

ABSTRACT
Objective: Long-term prognosis of hepatocellular carcinoma (HCC) remains poor owing to the lack of treatment options for advanced HCC. Cytokine-induced killer (CIK) cells are ex vivo expanded T lymphocytes expressing both NK– and T-cell markers. CIK cell therapy alone is insufficient for treating advanced HCC. Thus, this study aimed to determine whether treatment with CIK cells combined with valproic acid (VPA) could provide a synergistic effect to inhibit tumor growth in a mouse model of HCC. Methods: Upregulation of natural killer group 2D (NKG2D) ligands (retinoic acid early inducible 1 [RAE-1], mouse; major histocompatibility complex class I polypeptide-related sequence A [MIC-A], human) were evaluated by FACS. VPA concentrations that did not reduce tumor volume were calculated to avoid VPA cytotoxicity in a C3H mouse model of HCC. CIK cells were generated from mouse splenocytes using interferon gamma, a CD3 monoclonal antibody, and interleukin 2. The potential synergistic effect of CIK cells combined with VPA was evaluated in the mouse model and tissue pathology was investigated. Results: After 40 h of incubation with VPA, RAE-1 and MIC-A expression were increased in 4 HCC cell lines compared with that in control (2.3-fold in MH-134, 2.4-fold in Huh-7, 3.7-fold in SNU-761, and 6.5-fold in SNU-475). The maximal in vivo VPA dosage that showed no significant cytotoxicity compared with control was 10 mg/kg/day. CIK cells were well generated from C3H mouse splenocytes. After 7 d of treatment with CIK cells plus VPA, a synergistic effect was observed on relative tumor volume in the mouse model of HCC. While the relative tumor volume in untreated control mice increased to 11.25, that in the combination treatment group increased to only 5.20 (P = 0.047). Conclusions: The VPA-induced increase in NKG2D ligands expression significantly enhanced the effects of CIK cell therapy in a mouse model of HCC.
KEYWORDS: Cancer immunology, hepatocellular carcinoma, immunotherapy
Introduction
Hepatocellular carcinoma (HCC) is a common and fatal cancer, with increasing incidence worldwide.1,2 Even though many curative therapies have been developed, the long-term prognosis for patients with HCC remains poor because of its high recurrence rates.3 We recently determined that adjuvant immunotherapy using cytokine-induced killer (CIK) cells appeared to reduce the recurrence of HCC.4 Although adjuvant CIK cell-based immunotherapy is a promising method, it is not enough to treat primary HCC and block HCC recurrence.
CIK cells are ex vivo expanded T lymphocytes expressing both NK– and T-cell markers. CIK cell therapy alone is insufficient for treating primary and advanced HCC. CIK cells detect major histocompatibility complex class I polypeptide-related sequence A (MIC-A) and sequence B (MIC-B) through the natural killer group 2D (NKG2D).5 Adjuvant therapy with CIK cells offered better prognosis in patients with gastric cancer who overexpressed MIC-A than in those with low MIC-A-expressing tumors. Thus, MIC-A status is associated with outcome and may be a predictive factor in CIK cell therapy.6
Valproic acid (VPA) is reported to reduce tumor growth in vivo with several mechanisms. One of these mechanisms is an inhibition of histone deacetylase (HDAC) and this can modulate the biology of diverse tumor cells.7 In previous in vitro and in vivo studies, the antitumor effect of VPA alone was insufficient for many kinds of tumors, including HCC. However, a synergistic effect of VPA with other agents has been reported in some promising results.8-10 VPA upregulates MIC-A and MIC-B,11 and this upregulation on the tumor surface enhances CIK cells effects. Moreover, VPA has been safely used as an antiepileptic drug for decades, it is relatively inexpensive, and its adverse effects have been regarded as tolerable. In the present study, we determined whether treatment with CIK cells combined with VPA may produce a synergistic effect to inhibit the growth of HCC cells in mice.
