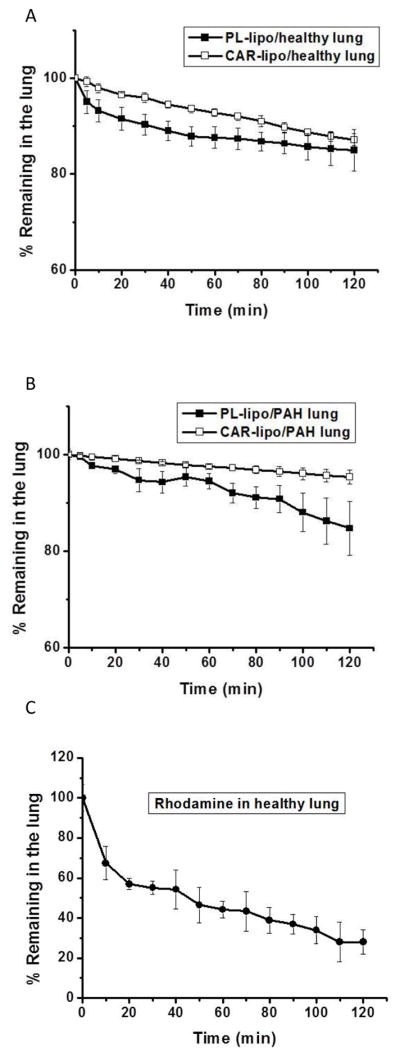
The percentage of unmodified liposomes, CAR-liposomes, and rhodamine accumulated in healthy (A and B) and PAH lungs (C) upon perfusion in an isolated rat lung. Both plain and CAR-liposome contained DPPC:CH:DSPE-PEG2000:DPPE. The data represent mean ± SD, n=3.
3.7. DN-loaded CAR-liposomes are safe to lung tissues
The safety of the formulation for using in the lung was evaluated both in vitro and in vivo. When positive control (0.1% SDS) was used, 85% of the cells, alveolar epithelial (Calu-3) and pulmonary arterial cells, died after 24 h treatment; but only 10% cells died when incubated with 10–100 μM DN-liposomes for 24 h. The percentage of live cells in liposome treated samples was similar to that of the samples treated with the saline (Fig. 10), suggesting that the liposomes was as innocuous as that of a physiological saline. Like unmodified liposomes, CAR modified liposomes was also as safe as the physiological saline; CAR had no deleterious effect on the cells. The broncho-alveolar lavage (BAL) study was performed to further establish the safety of DN or DN-liposomes for delivery into the lungs. Upon LPS treatment, the weight of saline treated lungs increased from 0.44±0.043 to 0.63±0.61 g for each 100 g of body weight; increased lung weight means fluid accumulation and edema formation (39). However, intra-tracheal administration of 1 mg/Kg DN or equivalent dose of DN-liposome with or without CAR did not increase the lung weight, pointing to no damage or injury to the lungs (Fig. 11A). The total protein content in BAL fluid showed that DN treatments (plain DN, DN-liposomes, and DN-CAR liposomes) did not increase the protein level in BAL fluid when compared with the positive control, i.e. LPS (Fig. 11B). Two injury markers, lactate dehydrogenase (LDH) and alkaline phosphatase (ALP), was quantified. The levels of LDH and ALP in DN, DN-plain liposomes and DN CAR-liposomes treated lungs were the same as that of saline treated lungs (Figs. 11C and 11D). Altogether, the liposomal formulations of DN are safe for inhalational administration.
Fig. 10.

The percentage of live lung epithelial (Calu-3) (A) and pulmonary arterial smooth muscle cells (B) 24 h after treatment with plain DN and DN encapsulated in liposomes and CAR-liposomes. The data represent mean ± SD, n=8.
Fig. 11.

The effect of plain DN, DN in unmodified liposomes and in CAR-liposomes on the wet lung weight and total protein content (A); and on the levels of lactate dehydrogenase (LDH) and alkaline phosphatase (ALP) (B) in bronchoalveolar lavage (BAL) fluid. The data represent mean ± SD, n=3.
