

Sherrard et al. demonstrate that, during embryogenesis, miR-35 and miR-58 bantam family miRNAs cooperate to prevent the precocious death of mothers of cells programmed to die by repressing the gene egl-1, which encodes a proapoptotic BH3-only protein.
Keywords: miRNA, programmed cell death, BH3-only, development, embryo, C. elegans
Abstract
Programmed cell death occurs in a highly reproducible manner during Caenorhabditis elegans development. We demonstrate that, during embryogenesis, miR-35 and miR-58 bantam family microRNAs (miRNAs) cooperate to prevent the precocious death of mothers of cells programmed to die by repressing the gene egl-1, which encodes a proapoptotic BH3-only protein. In addition, we present evidence that repression of egl-1 is dependent on binding sites for miR-35 and miR-58 family miRNAs within the egl-1 3′ untranslated region (UTR), which affect both mRNA copy number and translation. Furthermore, using single-molecule RNA fluorescent in situ hybridization (smRNA FISH), we show that egl-1 is transcribed in the mother of a cell programmed to die and that miR-35 and miR-58 family miRNAs prevent this mother from dying by keeping the copy number of egl-1 mRNA below a critical threshold. Finally, miR-35 and miR-58 family miRNAs can also dampen the transcriptional boost of egl-1 that occurs specifically in a daughter cell that is programmed to die. We propose that miRNAs compensate for lineage-specific differences in egl-1 transcriptional activation, thus ensuring that EGL-1 activity reaches the threshold necessary to trigger death only in daughter cells that are programmed to die.
Programmed cell death (apoptosis) is crucial for proper animal development, and perturbations in the cell death pathway or its regulation have been implicated in disorders such as autoimmune diseases and cancer (for review, see Conradt 2009; Fuchs and Steller 2011). In the nematode Caenorhabditis elegans, 131 cells are eliminated by programmed cell death in the developing hermaphrodite, and 113 of these deaths occur during embryogenesis (Sulston and Horvitz 1977; Sulston et al. 1983). Moreover, 98 of the 113 embryonic cell deaths affect cells derived from the AB blastomere (i.e., occur in the “AB lineage”). Cells programmed to die during C. elegans development are generally the result of an asymmetric cell division and exhibit distinct characteristics, such as detachment from surrounding cells and formation of a round, optically refractile cell corpse. Ultimately, cell corpses are engulfed and degraded by a neighboring cell (Hedgecock et al. 1983; Sulston et al. 1983). The core genetic pathway that triggers apoptotic cell death is conserved from nematodes to mammals and, in C. elegans, consists of four genes, egl-1, ced-9, ced-4, and ced-3, which act in a simple linear pathway (for review, see Horvitz 2003; Lettre and Hengartner 2006; Conradt et al. 2016). The egl-1 gene encodes a proapoptotic BH3-only protein, which binds to the anti-apoptotic Bcl-2-like protein CED-9 in cells programmed to die. This binding causes the release of the Apaf-1-like adaptor protein CED-4 from CED-9. CED-4 then forms the apoptosome, which mediates activation of the caspase CED-3. CED-3 then induces the processes necessary for the swift demise of the cell. Loss-of-function mutations in either of the proapoptotic genes (egl-1, ced-4, or ced-3) or a gain-of-function mutation in the anti-apoptotic gene ced-9 block nearly all programmed cell deaths in C. elegans (Ellis and Horvitz 1986; Hengartner et al. 1992; Conradt and Horvitz 1998).
As the most-upstream component of the central apoptotic pathway, the BH3-only protein EGL-1 must be tightly controlled to avoid inappropriate cell death (for review, see Nehme and Conradt 2008). One level at which EGL-1 activity is controlled is the transcriptional level. Mutations in cis-acting elements of the egl-1 locus result in the misexpression of egl-1 and, consequently, changes in the highly reproducible pattern of programmed cell death during development (Conradt and Horvitz 1999; Hirose et al. 2010). In addition, transcriptional reporters have demonstrated that the egl-1 gene is most highly expressed in cells programmed to die (Conradt and Horvitz 1999; Thellmann et al. 2003; Liu et al. 2006; Potts et al. 2009; Hirose et al. 2010; Winn et al. 2011; Hirose and Horvitz 2013; Jiang and Wu 2014; Wang et al. 2015). Furthermore, genetic studies revealed that egl-1 transcription is governed by lineage-specific transcription factors, which control the death of individual, often bilaterally symmetric, cells (for review, see Conradt et al. 2016). For example, a heterodimer of the basic helix–loop–helix (bHLH) transcription factors HLH-2 Daughterless and HLH-3 Achaete-scute (HLH-2/HLH-3) binds to a specific cis-acting element of the egl-1 locus [referred to as B(egl-1)] and is required for egl-1 transcriptional activation in the left and right NSM sister cells, which are programmed to die (Thellmann et al. 2003). In the left and right NSMs, which are programmed to survive, the Snail-like zinc finger transcription factor CES-1 antagonizes HLH-2/HLH-3 function, thereby preventing egl-1 transcriptional activation (Metzstein and Horvitz 1999; Thellmann et al. 2003; Hatzold and Conradt 2008).
Little is known about the post-transcriptional or post-translational regulation of EGL-1 activity. In mammals, at least eight BH3-only proteins exist, and their activities are regulated at various levels (for review, see Happo et al. 2012), including transcriptional (Oda et al. 2000; Nakano and Vousden 2001) and post-translational (Zha et al. 1996; Li et al. 1998) mechanisms. Mammalian BH3-only genes are also subject to post-transcriptional regulation via microRNAs (miRNAs). It has been demonstrated, for example, that the 3′ untranslated region (UTR) of Bim mRNA is the target of several miRNAs, which act to down-regulate and fine-tune Bim expression (Ventura et al. 2008; Kole et al. 2011; Qian et al. 2011; Pernaute et al. 2014; for review, see Sionov et al. 2015).
In this study, we investigate the roles in programmed cell death during C. elegans development of the miR-35 and miR-58 families of miRNAs (Lau et al. 2001; Kato et al. 2009; Alvarez-Saavedra and Horvitz 2010; Wu et al. 2010). The miR-35 family consists of eight members, collectively referred to as miR-35-42, which are found at high levels in oocytes and throughout early and mid-stage embryos but whose levels decline post-gastrulation (>350 embryonic nuclei) (Stoeckius et al. 2009; Alvarez-Saavedra and Horvitz 2010; Isik et al. 2010; Wu et al. 2010). The loss of all eight members in mir-35-41(nDf50) mir-42(nDf49) mutants (referred to as “mir-35 family mutants”) results in 100% embryonic lethality (Alvarez-Saavedra and Horvitz 2010). Animals lacking only the seven miRNAs miR-35-41 are viable but display altered RNAi sensitivity, reduced fecundity, and impaired responsiveness to hypoxia (Massirer et al. 2012; McJunkin and Ambros 2014; Kagias and Pocock 2015). The miR-58 family is homologous to bantam miRNA in Drosophila and consists of six members: four members that are found at increasing levels throughout embryos starting from mid-stage embryogenesis (miR-80, miR-58.1, miR-81, and miR-82) and two members that are found only at low levels at any stage of development (miR-58.2 and miR-2209.1) (Stoeckius et al. 2009; Alvarez-Saavedra and Horvitz 2010; Isik et al. 2010; Wu et al. 2010). The loss of all four abundant members in mir-80(nDf53); mir-58.1(n4640); mir-81-82(nDf54) mutants (referred to as “mir-58 family mutants”) results in reduced body and brood size as well as defects in locomotion and dauer formation (Alvarez-Saavedra and Horvitz 2010). A previous study demonstrated that egl-1 mRNA is an in vitro target of both miR-35 and miR-58 family miRNAs (Wu et al. 2010). Furthermore, it was proposed that miR-35 and miR-58 family miRNAs cause translational repression of egl-1 mRNA by mediating its deadenylation (Wu et al. 2010). Another study reported that the loss of the mir-58 family causes impairment of germline apoptosis; however, a target was not identified (Subasic et al. 2015). Therefore, to date, direct regulation of the central apoptotic pathway by miRNAs in vivo has not been demonstrated.
Here, we show that loss of the mir-35 family of miRNAs leads to the presence of abnormally large cell corpses in embryos and that this phenotype is enhanced in mir-35 mir-58 double-family mutants. These large corpses correspond to mothers and sisters of cells programmed to die and hence cells that normally survive. Furthermore, we demonstrate that the “precocious” and “collateral” death of mothers and sisters, respectively, is dependent on egl-1 BH3-only and that egl-1 mRNA is present in these mothers and sisters. Finally, we examined the temporal dynamics of egl-1 mRNA in cell death lineages and propose that egl-1 mRNA is targeted by miR-35 and miR-58 family miRNAs in order to avoid traversal of a cellular threshold that would trigger death.