Results
VPA increases NKG2D ligands expression in vitro
Fluorescence-activated cell sorting (FACS) was performed to confirm the molecular mechanism of VPA in vitro. The retinoic acid early inducible 1 (RAE-1) in MH-134 cells and MIC-A expression levels in 3 human HCC cell lines (Huh-7, SNU-761, and SNU-475) were quantified by calculating the Specific fluorescence indices (SFI). In all HCC cell lines, the expression of RAE-1 or MIC-A was markedly increased after VPA treatment. After the cells were incubated for 40 h with VPA, the expression of RAE-1 was increased 2.3-fold in MH-134 cells (control group: 130.3 ± 38.7; VPA exposed group: 301 ± 5.54, P = 0.050) and MIC-A was increased 2.4-fold in Huh-7 cells, 3.7-fold in SNU-761 cells, and 6.5-fold in SNU-475 cells compared with control (Fig. 1a, 1b). As a positive control for HDAC inhibition, we also evaluated whether vorinostat increases MIC-A expression in HCC cells. As shown in Fig. 1c, the expression of MIC-A was increased 3.4-fold by 1 μM vorinostat and 19.4-fold by 2 μM vorinostat treatment. These results are consistent with the assertion that inhibition of HDAC by VPA or vorinostat increased the expression of RAE-1 or MIC-A protein by accelerating the transcription of its messenger-RNA (mRNA). We additionally evaluated if VPA and/or vorinostat alters the expression levels of immunosuppressive molecules such as PD-L1 (programmed cell death ligand 1) and PD-L2. As shown in Fig. 2a, VPA and vorinostat decreased the transcription of PD-L1 (control: 1.00 ± 0.01; vorinostat: 0.95 ± 0.01, P = 0.047; VPA: 0.56 ± 0.03, P = 0.017) and PD-L2 (control: 1.04 ± 0.10; vorinostat: 0.71 ± 0.04, P = 0.005; VPA: 0.43 ± 0.01, P < 0.001). Protein expression levels were also decreased by VPA and vorinostat (Fig. 2b). Altogether, these results indicate that HDAC inhibitors may potentiate the efficacy of immunotherapies against HCC.
Figure 1.
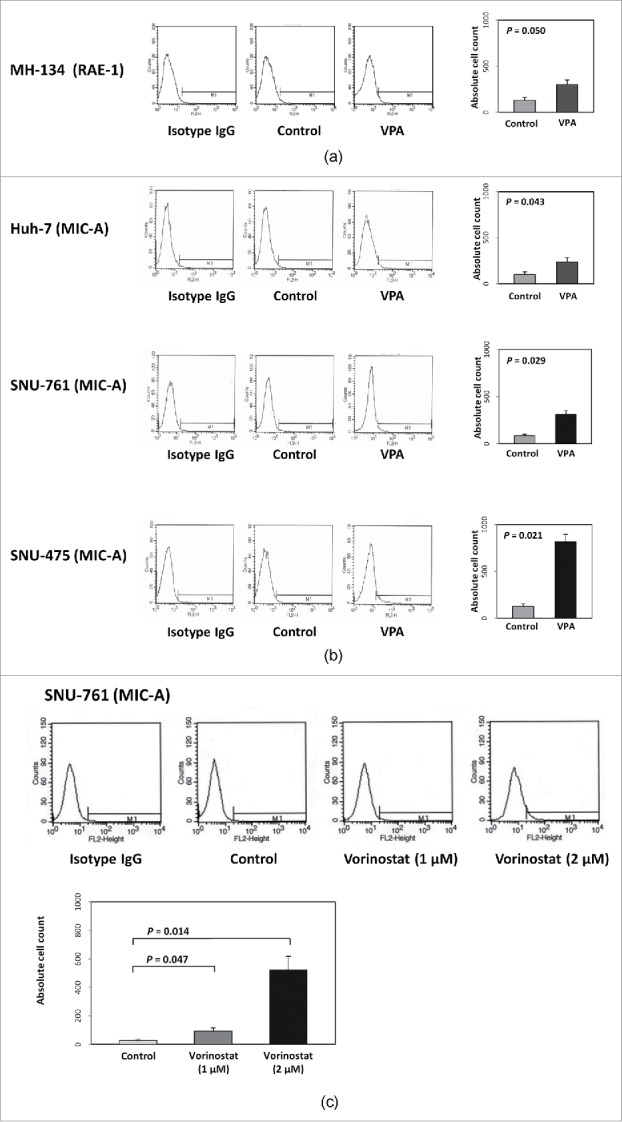