3.8. CAR-conjugated DN liposomes elicited pulmonary specific vasodilation in PAH rats
We assessed the pharmacological efficacy of the optimized DN formulations in an MCT-induced acute rodent model of PAH. Four weeks after a subcutaneous injection of MCT, the baseline mPAP in PAH rats was ~37±10 mm Hg. Plain DN, administered via IV and IT, reduced the mPAP by 20%; this action lasted for ~90 min and 120 min for IV and IT routes, respectively (Figs. 12A and 12B). However, similar to reducing the mPAP, plain DN also reduced the mSAP, although the mSAP went down more in rats treated with plain DN IV than in the rats treated with plain DN IT. The later form (DN IT) elicited a milder systemic vasodilation than did DN IV, this pattern is akin to that of IPRL data presented in Fig. 9. DN liposomes, with no surface CAR, reduced the mPAP by 25–30% and the vasodilatory duration for this formulation was ~ 120 minutes (Fig. 12C). The magnitude and duration of reduction in mPAP, produced by liposomal DN, were greater than those produced by plain DN. But liposomal DN-with-no-CAR also reduced the mSAP to the same extent as that of mPAP. DN-CAR liposomes, on the other hand, produced a 40% reduction in mPAP that persisted over 180 minutes (Fig. 12D); this formulation (DN in CAR-liposome) had minimal effect on the systemic pressure, suggesting a pulmonary vasculature specific action. We further confirmed whether CAR peptide or liposomes itself, with no drug, reduce mPAP. CAR liposomes-with-no-DN, changed neither mPAP nor mSAP (Fig. 12E), suggesting that DN, not the peptide or blank liposomes, evokes the therapeutic effect. A point-by-point comparison among the magnitudes in reduction of mPAPs (Fig. 12F) demonstrate that liposomal DN (both unmodified liposomes and CAR-modified liposomes) produced more pronounced vasodilation than that produced by the plain drug. Of the three forms, CAR-liposomes caused the greatest reduction in mPAP.
Fig. 12.

The percent reduction in initial mean pulmonary arterial pressure (mPAP) and mean systemic arterial pressure (mSAP) upon administration of 3 mg/kg DN as a single dose in the following forms: (A) plain DN IV, (B) plain DN IT, (C) plain DN liposomes, (D) DN entrapped CAR conjugated liposomes, (E) CAR conjugated liposomes with no drug; (F) the curves comparing the extent of reduction in mPAP produced by three formulations. The data represent mean ± SD, n=3–4.
To quantify the targeting specificity, we calculated the targeting index (TI) from the ratio of the area above the efficacy curve (AAC) for mPAP and mSAP (TI = AAC mPAP/AAC mSAP). The TIs for DN IV, DN IT, DN-liposomes and DN-CAR liposomes were 0.36, 0.79, 1.47 and 4.98, respectively (Fig. 13); the TI of CAR-liposomes was 6.3 and 3.4-fold greater than the TIs of plain DN and DN-liposomes-with-no-CAR, respectively. A larger TI of CAR-liposome points to the assumption that CAR liposomes concentrate over the PAH lesions in the lungs and this formulation had a modest effect on the systemic vasodilation. Overall, the pharmacological efficacy study agrees with our belief that DN-loaded CAR-liposomes is pulmonary selective.
Fig. 13.

The targeting indices of DN IV, DN IT, DN plain liposomes and DN CAR-liposomes in MCT induced PAH rats. The data represent mean ± SD (n=3–4). *p<0.05, **p<0.01, ***p<0.001.
4. CONCLUSIONS
This study reports that liposomes of DETA NONOate can be used as a long acting nitric oxide therapy and that can elicit lung specific vasodilation. The formulation protects the drug from degradation when refrigerated at 4°C; CAR-peptide concentrate over the proliferative smooth muscle cells and extends the residence time of liposomes in PAH rat lungs. The formulations, when instilled into the lungs, reduce the pulmonary arterial pressure without altering the systemic pressure. This non-invasive delivery strategy addresses the key challenges of the current ultra-short acting inhaled nitric oxide therapy that is cumbersome and requires hospitalization.