Results
mir-35 family mutants exhibit a large cell corpse phenotype
To identify cell death abnormalities in miRNA family mutants, we analyzed embryonic development using four-dimensional (4D) microscopy (Schnabel et al. 1997, 2006). We first analyzed wild-type (+/+) embryos and detected the 13 cell deaths of the “first wave of AB cell death” (Figs. 1A, 2A; Supplemental Fig. S1). As expected, dying cells rounded up to form refractile corpses ∼2.5 µm in diameter that were engulfed by neighboring cells. Next, we analyzed mir-35 family mutants [mir-35-41(nDf50) mir-42(nDf49)] prior to embryonic arrest. We identified the 13 cell deaths of the first wave of AB cell death (Figs. 1B, 2A); however, in addition to the normal cell corpses, we also detected corpses with diameters ∼1.5-fold larger than normal (Fig. 1B). These large corpses displayed the typical attributes of cell corpses; however, not all were eventually engulfed by neighboring cells. Instead, they often persisted or were extruded from the developing embryo (Supplemental Fig. S2). We quantified the number of large cell corpses per embryo until a predetermined developmental endpoint (ventral enclosure of the epidermal cells; ∼330 min at 25°C) and detected an average of 4.6 large corpses per embryo (Fig. 2A). Large cell corpses were never detected earlier than ∼180 min of embryonic development, and their formation became more frequent as embryogenesis progressed, peaking at ∼225 min and again at ∼300 min (Fig. 2B).
Figure 1.
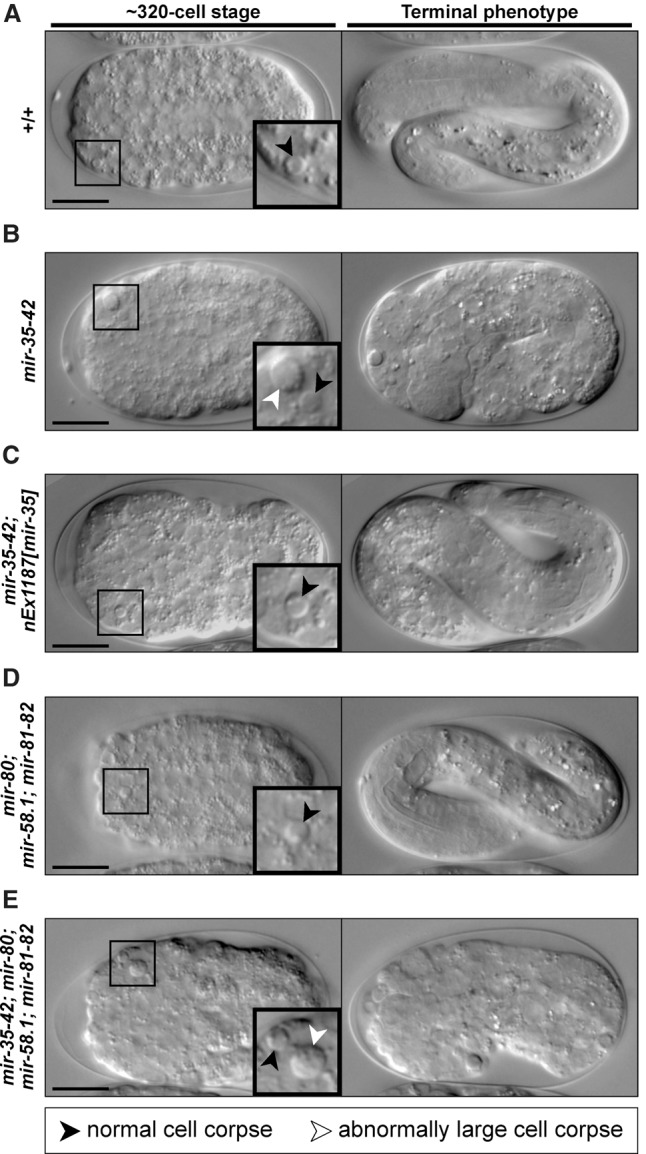
Embryos lacking miR-35 family miRNAs exhibit a large cell corpse phenotype. (A–E) Differential interface contrast (DIC) images of embryos of the genotypes +/+ (A), mir-35-41(nDf50) mir-42(nDf49) (B), mir-35-41(nDf50) mir-42(nDf49); nEx1187 [mir-35 (genomic)+sur-5::gfp] (C), mir-80(nDf53); mir-58.1(n4640); mir-81-82(nDf54) (D), and mir-35-41(nDf50) mir-42(nDf49); mir-80(nDf53); mir-58.1(n4640); mir-81-82(nDf54) (E). For each panel, a developing embryo (∼320-cell stage) is depicted at the left, with insets showing representative cell corpses. Normal cell corpses (black arrowheads) are present in all genetic backgrounds, but abnormally large cell corpses (white arrowheads) are also present (see B,E). The terminal phenotype of each embryo is shown at the right; those in A, C, and D survived to hatching, whereas those in B and E arrested. Bars, 10 µm.
Figure 2.

Inappropriate cell death in mir-35 family mutants is enhanced upon loss of the mir-58 family and is dependent on the central apoptotic pathway. (A) The number of first-wave AB cell deaths was scored per embryo of each genotype. n = 5 or 10, as indicated. The number of abnormally large corpses per embryo was also scored until ventral enclosure. Values are averages ± SEM when applicable. (****) P ≤ 0.0001 via Student's t-test. (B,C) The number of large cell corpses forming over time in mir-35-41(nDf50) mir-42(nDf49) mutants (B) and mir-35-41(nDf50) mir-42(nDf49); mir-80(nDf53); mir-58.1(n4640); mir-81-82(nDf54) mutants (C) until ventral enclosure. Data are based on the time of initial detection for each of the 46 and 107 large cells corpses in B and C, respectively. Each circle represents the detection of a single large cell corpse. Detection times were grouped into 15-min bins (i.e., intervals). n = 10 embryos for each genotype.
This large cell corpse phenotype has not been characterized previously in mir-35 family mutants. Therefore, we tested whether the phenotype could be rescued by a transgene (nEx1187) that carries mir-35 (Alvarez-Saavedra and Horvitz 2010). Since transgenic embryos contained only normal-sized cell corpses (Figs. 1C, 2A), we attribute the large cell corpse phenotype specifically to the loss of miR-35 family miRNAs.
The large cell corpse phenotype of mir-35 family mutants is enhanced by the loss of the mir-58 family
We next assessed cell death in mir-58 family mutants [mir-80(nDf53); mir-58.1(n4640); mir-81-82(nDf54)]. Normal cell corpses were detected throughout embryogenesis, but no large cell corpses were observed, and all embryos survived to hatching (Figs. 1D, 2A). Finally, we generated a strain lacking both miR-35 and miR-58 family miRNAs [mir-35-41(nDf50) mir-42(nDf49); mir-80(nDf53); mir-58.1(n4640); mir-81-82(nDf54)] and examined embryos for the large cell corpse phenotype. As observed in mir-35 family mutants, both normal and large cell corpses were present in mir-35 mir-58 double-family mutants, and all embryos arrested (Fig. 1E). We quantified the number of large cell corpses per embryo prior to ventral enclosure (∼390 min at 25°C in mir-35 mir-58 double-family mutants) and detected an average of 10.7 large corpses per embryo (Fig. 2A), a 2.3-fold increase over mir-35 family mutants. Large corpses were never detected earlier than ∼160 min of embryonic development, and, as in mir-35 family mutants, the formation of large corpses increased over time, peaking at ∼240 min and again at ∼285 min (Fig. 2C). Therefore, the large cell corpse phenotype in the mir-35 family mutant is enhanced by the loss of the mir-58 family.
miR-35 family miRNAs act upstream of or in parallel to egl-1 BH3-only
The large cell corpses observed in mir-35 family mutants and mir-35 mir-58 double-family mutants displayed the typical attributes of cells programmed to die in C. elegans. Therefore, we tested whether the formation of these large cell corpses requires the central apoptotic pathway. The loss of either ced-3 caspase or egl-1 BH3-only blocks most programmed cell deaths in C. elegans (Ellis and Horvitz 1986; Conradt and Horvitz 1998), including the 13 cell deaths of the first wave of AB cell death (Fig. 2A). Our analysis of mir-35-42; ced-3 and mir-35-42; egl-1 double mutants revealed that both normal-sized and large cell corpses were absent during embryogenesis. This indicates that miR-35 family miRNAs act upstream of or in parallel to egl-1 to suppress the appearance of abnormally large cell corpses. Of note, the loss of either ced-3 or egl-1 did not rescue embryonic lethality in mir-35 family mutants (Supplemental Fig. S3), indicating that the large cell corpse phenotype is not the underlying cause of embryonic arrest.