Acknowledgments
This work was supported in part by an American Recovery and Reinvestment Act Fund, NIH 1R15HL103431 and 5 R01 HL114677-02.
ABBREVIATIONS
- AAC
Area above the curve
- AUC
Area under the curve
- BCA
Bicinchoninic Acid
- DAPI
4′,6-diamidino-2-phenylindole
- DETA NONOate
Diethylenetriamine NONOate
- DMEM
Dulbecco’s Modified Eagle Medium
- DMSO
Dimethyl Sulfoxide
- DPPC
1,2-dipalmitoyl-sn-glycero-3-phosphocholine
- DPPE
1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine
- DSPE-PEG2000
1,2 disteroyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000]
- FBS
Fetal Bovine Serum
- IPRL
Isolated Perfused Rat Lung
- MCT
Monocrotaline
- MTT
(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NO
Nitric oxide
- PAH
Pulmonary Arterial Hypertension
- PASMC
Pulmonary Arterial Smooth Muscle Cells
- PBS
Phosphate Buffered Saline
- PDI
Polydispersity Index
- SPDP
N-Succinimidyl1,3-(2-pyridyldithio) propionate
- TEM
Transmission Electron Microscopy
- TGF-β
Transforming growth factor beta
- TI
Targeting index
References
- 1.McHugh J, Cheek DJ. Nitric oxide and regulation of vascular tone: pharmacological and physiological considerations. Am J Crit Care. 1998;7(2):131–140. [PubMed] [Google Scholar]
- 2.Ghofrani HA, Pepke-Zaba J, Barbera JA, Channick R, Keogh AM, Gomez-Sanchez MA, Kneussl M, Grimminger F. Nitric oxide pathway and phosphodiesterase inhibitors in pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):68S–72S. doi: 10.1016/j.jacc.2004.02.031. [DOI] [PubMed] [Google Scholar]
- 3.O’Callaghan DS, Savale L, Montani D, Jais X, Sitbon O, Simonneau G, Humbert M. Treatment of pulmonary arterial hypertension with targeted therapies. Nat Rev Cardiol. 2011;8(9):526–538. doi: 10.1038/nrcardio.2011.104. [DOI] [PubMed] [Google Scholar]
- 4.Barst RJ, Channick R, Ivy D, Goldstein B. Clinical perspectives with long-term pulsed inhaled nitric oxide for the treatment of pulmonary arterial hypertension. Pulm Circ. 2012;2(2):139–147. doi: 10.4103/2045-8932.97589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinberger B, Fakhrzadeh L, Heck DE, Laskin JD, Gardner CR, Laskin DL. Inhaled nitric oxide primes lung macrophages to produce reactive oxygen and nitrogen intermediates. Am J Respir Crit Care Med. 1998;158(3):931–938. doi: 10.1164/ajrccm.158.3.9708014. [DOI] [PubMed] [Google Scholar]
- 6.Clutton-Brock J. Two cases of poisoning by contamination of nitrous oxide with higher oxides of nitrogen during anaesthesia. Br J Anaesth. 1967;39(5):388–392. doi: 10.1093/bja/39.5.388. [DOI] [PubMed] [Google Scholar]
- 7.Hagan G, Pepke-Zaba J. Pulmonary hypertension, nitric oxide and nitric oxide-releasing compounds. Expert Rev Respir Med. 2011;5(2):163–171. doi: 10.1586/ers.11.5. [DOI] [PubMed] [Google Scholar]
- 8.Miller OI, Tang SF, Keech A, Celermajer DS. Rebound pulmonary hypertension on withdrawal from inhaled nitric oxide. Lancet. 1995;346(8966):51–52. doi: 10.1016/s0140-6736(95)92681-x. [DOI] [PubMed] [Google Scholar]
- 9.Bernasconi A, Beghetti M. Inhaled nitric oxide applications in paediatric practice. Images Paediatr Cardiol. 2002;4(1):4–29. [PMC free article] [PubMed] [Google Scholar]