The 3′ UTR of egl-1 causes repression of reporter gene expression in a mir-35 and mir-58 family-dependent manner
The 3′ UTR of egl-1 is predicted to harbor multiple miRNA-bindings sites, including one for miR-35 family miRNAs and one for miR-58 family miRNAs (Fig. 3A; TargetScanWorm release 6.2; Jan et al. 2011). In addition, both sites are conserved across at least three other nematode species and were found to associate with C. elegans Argonaut ALG-1 in iPAR-CLIP (in vivo photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation) experiments, suggesting that they are likely targets of the RNA-induced silencing complex (RISC) (Grosswendt et al. 2014). To determine whether the egl-1 3′ UTR can suppress the expression of a reporter gene in vivo, we generated five different reporter constructs (Fig. 3B). Each of the reporters contained a gfp::histone h2b fusion gene (gfp::h2b) driven by the mai-2 promoter (Pmai-2gfp::h2b) (see the Materials and Methods; Ichikawa et al. 2006). The five reporters differed only in their 3′ UTRs. The first reporter contained the 3′ UTR of the mai-2 gene, which lacks predicted binding sites for miR-35 and miR-58 family miRNAs (mai-2 3′ UTR). The remaining reporters contained the wild-type egl-1 3′ UTR (egl-1wt 3′ UTR) or mutant egl-1 3′ UTRs, in which the predicted miR-35 family miRNA-binding site (egl-1mut mir-35 3′ UTR), the predicted miR-58 family miRNA-binding site (egl-1mut mir-58 3′ UTR), or both (egl-1mut mir-35 mir-58 3′ UTR) were mutated (Fig. 3A, B). The expression pattern of each transgenic construct was then analyzed at the single-copy level (MosSCI [Mos1 transposase-mediated single-copy gene insertion] alleles) in four-cell, ∼320-cell (mid), and ∼550-cell (late) stage embryos. The mai-2 3′ UTR transgene displayed ubiquitous expression at all embryonic stages examined (Fig. 3C). In contrast, the expression of the egl-1wt 3′ UTR transgene was markedly suppressed in all cells in four-cell and ∼320-cell stage embryos; however, expression was detected in ∼550-cell stage embryos (Fig. 3C). (This derepression of transgene expression in older embryos most likely reflects the decrease in mir-35-42 expression observed after gastrulation [Stoeckius et al. 2009; Alvarez-Saavedra and Horvitz 2010; Isik et al. 2010; Wu et al. 2010].) The egl-1mut mir-35 3′ UTR transgene as well as the egl-1wt 3′ UTR transgene in a mir-35-42 mutant background exhibited ubiquitous expression that was indistinguishable from that of the mai-2 3′ UTR transgene. Finally, mutating the binding site for miR-58 family miRNAs had no detectable effect on the repression of an otherwise wild-type transgene (egl-1mut mir-58) but slightly increased expression of the egl-1mut mir-35 transgene (egl-1mut mir-35 mir-58). Therefore, we conclude that miRNAs of the miR-35 family target the egl-1 3′ UTR in a site-specific manner to repress expression of the egl-1 gene and that miRNAs of the miR-58 family may act to enhance this repression.
Figure 3.
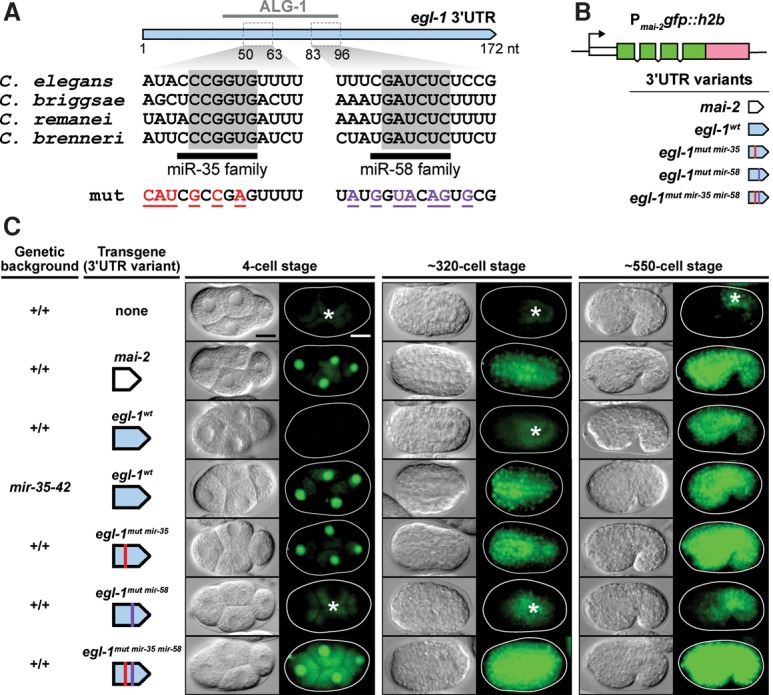
The 3′ UTR of egl-1 is a target of miR-35 and miR-58 family miRNAs in vivo. (A) The 3′ UTR of egl-1 is illustrated in blue, with a reported C. elegans Argonaut ALG-1-binding site from a recent study (Grosswendt et al. 2014) indicated above. Sequences are given for predicted miR-35 family-binding sites and miR-58 family-binding sites, and conserved bases within each site are highlighted in gray across four Caenorhabditis species. The mutated sequences corresponding to egl-1mut mir-35 (red) and egl-1mut mir-58 (purple) are shown (for complete mutated sequences, see the Materials and Methods). (B) A schematic representation of the 3′ UTR reporters constructed for this study. (C) Analysis of single-copy 3′ UTR reporter expression during embryogenesis. The genetic background and transgene under investigation are indicated at the left of each image sequence, which shows representative embryos from three developmental stages. For each embryo, a DIC image (left) and GFP image (right) are shown. The following alleles were used: bcSi25 [Pmai-2gfp::h2b::mai-2 3′ UTR], bcSi26 [Pmai-2gfp::h2b::egl-1wt 3′ UTR], bcSi27 [Pmai-2gfp::h2b::egl-1mut mir-35 3′ UTR], bcSi46 [Pmai-2gfp::h2b::egl-1mut mir-35 3′ UTR], and bcSi47 [Pmai-2gfp::h2b::egl-1mut mir-35 mir-58 3′ UTR]. Asterisks indicate autofluorescence. Transgenic strains were homozygous for unc-119(ed3) and the cb-unc-119(+) selection marker. Bars, 10 µm.
miRNAs repress target gene expression by causing translational inhibition and/or mRNA decay (for review, see Huntzinger and Izaurralde 2011). To determine which of these two mechanisms is responsible for repression of gfp::h2b expression in the case of the egl-1wt 3′ UTR transgene, we used single-molecule RNA fluorescent in situ hybridization (smRNA FISH) to directly probe gfp::h2b mRNA in transgenic embryos. Compared with embryos carrying the mai-2 3′ UTR transgene, we found that embryos carrying the egl-1wt 3′ UTR transgene had significantly reduced copy numbers of gfp::h2b mRNA (Supplemental Fig. S4A,B). Based on this observation, we propose that miR-35 and miR-58 family miRNAs repress egl-1 expression by causing both translational inhibition and decay of egl-1 mRNA.
Mothers and sisters of cells normally programmed to die undergo apoptosis in mir-35 family mutants
Abnormally large cell corpses have been observed previously in some C. elegans mutants (Hengartner et al. 1992; Sugimoto et al. 2001; Frank et al. 2005), and their appearance has been attributed to the death of physically larger cells, which might result from a prematurely active cell death program or a reversal of polarity in the mother of a cell programmed to die. Therefore, using 4D microscopy and lineage analyses, we determined the identities of all 46 large cell corpses that we had detected in 10 mir-35 family mutant embryos (Fig. 2A). In each case, we found that the large corpse was formed by a cell that was not normally programmed to die; however, each was closely related to a programmed cell death (Fig. 4A). Specifically, all large cell corpses could be classified as either the mother of a cell death (37 out of 46; ∼80%) or the sister of a cell death (13 out of 46; ∼28%). (One inappropriately dying cell, ABalappaap, which died in four of 10 embryos [40% inappropriate death in mir-35-42 embryos], is both a sister and a mother of a cell death and is therefore considered in both classifications.) Hence, we consider these inappropriate deaths to be “precocious” in the case of mother cells and “collateral” in the case of sister cells (Fig. 4B). The precocious death of mother cells always occurred prior to the expected time of division, and divisions giving rise to collaterally dying sister cells exhibited normal timing and polarity (data not shown). Thus, our data support a prematurely or collaterally active central apoptotic pathway as the cause of inappropriate cell death in mir-35 family mutants, affecting mothers and sisters of programmed cell deaths.
Figure 4.
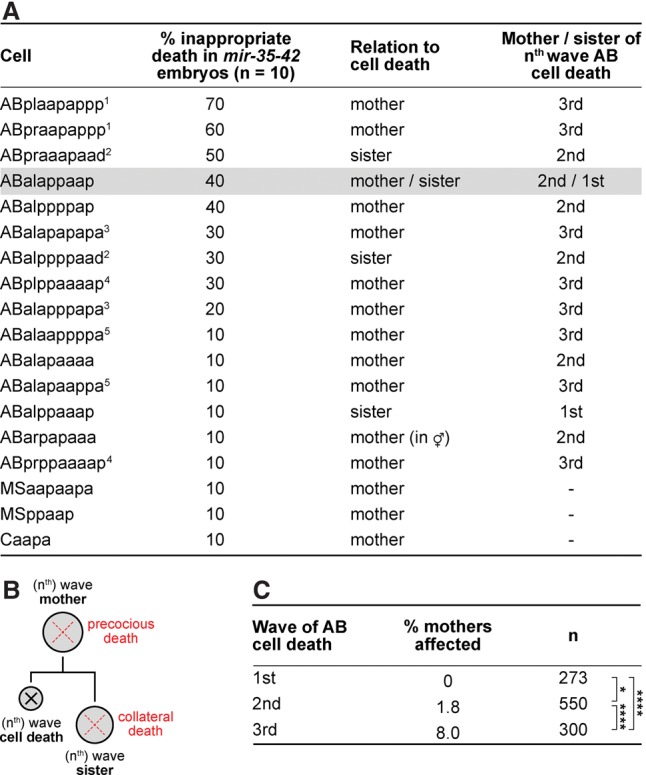
Abnormal cell death in mir-35 family mutants affects mothers and sisters of programmed cell death. (A) The identities of 46 larges corpses present across 10 mir-35-41(nDf50) mir-42(nDf49) embryos were determined as described in the Materials and Methods. Numerical superscripts 1–5 indicate pairs of bilaterally symmetric cells. (B) Lineage representation of “precocious death” affecting mother cells and “collateral death” affecting sister cells. (C) The average percentage of precociously dying mothers was determined per embryo for the three waves of AB cell death according to data presented in A. (*) P < 0.05; (****) P < 0.0001 via Fisher’s exact test.
Lineage and developmental stage affect the penetrance of inappropriate cell deaths in mir-35 family mutants
ABalappaap dies in 40% of mir-35 family mutants (Fig. 4A). We specifically examined ABalappaap in mir-35 mir-58 double-family mutants and discovered that this cell dies precociously in 80% of embryos (n = 10), which is double the penetrance observed in mir-35 family mutants (Fig. 4A). This provides further evidence that inappropriate death, resulting in the formation of large cell corpses, is enhanced in mir-35 mir-58 double-family mutants.
The 98 programmed cell deaths of the AB lineage can be classified into three temporal “waves” (Supplemental Fig. S1). The first wave consists of 13 cells that die after the ninth round of embryonic cell division. The second and third waves consist of 55 and 30 cells that die after the 10th and 11th rounds of embryonic cell division, respectively. Notably, mothers of first-wave cell deaths were never found to die precociously (zero of 273) in mir-35 family mutants (Fig. 4C). Mothers of second-wave cell deaths were modestly affected, with an average of 1.8% dying precociously (10 of 550). However, the greatest impact was on mothers of third-wave cell deaths, with an average of 8% dying precociously (24 of 300). Thus, we conclude that a loss of the mir-35 family does not trigger the precocious death of mothers of the first wave of cell deaths in the AB lineage, and mothers of the third wave are about four times more likely to die precociously than mothers of the second wave.
miRNA-dependent control of egl-1 mRNA copy number is crucial for the survival of ABalappaap
Using smRNA FISH, we directly probed egl-1 mRNA in the ABalappaap lineage in situ. To identify ABalappaap in fixed whole-mount embryos, we used an unc-3 reporter (Punc-3unc-3::gfp) (Wang et al. 2015) and concurrently stained gfp mRNA (Fig. 5B). Single-cell copy numbers of egl-1 mRNA were quantified from image stacks obtained by confocal microscopy. Briefly, the total smRNA FISH signal intensity corresponding to ABalappaap was quantified and then divided by the intensity of a single mRNA molecule, thereby yielding a measure of mRNA copy number (see the Materials and Methods; Supplemental Fig. S5). We also measured the volumes of ABalappaap and its daughters (Fig. 5A) and calculated the mean concentrations of egl-1 mRNA.
Figure 5.

egl-1 mRNA copy number crosses a threshold to trigger the precocious death of ABalappaap in mir-35 mir-58 double-family mutants. (A) smRNA FISH analysis in the ABalappaap lineage. The lineage is illustrated at the right, with the approximate time of each analysis (pre- and post-division) indicated. Representative images of the ABalappaap cell and its daughters are shown for each time point in four genetic backgrounds, as indicated. The egl-1 mRNA copy number corresponding to each image is given. Nuclei are labeled as belonging to the surviving daughter (anterior [a]) or the dying daughter (posterior [p]). Approximate cellular volumes are given at the right. Bars, 2 µm. (B) A fixed wild-type embryo showing the ABalappaap cell (arrowhead) as identified by smRNA FISH staining against mRNA transcribed from the Punc-3unc-3::gfp reporter. The anterior–posterior axis (A ↔ P) is indicated. Bar, 10 µm. (C) Quantification of egl-1 mRNA copy number in the ABalappaap cell and its two daughters (ABalappaapa and ABalappaapp [X]) for each of the indicated genotypes. An estimated threshold for triggering cell death in ABalappaap is represented by the red line. The mean value is given above each data set. Box and whiskers were plotted according to Tukey's test. (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001 via Mann-Whitney test. The following alleles were used: mir-35-41(nDf50), mir-42 (nDf49), mir-80(nDf53), mir-58.1(n4640), and mir-81-82(nDf54). All strains analyzed carried the extrachromosomal array xdEx1091 [Punc-3unc-3::gfp+Psur-5rfp].
We first analyzed ABalappaap in the two genetic backgrounds in which this cell does not die precociously. In both wild-type and mir-58 family mutant embryos, we found egl-1 mRNA to be largely absent from ABalappaap, with a mean number of 0.2 and 0.5 copies, respectively (0.01 mRNA copies per cubic micrometer) (Fig. 5A,C; Supplemental Fig. S6A). In the two genetic backgrounds in which ABalappaap dies precociously (mir-35 family mutants and mir-35 mir-58 double-family mutants), we measured a significantly larger number of copies of egl-1 mRNA in ABalappaap, with the highest value found in mir-35 mir-58 double-family mutants (mean 5.1 copies; 0.13 mRNA copies per cubic micrometer). Since ABalappaap dies in 40% of mir-35 family mutants (Fig. 2A) and 80% of mir-35 mir-58 double-family mutants, a corresponding percentage of embryos would be expected to contain copy numbers of egl-1 mRNA sufficient to trigger death. Therefore, our data suggest that two to three copies of egl-1 mRNA are sufficient to trigger the precocious death of ABalappaap and hence represent “above threshold” copy numbers of egl-1 mRNA in this cell (Fig. 5C). In addition, our data demonstrate that miR-35 family miRNAs and miR-58 family miRNAs cooperate in ABalappaap to keep the copy number of egl-1 mRNA below this threshold.
Next, we analyzed egl-1 mRNA copy numbers in the daughters of ABalappaap: ABalappaapa, which survives, and ABalappaapp (X), which dies (Fig. 5A,C). (The programmed death of ABalappaapp [X] is dependent on egl-1 [Wang et al., 2015].) In the surviving daughter, we found similar “below threshold” egl-1 mRNA copy numbers in all genotypes (Fig. 5C). In contrast, in the dying daughter, we observed similar “above threshold” egl-1 mRNA copy numbers in all genotypes (Fig. 5C).
miR-35 and miR-58 family miRNAs affect the level of egl-1 mRNA in cell death lineages
We investigated the copy numbers of egl-1 mRNA in a second cell death lineage, this time in a lineage in which no effect was observed upon loss of the mir-35 and/or mir-58 family (i.e., no inappropriate death). For this purpose, we chose the MSpaap cell and its daughters, since these cells can be identified in fixed whole-mount embryos by DAPI and egl-1 smRNA FISH signals alone (Fig. 6B). The MSpaap cell divides at the 180-cell stage, giving rise to MSpaapp (X), which is the first cell to undergo programmed cell death during C. elegans development. (The programmed death of MSpaapp [X] is dependent on egl-1 [data not shown].)
Figure 6.

egl-1 mRNA levels are temporally dynamic in the MSpaap lineage and more abundant in mir family mutants. (A) smRNA FISH analysis in the MSpaap lineage. The lineage is illustrated at the right, with the approximate time of each analysis (predivision, dividing, and post-division) indicated. Representative images of the MSpaap cell and its daughters are shown for each time point in four genetic backgrounds, as indicated. Nuclei are labeled as belonging to the surviving daughter (anterior [a]) or the dying daughter (posterior [p]). The egl-1 mRNA copy number corresponding to each image is given. Approximate cellular volumes are given at the right. Bars, 2 µm. (B) A fixed wild-type embryo containing 167 nuclei showing the MSpaap cell (inset). The anterior–posterior axis (A ↔ P) is indicated Bar, 10 µm. (C) Quantification of egl-1 mRNA in the MSpaap cell and its two daughters (MSpaapa and MSpaapp [X]) for each of the indicated genotypes. The mean value is given above each data set. Box and whiskers were plotted according to Tukey's test. (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001 via Mann-Whitney test. (D) Developmental time course showing egl-1 mRNA concentration in cells of the MSpaap lineage for the indicated genotypes. Division of the MSpaap cell is indicated by a vertical dashed line. Insets show a 2× zoom of the graph at the time of division. Graphs were generated from raw data as a centered moving average of order 5. Shaded areas represent SEM, when applicable. The following alleles were used: mir-35-41(nDf50), mir-42 (nDf49), mir-80(nDf53), mir-58.1(n4640), and mir-81-82(nDf54).