- 10.Wessel DL, Adatia I, Thompson JE, Hickey PR. Delivery and Monitoring of Inhaled Nitric-Oxide in Patients with Pulmonary-Hypertension. Crit Care Med. 1994;22(6):930–938. doi: 10.1097/00003246-199406000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Ashutosh K, Phadke K, Jackson JF, Steele D. Use of nitric oxide inhalation in chronic obstructive pulmonary disease. Thorax. 2000;55(2):109–113. doi: 10.1136/thorax.55.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keefer LK, Nims RW, Davies KM, Wink DA. “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol. 1996;268:281–293. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 13.Lam CF, Caterina P, Filion P, Ilett KF, van Heerden PV. The safety of aerosolized diethylenetriamine nitric oxide adduct after single-dose administration to anesthetized piglets and multiple-dose administration to conscious rats. Toxicol Appl Pharmacol. 2003;190(1):65–71. doi: 10.1016/s0041-008x(03)00193-5. [DOI] [PubMed] [Google Scholar]
- 14.Mooradian DL, Hutsell TC, Keefer LK. Nitric oxide (NO) donor molecules: effect of NO release rate on vascular smooth muscle cell proliferation in vitro. J Cardiovasc Pharmacol. 1995;25(4):674–678. [PubMed] [Google Scholar]
- 15.Lam CF, van Heerden PV, Ilett KF, Caterina P, Filion P. Two aerosolized nitric oxide adducts as selective pulmonary vasodilators for acute pulmonary hypertension. Chest. 2003;123(3):869–874. doi: 10.1378/chest.123.3.869. [DOI] [PubMed] [Google Scholar]
- 16.Kandadai MA, Meunier JM, Hart K, Holland CK, Shaw GJ. Plasmin-loaded echogenic liposomes for ultrasound-mediated thrombolysis. Transl Stroke Res. 2015;6(1):78–87. doi: 10.1007/s12975-014-0376-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tahover E, Patil YP, Gabizon AA. Emerging delivery systems to reduce doxorubicin cardiotoxicity and improve therapeutic index: focus on liposomes. Anti-cancer drugs. 2015;26(3):241–258. doi: 10.1097/CAD.0000000000000182. [DOI] [PubMed] [Google Scholar]
- 18.Yang YJ, Zhao PS, Zhang T, Wang HL, Liang HR, Zhao LL, Wu HX, Wang TC, Yang ST, Xia XZ. Small interfering RNAs targeting the rabies virus nucleoprotein gene. Virus Res. 2012;169(1):169–174. doi: 10.1016/j.virusres.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koide H, Asai T, Kato H, Yonenaga N, Yokota M, Ando H, Dewa T, Nango M, Maeda N, Oku N. Susceptibility of PTEN-positive metastatic tumors to small interfering RNA targeting the mammalian target of rapamycin. Nanomedicine. 2015;11(1):185–194. doi: 10.1016/j.nano.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Magalhaes M, Farinha D, Pedroso de Lima MC, Faneca H. Increased gene delivery efficiency and specificity of a lipid-based nanosystem incorporating a glycolipid. Int J Nanomedicine. 2014;9:4979–4989. doi: 10.2147/IJN.S69822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weecharangsan W, Opanasopit P, Yingyongnarongkul BE, Kewsuwan P, Lee RJ. Co-delivery of Plasmid DNA and Antisense Oligodeoxyribonucleotide into Human Carcinoma Cells by Cationic Liposomes. Curr Pharm Biotechnol. 2014;15(9):790–799. doi: 10.2174/1389201015666141020154352. [DOI] [PubMed] [Google Scholar]
- 22.Varypataki EM, van der Maaden K, Bouwstra J, Ossendorp F, Jiskoot W. Cationic liposomes loaded with a synthetic long peptide and poly(I:C): a defined adjuvanted vaccine for induction of antigen-specific T cell cytotoxicity. AAPS J. 2015;17(1):216–226. doi: 10.1208/s12248-014-9686-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 24.Nahire R, Hossain R, Patel R, Paul S, Meghnani V, Ambre AH, Gange KN, Katti KS, Leclerc E, Srivastava DK, Sarkar K, Mallik S. pH-Triggered Echogenicity and Contents Release from Liposomes. Mol Pharm. 