We quantified single-cell copy numbers of egl-1 mRNA at three distinct time points: predivision (MSpaap), dividing (MSpaap), and post-division (MSpaapa and MSpaapp [X]). In addition, we measured the volumes of the cells (Fig. 6A) and calculated the mean concentrations of egl-1 mRNA. Predivision, egl-1 mRNA was detected in MSpaap across all genotypes (Fig. 6A). The mean copy number of egl-1 mRNA in MSpaap in wild type was 12.5 (0.11 mRNA copies per cubic micrometer), and this number increased to as much as 18.0 (0.16 mRNA copies per cubic micrometer) in mir-35 mir-58 double-family mutants (Fig. 6C; Supplemental Fig. S6B). Since MSpaap was not found to die precociously in any of the mutant backgrounds, we conclude that 0.16 egl-1 mRNA copies per cubic micrometer is not sufficient to trigger its precocious death. (For comparison, our data suggest that 0.13 egl-1 mRNA copies per cubic micrometer is sufficient to trigger the precocious death of 80% of ABalappaap cells.) Slight differences in egl-1 mRNA copy numbers were also observed in the surviving daughter MSpaapa, which also was not found to die collaterally in any of the mutant backgrounds. In contrast, larger and significant differences between the genotypes were observed in the dying daughter MSpaapp (X) (Fig. 6C; Supplemental Fig. S6B). In wild type, the mean concentration of egl-1 mRNA in this cell was 0.53 mRNA copies per cubic micrometer, which is similar to the mean concentration that we determined for ABalappaapp (X) (0.86 mRNA copies per cubic micrometer) (Supplemental Fig. S6A). egl-1 mRNA copy numbers and concentration were elevated in the mir mutant backgrounds, increasing to as high as 33.8 copies and 1.5 mRNA copies per cubic micrometer in mir-35 mir-58 double-family mutants. Since our data demonstrate that, in mir-35 family and/or mir-58 family mutants, the majority of MSpaapp (X) cells contains copy numbers of egl-1 mRNA that are significantly larger than in wild type (Fig. 6C), we asked whether this affected their deaths. We measured the time needed for MSpaapp (X) to form a cell corpse in each genetic background and found no difference in the timing of the cell death process (Supplemental Fig. S7).
Next, we plotted egl-1 mRNA copy numbers (Supplemental Fig. S8) and egl-1 mRNA concentrations (Fig. 6D) in MSpaap, MSpaapa, and MSpaapp (X) according to the developmental stage of the respective embryo. This revealed the temporal dynamics of egl-1 mRNA in the three cells. In the mother MSpaap, egl-1 mRNA levels increased just prior to cell division in all genotypes, presumably reflecting the start of transcriptional up-regulation of the egl-1 gene (Fig. 6D; Supplemental Fig. S8). In wild type, this amounted to an ∼50% increase, but the increase was larger in mir-35 and/or mir-58 family mutants (Supplemental Fig. S8). Furthermore, plotting egl-1 mRNA concentrations according to developmental stage also revealed that the distribution of egl-1 mRNA among the descendants of MSpaap reflects the inherent difference in sizes of the two daughter cells and that each “inherits” ∼0.2 egl-1 mRNA copies per cubic micrometer (Fig. 6D). Therefore, at least in the MSpaap lineage, egl-1 mRNA does not appear to be segregated asymmetrically into the daughter that is programmed to die.
After the division of MSpaap, the levels of egl-1 mRNA diminished in the surviving daughter MSpaapa in all genotypes, reaching negligible levels around the 190-cell stage (Fig. 6D; Supplemental Fig. S8). This suggests that the degradation of egl-1 mRNA in MSpaapa is not dependent on miR-35 and miR-58 family miRNAs. In contrast, in all genotypes, the levels of egl-1 mRNA increased significantly in the dying daughter Mspaapp (X), presumably as a consequence of egl-1 transcriptional up-regulation (Fig. 6D; Supplemental Fig. S8). However, the magnitude of this increase varied significantly among the different genetic backgrounds and was most dramatic in mir-35 mir-58 double-family mutants, peaking at ∼37 copies of egl-1 mRNA (compared with ∼19 copies of egl-1 mRNA in the wild type) or ∼1.7 egl-1 mRNA copies per cubic micrometer (compared with ∼0.9 egl-1 mRNA copies per cubic micrometer in the wild type) at the 190- to 200-cell stage (Supplemental Fig. S8). Based on these results, we conclude that miR-35 family and miR-58 family miRNAs target egl-1 mRNA in the MSpaap lineage, although the loss of these miRNA families is not sufficient to trigger inappropriate death.
Discussion
miR-35 and miR-58 family miRNAs cooperate to directly repress expression of the BH3-only gene egl-1 in vivo
We demonstrate that miR-35 and miR-58 family miRNAs cooperate in the ABalappaap and most probably other cell lineages to directly control egl-1 expression in mothers of cells programmed to die during embryogenesis, thereby preventing their precocious deaths (Fig. 7). “miRNA cooperativity” occurs when the simultaneous targeting of two miRNA-binding sites causes a more than additive effect, and this phenomenon is observed predominantly for closely linked miRNA-binding sites (Grimson et al. 2007; Sætrom et al. 2007). With a distance of 33 nucleotides (nt) separating the miR-35 family-binding site and miR-58 family-binding site, the egl-1 3′ UTR is likely the target of cooperative miRNA function, and this is confirmed by our in vivo studies of the ABalappaap lineage. Direct targets have been described recently for the miR-35 family of miRNAs (Liu et al. 2011; Kagias and Pocock 2015) as well as the miR-58 family of miRNAs (de Lucas et al. 2015; Pagano et al. 2015). Furthermore, it has been demonstrated previously that, in C. elegans, miRNAs of a particular family can function redundantly (Miska et al. 2007) and/or in combination (Abbott et al. 2005) to control target gene expression. However, at least to our knowledge, the finding that members of the miR-35 and miR-58 family cooperate to directly repress the expression of the BH3-only gene egl-1 is the first in vivo demonstration of miRNA cooperativity between members of two miRNA families in C. elegans.
Figure 7.
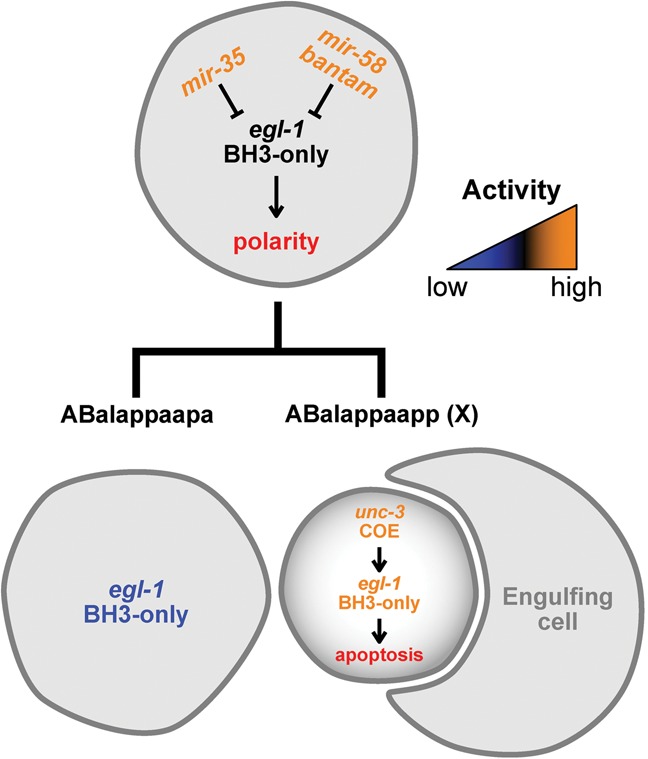
Genetic model of mir-35- and mir-58-dependent control of egl-1 BH3-only in the ABalappaap lineage. See the text for details.
miR-35 and miR-58 family miRNAs cause egl-1 mRNA decay in the ABalappaap and MSpaap cell death lineages
Based on in vitro studies, it has been proposed that, by mediating its deadenylation, miR-35 and miR-58 family miRNAs can cause translational repression of egl-1 mRNA (Wu et al. 2010). This is confirmed by our in vivo studies of animals carrying the Pmai-2gfp::h2b::egl-1 3′ UTR transgene. (Whether miR-35 and miR-58 family miRNAs also cause translational repression of egl-1 mRNA generated from the egl-1 locus is unknown, since we were unable to detect endogenous EGL-1 protein in any genetic background.) Based on our in vivo studies of both the Pmai-2gfp::h2b::egl-1 3′ UTR transgene and egl-1 mRNA generated from the egl-1 locus, we propose that miR-35 and miR-58 family miRNAs also mediate egl-1 mRNA decay. For example, we found that, in the ABalappaap lineage, miR-35 and miR-58 family miRNAs cooperate to control the copy number of egl-1 mRNA in the mother of a cell programmed to die and thus presumably maintain egl-1 expression below a threshold that is sufficient to trigger death (Fig. 7). In the MSpaap lineage, in which the loss of the mir-35 and/or mir-58 family did not induce precocious or collateral death, miR-35 and miR-58 family miRNAs cooperate to control the copy number of egl-1 mRNA in the daughter that is programmed to die [MSpaapp (X)] and thereby dampen the transcriptional boost of egl-1 that occurs in this cell.
Our results raise a number of interesting questions. For example, it is unclear why a mean concentration of 0.13 egl-1 mRNA copies per cubic micrometer is sufficient to trigger the precocious death of 80% of ABalappaap cells but 0% of MSpaap cells. One possible explanation is that, in the MSpaap lineage, factors other than egl-1 expression may be limiting. Furthermore, our results indicate that while in the ABalappaap lineage, miR-35 and miR-58 family miRNAs cause egl-1 mRNA decay predominantly in the mother, in the MSpaap lineage, they act predominantly in the daughter that is programmed to die. As discussed in more detail below, we propose that this reflects the necessity of precisely controling egl-1 expression and hence EGL-1 activity in a lineage-specific manner. Furthermore, this observation suggests that the activities of miR-35 and miR-58 family miRNAs themselves are precisely controlled in a spatial as well as temporal manner, at least in cell death lineages. How this is accomplished remains to be determined.
miR-35 and miR-58 family miRNAs adjust lineage-specific differences in egl-1 transcriptional control
We provide the first in vivo evidence that the activity of the BH3-only gene egl-1 is regulated at the post-transcriptional level through miRNAs. It is well established that EGL-1 activity is controlled at the transcriptional level (for review, see Nehme and Conradt 2008; Conradt et al. 2016), and it has been proposed that transcription factors that are critical for differentiation in a particular lineage may be used in that same lineage to also cause transcriptional up-regulation of egl-1 in the daughter that is programmed to die. For example, in the ABalappaap lineage, the COE transcription factor UNC-3 of C. elegans is required for both the complete differentiation of the daughter programmed to survive (ABalappaapa) and the death of the daughter that is programmed to die [ABalappaapp (X)] (Wang et al. 2015). [How UNC-3 affects differentiation in ABalappaapa is currently unclear; however, in ABalappaapp (X), UNC-3 triggers cell death by directly activating egl-1 transcription.] Interestingly, the unc-3 gene is expressed (and hence UNC-3 protein is presumably present) in not only ABalappaapa and ABalappaapp (X) but also their mother, ABalappaap (Wang et al. 2015). Therefore, we propose that UNC-3 starts to activate egl-1 transcription in the mother and that miR-35 and miR-58 family miRNAs keep the copy number of egl-1 mRNAs below the threshold necessary to trigger death in this cell (Fig. 7). The mothers that are affected by the loss of the mir-35 family are affected to varying degrees (penetrance between 10% and 70%). Furthermore, in different lineages, different transcription factors are responsible for egl-1 transcriptional regulation. Therefore, we propose that the varying penetrance observed in different lineages of mir-35 family mutants is a reflection of the lineage-specific regulators of egl-1 transcription and their functional status in the mothers. Hence, the normal function of miR-35 and miR-58 family miRNAs is to adjust these lineage-specific differences. In support of this notion, the two mothers that die with the highest penetrance, ABplaapappp and ABpraapappp, are mothers of two bilaterally symmetric lineages that most probably use the same transcription factors for egl-1 transcriptional control.
miR-35 and miR-58 family miRNAs critically contribute to the precise control of programmed cell death during C. elegans development
We demonstrate that miR-35 and miR-58 family miRNAs fine-tune EGL-1 activity and therefore critically contribute to the precise temporal control of programmed cell death during C. elegans development. We recently showed that, in the NSM lineage, EGL-1 activity is already present at a certain level in the NSM neuroblast and that this activity is necessary for cellular polarization of the NSM neuroblast and the segregation of apoptotic potential into the daughter that is programmed to die (for review, see Chakraborty et al. 2015; Lambie and Conradt 2016). Although we have not yet established that egl-1 is necessary for cellular polarization in ABalappaap, we suggest that the copy number of egl-1 mRNA needs to be kept not only below the threshold sufficient to trigger the mother's death but also above the threshold necessary to cause cellular polarization.
The observation that, apart from mothers, certain sisters inappropriately die in mir-35 family mutants (“collateral” deaths) suggests that miR-35 and miR-58 family miRNAs may also be required for precise spatial control of programmed cell death during C. elegans development. In the ABalappaap and MSpaap lineages, the turnover of egl-1 mRNA in the sisters programmed to live appears to occur mainly in a mir-35 and mir-58 family-independent manner; however, in some lineages, it may depend on miR-35 and miR-58 family miRNAs. Alternatively, collateral deaths may reflect the activity of lineage-specific regulators of egl-1 transcription and their functional status, in this case, in the daughters programmed to survive.
Overall, we found 0%, 1.8%, and 8.0% of mothers of the first, second, and third waves of AB cell death to be affected by the loss of the mir-35 family. Furthermore, based on our analysis of the ABalappaap lineage, the percentages for the second and third waves of cell death presumably increase twofold by the simultaneous loss of the mir-58 family. Why are mothers of the first wave not affected and why are not more mothers of the second and third wave affected? Mothers of first-wave cell deaths may be resistant to the loss of the mir-35 and mir-58 family because other families of miRNAs may act to repress egl-1 expression. Alternatively, an increase in egl-1 expression at this developmental stage might not be sufficient to trigger deaths because the activities of other proapoptotic proteins (i.e., CED-4 and CED-3) might be limiting or because EGL-1 protein might be inactive due to post-translational control. The involvement of other families of miRNAs could also explain why not more mothers of second- and third-wave cell deaths die in mir-35 mir-58 double-family mutants. Alternatively, in “resistant” mothers, as a result of lineage-specific control of egl-1 transcriptional activation, the egl-1 locus may simply not yet be transcribed. Finally, the cytoplasmic volume of mothers may play a critical role. The relatively large volume of mothers of first-wave cell deaths might confer a buffering capacity to modest changes in the transcriptome or proteome, and this buffering capacity might decrease with decreasing volume. In support of this notion, mothers of second-wave cell deaths, which are larger than mothers of third-wave cell deaths, appear to be more “resistant” than mothers of third-wave cell deaths.
Conservation of miRNA-dependent control of programmed cell death
Through studies at single-cell resolution, we uncovered another aspect of the regulation of programmed cell death during C. elegans development: miRNA-dependent control of the activity of the BH3-only protein EGL-1. Interestingly, the mir-35 family of miRNA genes is found only in other nematodes, although miRNAs with homologous functions might very well exist in higher animals, such as those reported to repress the mammalian BH3-only gene Bim (for review, see Sionov et al. 2015). However, the miR-58 family of miRNAs is homologous in both sequence and function to the bantam miRNA in Drosophila, which controls the expression of the proapoptotic gene hid (Brennecke et al. 2003). However, this conservation does not extend to higher animals. The conservation of miR-58 family miRNAs between nematodes and Drosophila hints at a similar developmental control of programmed cell death between the two model systems, which has been studied recently in depth (Li et al. 2014). It remains to be determined whether components of the core apoptosis pathway other than egl-1 are also controlled by miRNA function during C. elegans development and whether single-cell studies will uncover additional aspects of the regulation of egl-1 activity that may be conserved in Drosophila and mammals.
Materials and methods
General C. elegans maintenance and strains
C. elegans strains were cultured and maintained as described previously (Brenner 1974). The Bristol N2 strain was used as wild type, and the following transgenes and alleles were used in this study: LGI: bcSi25 [Pmai-2gfp::h2b::mai-2 3′ UTR] (this study), bcSi26 [Pmai-2gfp::h2b::egl-1wt 3′ UTR] (this study), bcSi27 [Pmai-2gfp::h2b::egl-1mut mir-35 3′ UTR] (this study), and bcSi46 [Pmai-2gfp::h2b::egl-1mut mir-58 3′ UTR] (this study); LGII: mir-35-41(nDf50) (Miska et al. 2007), mir-42-44(nDf49) (Miska et al. 2007), mIn1 [mIs14 dpy-10(e128)] (Edgley and Riddle 2001), and bcSi47 [Pmai-2gfp::h2b::egl-1mut mir-35 mir-58 3′ UTR] (this study); LGIII: mir-80(nDf53) (Miska et al. 2007) and unc-119(ed3) (Maduro and Pilgrim 1995); LGIV: mir-58.1(n4640) (Miska et al. 2007) and ced-3(n717) (Shaham et al. 1999); LGV: egl-1(n3330) (B. Conradt); and LGX: mir-81-82(nDf54) (Miska et al. 2007). In addition, the following multicopy transgenes and extrachromosomal arrays were used: nEx1187 [mir-35 (genomic)+sur-5::gfp] (Alvarez-Saavedra and Horvitz 2010) and xdEx1091 [Punc-3unc-3::gfp+ Psur-5rfp] (Wang et al. 2015).
The single-copy integration alleles bcSi25, bcSi26, bcSi27, bcSi46, and bcSi47 were generated using MosSCI (Frøkjær-Jensen et al. 2008) and the plasmids pBC1483, pBC1484, pBC1485, pBC1653, and pBC1654, respectively. The strain EG8078 [oxTi185; unc-119(ed3)] was used for targeted insertion on LGI, and EG6699 [ttTi5605; unc-119(ed3)] was used for targeted insertion on LGII (only pBC1654) (Frøkjær-Jensen et al. 2014).