2014;11(11):4059–4068. doi: 10.1021/mp500186a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nahar K, Absar S, Patel B, Ahsan F. Starch-coated magnetic liposomes as an inhalable carrier for accumulation of fasudil in the pulmonary vasculature. Int J Pharm. 2014;464(1–2):185–195. doi: 10.1016/j.ijpharm.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nahar K, Absar S, Gupta N, Kotamraju VR, McMurtry IF, Oka M, Komatsu M, Nozik-Grayck E, Ahsan F. Peptide-coated liposomal fasudil enhances site specific vasodilation in pulmonary arterial hypertension. Mol Pharm. 2014;11(12):4374–4384. doi: 10.1021/mp500456k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo Z, Li Y, Fu Y, Guo T, Li X, Yang S, Xie J. Enhanced antisense oligonucleotide delivery using cationic liposomes incorporating Fatty Acid-modified polyethylenimine. Curr Pharm Biotechnol. 2014;15(9):800–805. doi: 10.2174/138920101509141107122927. [DOI] [PubMed] [Google Scholar]
- 28.Alshaer W, Hillaireau H, Vergnaud J, Ismail S, Fattal E. Functionalizing Liposomes with anti-CD44 Aptamer for Selective Targeting of Cancer Cells. Bioconjugate Chem. 2015;26(7):1307–1313. doi: 10.1021/bc5004313. [DOI] [PubMed] [Google Scholar]
- 29.Kliment CR, Tobolewski JM, Manni ML, Tan RJ, Enghild J, Oury TD. Extracellular superoxide dismutase protects against matrix degradation of heparan sulfate in the lung. Antioxid Redox Signal. 2008;10(2):261–268. doi: 10.1089/ars.2007.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Urakami T, Jarvinen TA, Toba M, Sawada J, Ambalavanan N, Mann D, McMurtry I, Oka M, Ruoslahti E, Komatsu M. Peptide-directed highly selective targeting of pulmonary arterial hypertension. Am J Pathol. 2011;178(6):2489–2495. doi: 10.1016/j.ajpath.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Absar S, Nahar K, Kwon YM, Ahsan F. Thrombus-targeted nanocarrier attenuates bleeding complications associated with conventional thrombolytic therapy. Pharm Res. 2013;30(6):1663–1676. doi: 10.1007/s11095-013-1011-x. [DOI] [PubMed] [Google Scholar]
- 32.Bai S, Ahsan F. Inhalable liposomes of low molecular weight heparin for the treatment of venous thromboembolism. J Pharm Sci. 2010;99(11):4554–4564. doi: 10.1002/jps.22160. [DOI] [PubMed] [Google Scholar]
- 33.Moss OR. Simulants of lung interstitial fluid. Health Phys. 1979;36(3):447–448. [PubMed] [Google Scholar]
- 34.Margareth R, Marques1 CRL, Almukainzi May. Simulated Biological Fluids with Possible Application in Dissolution Testing. Dissolut Technol. 2011;18(3):15–28. [Google Scholar]
- 35.Chen JK, Hoshi H, McKeehan WL. Stimulation of human arterial smooth muscle cell chondroitin sulfate proteoglycan synthesis by transforming growth factor-beta. In Vitro Cell Dev Biol. 1991;27(1):6–12. doi: 10.1007/BF02630888. [DOI] [PubMed] [Google Scholar]
- 36.Gupta V, Ahsan F. Influence of PEI as a core modifying agent on PLGA microspheres of PGE(1), a pulmonary selective vasodilator. Int J Pharm. 2011;413(1–2):51–62. doi: 10.1016/j.ijpharm.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nahar K, Gupta N, Gauvin R, Absar S, Patel B, Gupta V, Khademhosseini A, Ahsan F. In vitro, in vivo and ex vivo models for studying particle deposition and drug absorption of inhaled pharmaceuticals. Eur J Pharm Sci. 2013;49(5):805–818. doi: 10.1016/j.ejps.