Plasmid construction
The plasmids pBC1483, pBC1484, and pBC1485 were constructed using a two-step overlap extension PCR and restriction enzyme cloning. First, Pmai-2 was amplified as the 1763 base pairs (bp) immediately upstream of the mai-2 start codon (Ichikawa et al. 2006), stopping at the transcription unit for B0546.4. The primers used for this were 5′-agatctGGAAAAAATCGATA-3′ (lowercase letters indicate BglII site) and 5′-GAAAAGTTCTTCTCCTTTACTCATTCTGAAAATTGAGTGAATTAG-3′. Second, this Pmai-2 fragment was fused to the 1287-bp coding sequence of gfp::h2b as amplified from the plasmid pCM1.35 (a gift from G. Seydoux; Addgene plasmid no. 17248) using the primers 5′-CTAATTCACTCAATTTTCAGAATGAGTAAAGGAGAAGAACTTTT-3′ and 5′-GCTCGCGTTCTTGTACTGCAAATTACTTGCTGGAAGTGTACTTG-3′. Finally, the 3′ UTR from either mai-2 (142 bp) or egl-1wt (172 bp) was fused to Pmai-2gfp::h2b to yield the final transgene. The mai-2 3′ UTR was amplified using the primers 5′-CAAGTACACTTCCAGCAAGTAATTTGCAGTACAAGAACGCGAGC-3′ and 5′-TcttaagTTCGCTAAAAACTA-3′ (lowercase letters indicate AflII site). The egl-1wt 3′ UTR was amplified using the primers 5′-CAAGTACACTTCCAGCAAGTAAGTGATCAAAATCTCCAACTTTTC-3′ and 5′-TcttaagAAAAAAAACCATATTTATTATTAG-3′ (lowercase letters indicate AflII site). For the egl-1mut mir-35 variation, the wild-type sequence 5′-GATTTCTCATAATACCCGGT-3′ was mutated to 5′-CTAATCTCATACATCGCCGA-3′ using PCR site-directed mutagenesis and then validated by sequencing. The mutagenic primers used for this were 5′-TCTAATCTCATACATCGCCGAGTTTTTTCTTCATTTGTGATTATTTTTC-3′ and 5′-CTCGGCGATGTATGAGATTAGATGGTACAAATTGGAGAAAAG-3′. Each transgene was then ligated into the EcoRV site of pBluescript II KS+, subsequently excised using BglII/AflII, and ligated into the BglII/AflII site of the final destination MosSCI vector pCFJ350 (a gift from Erik Jorgensen; Addgene plasmid no. 34866) (Frøkjær-Jensen et al. 2012), which harbors the cb-unc-119(+) rescue fragment. The final plasmids were named pBC1483 (mai-2 3′ UTR transgene), pBC1484 (egl-1wt 3′ UTR transgene), and pBC1485 (egl-1mut mir-35 3′ UTR transgene).
The plasmids pBC1653 (egl-1mut mir-58 3′ UTR transgene) and pBC1654 (egl-1mut mir-35 mir-58 3′ UTR transgene) were constructed using PCR site-directed mutagenesis with the templates pBC1484 and pBC1485, respectively. The wild-type miR-58 family miRNA-binding site in the egl-1 3′ UTR was mutated from 5′-GAGAGATCGAAAA-3′ to 5′-CACTGTACCATAT-3′ using mutagenic primers 5′-CACTGTACCATATATAATCACAAATGAAGAAAAAACAC-3′ and 5′-ATATGGTACAGTGCGTCTCCAACTCCCCT-3′.
4D microscopy and lineage analysis
C. elegans L4 animals were grown to the adult stage overnight at 25°C. Two- or four-cell embryos were harvested from the young adults and mounted on 4.5% agarose pads for differential interface contrast (DIC) and fluorescence microscopy. 4D recordings were made throughout development as described previously (Schnabel et al. 1997, 2006) using a Zeiss Axio Imager.M2 and Time to Live software (Caenotec), capturing 25 DIC and/or fluroescence z-stacks every 35 sec at 25°C. Lineage analysis of the 4D recordings was performed using SIMIBioCell software (SIMI Reality Motion Systems, http://www.simi.com).
smRNA FISH and image analysis
smRNA FISH was performed in C. elegans embryos, as described previously (Raj et al. 2008). Embryos were harvested by bleaching healthy adults and then permitted to develop in M9 buffer at 25°C until the desired stage was reached. Stellaris FISH probes (Biosearch Technologies) were designed against the mature mRNA of egl-1 or gfp. The egl-1 probe set contained 23 TAMRA-labeled oligonucleotides and was used at 250 nM. The gfp probe set to target unc-3::gfp contained 48 fluorescein-labeled oligonucleotides and was used at 500 nM. The gfp probe set to target gfp::h2b contained 30 TAMRA-labeled oligonucleotides and was used at 500 nM. Image stacks were obtained using a Leica LAS AF software and a Leica TCS SP5 II confocal microscope with a 63× oil immersion lens and a z-spacing of 500 nm to capture diffraction-limited mRNA spots over several z-slices.
Image analysis was performed using Fiji software (Schindelin et al. 2012). The number of nuclei in a given embryo was determined using Multiview Reconstruction software (Preibisch et al. 2010) to detect interest points in the DAPI channel, with manual corrections as required. The pipeline used to quantify the mRNA copy number in a cell of interest is outlined (Supplemental Fig. S5). Briefly, a three-dimensional region of interest (ROI) was defined for the cell of interest, and the background signal was subtracted. Next, the total smRNA FISH signal intensity was measured for this ROI from a z-projection summing the slices. Finally, the mRNA copy number for the cell of interest was calculated, dividing the total signal intensity of the ROI by the average intensity of a single diffraction-limited mRNA spot. For presentation, maximum intensity z-projection images were smoothed (Gaussian blur; radius, 1.5), and the DAPI signal of neighboring nuclei was removed. Quantification of gfp::h2b mRNA in transgenic embryos was performed on a maximum intensity z-projection of the smRNA FISH signal generated for the whole embryo. Next, a background subtraction (radius, 4) and Gaussian blur filter (radius, 1.5) were applied, and the resulting image was water-shedded. Finally, the number of particles was determined using the Analyze Particles tool (size, >15; circularity, 0–1). Cellular volumes were calculated by assuming sphericality and measuring the average diameter of the cell of interest from confocal image stacks.
Supplementary Material
Acknowledgments
We thank E. Lambie and S. Rolland for comments on the manuscript; V. Ambros, B. Derry, and members of the Conradt laboratory for discussion; L. Jocham, N. Lebedeva, and M. Schwarz for excellent technical support; M. Basch for help in cloning; and V. Ambros and M. Zhen for providing strains. Some strains used in this study were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). R.S. was supported by a Post-graduate Scholarship-Doctoral (PGS D) from the Natural Sciences and Engineering Research Council of Canada (NSERC), and K.M. was supported by a Charles King Trust Post-doctoral Research Fellowship. This work was supported by National Institutes of Health grants F32 GM097895 (to K.M.), K99 GM113063-01 (to K.M.), and R01 GM069950 (to B.C.), and the Deutsche Forschungsgemeinschaft (Center for Integrated Protein Science Munich [CIPSM; EXC 114] and SFB646 to B.C.).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.288555.116.
Freely available online through the Genes & Development Open Access option.
References
- Abbott AL, Alvarez-Saavedra E, Miska EA, Lau NC, Bartel DP, Horvitz HR, Ambros V. 2005. The let-7 microRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev Cell 9: 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Saavedra E, Horvitz HR. 2010. Many families of C. elegans microRNAs are not essential for development or viability. Curr Biol 20: 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. 2003. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113: 25–36. [DOI] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Lambie EJ, Bindu S, Mikeladze-Dvali T, Conradt B. 2015. Engulfment pathways promote programmed cell death by enhancing the unequal segregation of apoptotic potential. Nat Commun 6: 10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt B. 2009. Genetic control of programmed cell death during animal development. Annu Rev Genet 43: 493–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt B, Horvitz HR. 1998. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93: 519–529. [DOI] [PubMed] [Google Scholar]
- Conradt B, Horvitz HR. 1999. The TRA-1A sex determination protein of C. elegans regulates sexually dimorphic cell deaths by repressing the egl-1 cell death activator gene. Cell 98: 317–327. [DOI] [PubMed] [Google Scholar]
- Conradt B, Wu Y-C, Xue D. 2016. Programmed cell death during Caenorhabditis elegans development. Genetics 203: 1533–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lucas MP, Sáez AG, Lozano E. 2015. miR-58 family and TGF-β pathways regulate each other in Caenorhabditis elegans. Nucleic Acids Res 43: 9978–9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgley ML, Riddle DL. 2001. LG II balancer chromosomes in Caenorhabditis elegans: mT1(II;III) and the mIn1 set of dominantly and recessively marked inversions. Mol Genet Genomics 266: 385–395. [DOI] [PubMed] [Google Scholar]
- Ellis HM, Horvitz HR. 1986. Genetic control of programmed cell death in the nematode C. elegans. Cell 44: 817–829. [DOI] [PubMed] [Google Scholar]
- Frank CA, Hawkins NC, Guenther C, Horvitz HR, Garriga G. 2005. C. elegans HAM-1 positions the cleavage plane and regulates apoptosis in asymmetric neuroblast divisions. Dev Biol 284: 301–310. [DOI] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen S-P, Grunnet M, Jorgensen EM. 2008. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet 40: 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Davis MW, Ailion M. 2012. Improved Mos1-mediated transgenesis in C. elegans. Nat Methods 9: 117–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Davis MW, Sarov M, Taylor J, Flibotte S, LaBella M, Pozniakovsky A, Moerman DG, Jorgensen EM. 2014. Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon. Nat Methods 11: 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs Y, Steller H. 2011. Programmed cell death in animal development and disease. Cell 147: 742–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosswendt S, Filipchyk A, Manzano M, Klironomos F, Schilling M, Herzog M, Gottwein E, Rajewsky N. 2014. Unambiguous identification of miRNA:target site interactions by different types of ligation reactions. Mol Cell 54: 1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happo L, Strasser A, Cory S. 2012. BH3-only proteins in apoptosis at a glance. J Cell Sci 125: 1081–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzold J, Conradt B. 2008. Control of apoptosis by asymmetric cell division. PLoS Biol 6: e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock EM, Sulston JE, Thomson JN. 1983. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science 220: 1277–1279. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Ellis RE, Horvitz HR. 1992. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature 356: 494–499. [DOI] [PubMed] [Google Scholar]
- Hirose T, Horvitz HR. 2013. An Sp1 transcription factor coordinates caspase-dependent and -independent apoptotic pathways. Nature 500: 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose T, Galvin BD, Horvitz HR. 2010. Six and Eya promote apoptosis through direct transcriptional activation of the proapoptotic BH3-only gene egl-1 in Caenorhabditis elegans. Proc Natl Acad Sci 107: 15479–15484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz HR. 2003. Worms, life, and death (Nobel lecture). Chembiochem 4: 697–711. [DOI] [PubMed] [Google Scholar]
- Huntzinger E, Izaurralde E. 2011. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 12: 99–110. [DOI] [PubMed] [Google Scholar]
- Ichikawa N, Ando C, Fumino M. 2006. Caenorhabditis elegans MAI-1 protein, which is similar to mitochondrial ATPase inhibitor (IF1), can inhibit yeast F0F1-ATPase but cannot be transported to yeast mitochondria. J Bioenerg Biomembr 38: 93–99. [DOI] [PubMed] [Google Scholar]
- Isik M, Korswagen HC, Berezikov E. 2010. Expression patterns of intronic microRNAs in Caenorhabditis elegans. Silence 1: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Friedman RC, Ruby JG, Bartel DP. 2011. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 469: 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H-S, Wu Y-C. 2014. LIN-3/EGF promotes the programmed cell death of specific cells in Caenorhabditis elegans by transcriptional activation of the pro-apoptotic gene egl-1. PLoS Genet 10: e1004513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagias K, Pocock R. 2015. microRNA regulation of the embryonic hypoxic response in Caenorhabditis elegans. Sci Rep 5: 11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, de Lencastre A, Pincus Z, Slack FJ. 2009. Dynamic expression of small non-coding RNAs, including novel microRNAs and piRNAs/21U-RNAs, during Caenorhabditis elegans development. Genome Biol 10: R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole AJ, Swahari V, Hammond SM, Deshmukh M. 2011. miR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Genes Dev 25: 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambie EJ, Conradt B. 2016. Deadly dowry: how engulfment pathways promote cell killing. Cell Death Differ 23: 553–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858–862. [DOI] [PubMed] [Google Scholar]
- Lettre G, Hengartner MO. 2006. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol 7: 97–108. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. 1998. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491–501. [DOI] [PubMed] [Google Scholar]
- Li JJ, Huang H, Bickel PJ, Brenner SE. 2014. Comparison of D. melanogaster and C. elegans developmental stages, tissues, and cells by modENCODE RNA-seq data. Genome Res 24: 1086–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Strauss TJ, Potts MB, Cameron S. 2006. Direct regulation of egl-1 and of programmed cell death by the Hox protein MAB-5 and by CEH-20, a C. elegans homolog of Pbx1. Development 133: 641–650. [DOI] [PubMed] [Google Scholar]
- Liu M, Liu P, Zhang L, Cai Q, Gao G, Zhang W, Zhu Z, Liu D, Fan Q. 2011. mir-35 is involved in intestine cell G1/S transition and germ cell proliferation in C. elegans. Cell Res 21: 1605–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro MF, Pilgrim DB. 1995. Identification and cloning of unc-119, a gene expressed in the Caenorhabditis elegans nervous system. Genetics 141: 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massirer KB, Perez SG, Mondol V, Pasquinelli AE. 2012. The miR-35-41 family of microRNAs regulates RNAi sensitivity in Caenorhabditis elegans. PLoS Genet 8: e1002536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McJunkin K, Ambros V. 2014. The embryonic mir-35 family of microRNAs promotes multiple aspects of fecundity in Caenorhabditis elegans. G3 (Bethesda) 4: 1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzstein MM, Horvitz HR. 1999. The C. elegans cell death specification gene ces-1 encodes a snail family zinc finger protein. Mol Cell 4: 309–319. [DOI] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, Bartel DP, Ambros VR, Horvitz HR. 2007. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS Genet 3: e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694. [DOI] [PubMed] [Google Scholar]
- Nehme R, Conradt B. 2008. egl-1: a key activator of apoptotic cell death in C. elegans. Oncogene 27: S30–S40. [DOI] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058. [DOI] [PubMed] [Google Scholar]
- Pagano DJ, Kingston ER, Kim DH. 2015. Tissue expression pattern of PMK-2 p38 MAPK is established by the miR-58 family in C. elegans. PLoS Genet 11: e1004997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernaute B, Spruce T, Smith KM, Sánchez-Nieto JM, Manzanares M, Cobb B, Rodríguez TA. 2014. MicroRNAs control the apoptotic threshold in primed pluripotent stem cells through regulation of BIM. Genes Dev 28: 1873–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts MB, Wang DP, Cameron S. 2009. Trithorax, Hox, and TALE-class homeodomain proteins ensure cell survival through repression of the BH3-only gene egl-1. Dev Biol 329: 374–385. [DOI] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, Schindelin J, Tomancak P. 2010. Software for bead-based registration of selective plane illumination microscopy data. Nat Methods 7: 418–419. [DOI] [PubMed] [Google Scholar]
- Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. 2011. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med 208: 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. 2008. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5: 877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sætrom P, Heale BSE, Snøve O, Aagaard L, Alluin J, Rossi JJ. 2007. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res 35: 2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel R, Hutter H, Moerman D, Schnabel H. 1997. Assessing normal embryogenesis in Caenorhabditis elegans using a 4D microscope: variability of development and regional specification. Dev Biol 184: 234–265. [DOI] [PubMed] [Google Scholar]
- Schnabel R, Bischoff M, Hintze A, Schulz A-K, Hejnol A, Meinhardt H, Hutter H. 2006. Global cell sorting in the C. elegans embryo defines a new mechanism for pattern formation. Dev Biol 294: 418–431. [DOI] [PubMed] [Google Scholar]
- Shaham S, Reddien PW, Davies B, Horvitz HR. 1999. Mutational analysis of the Caenorhabditis elegans cell-death gene ced-3. Genetics 153: 1655–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sionov RV, Vlahopoulos SA, Granot Z. 2015. Regulation of Bim in health and disease. Oncotarget 6: 23058–23134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Maaskola J, Colombo T, Rahn H-P, Friedländer MR, Li N, Chen W, Piano F, Rajewsky N. 2009. Large-scale sorting of C. elegans embryos reveals the dynamics of small RNA expression. Nat Methods 6: 745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subasic D, Brümmer A, Wu Y, Pinto SM, Imig J, Keller M, Jovanovic M, Lightfoot HL, Nasso S, Goetze S, et al. 2015. Cooperative target mRNA destabilization and translation inhibition by miR-58 microRNA family in C. elegans. Genome Res 25: 1680–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto A, Kusano A, Hozak RR, Derry WB, Zhu J, Rothman JH. 2001. Many genomic regions are required for normal embryonic programmed cell death in Caenorhabditis elegans. Genetics 158: 237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR. 1977. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol 56: 110–156. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. 1983. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100: 64–119. [DOI] [PubMed] [Google Scholar]
- Thellmann M, Hatzold J, Conradt B. 2003. The Snail-like CES-1 protein of C. elegans can block the expression of the BH3-only cell-death activator gene egl-1 by antagonizing the function of bHLH proteins. Development 130: 4057–4071. [DOI] [PubMed] [Google Scholar]
- Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, et al. 2008. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132: 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chitturi J, Ge Q, Laskova V, Wang W, Li X, Ding M, Zhen M, Huang X. 2015. The C. elegans COE transcription factor UNC-3 activates lineage-specific apoptosis and affects neurite growth in the RID lineage. Development 142: 1447–1457. [DOI] [PubMed] [Google Scholar]
- Winn J, Carter M, Avery L, Cameron S. 2011. Hox and a newly identified E2F co-repress cell death in Caenorhabditis elegans. Genetics 188: 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E, Thivierge C, Flamand M, Mathonnet G, Vashisht AA, Wohlschlegel J, Fabian MR, Sonenberg N, Duchaine TF. 2010. Pervasive and cooperative deadenylation of 3′UTRs by embryonic microRNA families. Mol Cell 40: 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. 1996. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL. Cell 87: 619–628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.