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Gupta V, Gupta N, Shaik IH, Mehvar R, Nozik-Grayck E, McMurtry IF, Oka M, Komatsu M, Ahsan F. Inhaled PLGA particles of prostaglandin E(1) ameliorate symptoms and progression of pulmonary hypertension at a reduced dosing frequency. Mol Pharm. 2013;10(5):1655–1667. doi: 10.1021/mp300426u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta V, Gupta N, Shaik IH, Mehvar R, McMurtry IF, Oka M, Nozik-Grayck E, Komatsu M, Ahsan F. Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary preferential vasodilation in pulmonary arterial hypertension. J Control Release. 2013;167(2):189–199. doi: 10.1016/j.jconrel.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoo JW, Lee JS, Lee CH. Characterization of nitric oxide-releasing microparticles for the mucosal delivery. J Biomed Mater Res A. 2010;92(4):1233–1243. doi: 10.1002/jbm.a.32434. [DOI] [PubMed] [Google Scholar]
- 41.Maurer N, Fenske DB, Cullis PR. Developments in liposomal drug delivery systems. Expert Opin Biol Ther. 2001;1(6):923–947. doi: 10.1517/14712598.1.6.923. [DOI] [PubMed] [Google Scholar]
- 42.Mozafari MR, Johnson C, Hatziantoniou S, Demetzos C. Nanoliposomes and their applications in food nanotechnology. J Liposome Res. 2008;18(4):309–327. doi: 10.1080/08982100802465941. [DOI] [PubMed] [Google Scholar]
- 43.Chan SY, Loscalzo J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol. 2008;44(1):14–30. doi: 10.1016/j.yjmcc.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jarvinen TA, Ruoslahti E. Target-seeking antifibrotic compound enhances wound healing and suppresses scar formation in mice. Proc Natl Acad Sci. 2010;107(50):21671–21676. doi: 10.1073/pnas.1016233107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yue X, Li X, Nguyen HT, Chin DR, Sullivan DE, Lasky JA. Transforming growth factor-beta1 induces heparan sulfate 6-O-endosulfatase 1 expression in vitro and in vivo. J Biol Chem. 2008;283(29):20397–20407. doi: 10.1074/jbc.M802850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen G, Khalil N. TGF-beta1 increases proliferation of airway smooth muscle cells by phosphorylation of map kinases. Respir Res. 2006;7:2. doi: 10.1186/1465-9921-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chono S, Tanino T, Seki T, Morimoto K. Uptake characteristics of liposomes by rat alveolar macrophages: influence of particle size and surface mannose modification. J Pharm Pharmacol. 2007;59(1):75–80. doi: 10.1211/jpp.59.1.0010. [DOI] [PubMed] [Google Scholar]
- 48.Frohlich E, Salar-Behzadi S. Toxicological assessment of inhaled nanoparticles: role of in vivo, ex vivo, in vitro, and in silico studies. Int J Mol Sci. 2014;15(3):4795–4822. doi: 10.3390/ijms15034795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buzea C, Pacheco, Robbie K. Nanomaterials and nanoparticles: sources and toxicity. Biointerphases. 2007;2(4):MR17–71. doi: 10.1116/1.2815690. [DOI] [PubMed] [Google Scholar]
- 50.Lian T, Ho RJ. Trends and developments in liposome drug delivery systems. J Pharm Sci. 2001;90(6):667–680. doi: 10.1002/jps.1023. [DOI] [PubMed] [Google Scholar]
- 51.Pardridge WM. CNS drug design based on principles of blood-brain barrier transport. J Neurochem. 1998;70(5):1781–1792. doi: 10.1046/j.1471-4159.1998.70051781.x. [DOI] [PubMed] [Google Scholar]
- 52.Jarvinen TA, Ruoslahti E. Molecular changes in the vasculature of injured tissues. Am J Pathol. 2007;171(2):702–711. doi: 10.2353/ajpath.2007.061251. [DOI] [PMC free article] [PubMed] [Google Scholar]