Effect of VPA or vorinostat on NKG2D ligand (RAE-1 or MIC-A) expression in 4 HCC cell lines. (a) MH-134 cells were evaluated for RAE-1 expression. After 40 hours of incubation with VPA (5 mM), the expression of RAE-1 was increased 2.3-fold in MH-134 cells. (b) Huh-7, SNU-475, and SNU-761 cell lines were evaluated for MIC-A expression. After 40 hours of incubation with VPA (5 mM), the expression of MIC-A was increased 2.4-fold in Huh-7, 3.7-fold in SNU-761, and 6.5-fold in SNU-475 cells compared with control. (c) SNU-761 cells were evaluated for MIC-A expression following vorinostat treatment. After 40 hours of incubation with vorinostat (1 μM and 2 μM), the expression of MIC-A was increased 3.4-fold by 1 μM and 19.4-fold by 2 μM, respectively.
Figure 2.
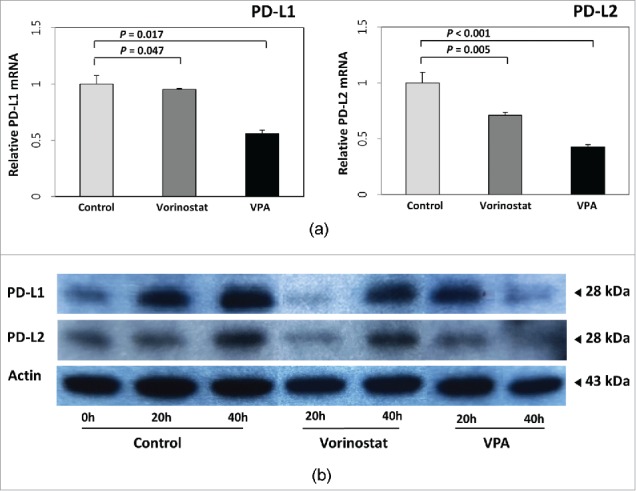
Effect of VPA and vorinostat on PD-L1 and PD-L2 expression in HCC cells. (a) The mRNA expressions of PD-L1 and PD-L2 in SNU-761 cells were quantified by real-time PCR and normalized to GAPDH expression levels. Both VPA (5 mM) and vorinostat (2 μM) decreased transcription of PD-L1 (control: 1.00 ± 0.01; vorinostat: 0.95 ± 0.01, P = 0.047; VPA: 0.56 ± 0.03, P = 0.017) and PD-L2 (control: 1.04 ± 0.10; vorinostat: 0.71 ± 0.04, P = 0.005; VPA: 0.43 ± 0.01, P < 0.001). The experiments were repeated 3 times. (b) Immunoblot analyses of PD-L1 and PD-L2 were also performed for SNU-761 cells treated with VPA (5 mM) or vorinostat (2 μM). Protein expression levels were also decreased following VPA or vorinostat treatment.
In vitro cytotoxicity assay
Since VPA and vorinostat increased MIC-A expression in HCC cells, we next evaluated whether VPA and vorinostat indeed enhance CIK cell cytotoxicity against HCC cells. Human CIK cells were obtained for this purpose, and in vitro cytotoxicity assays were performed. As shown in Fig. 3, VPA and vorinostat significantly increased CIK cell cytotoxicity against SNU-761 cells (control: 22.2 ± 7.6; VPA: 60.40 ± 10.16, P < 0.001; vorinostat: 56.54 ± 9.73, P < 0.001).
Figure 3.
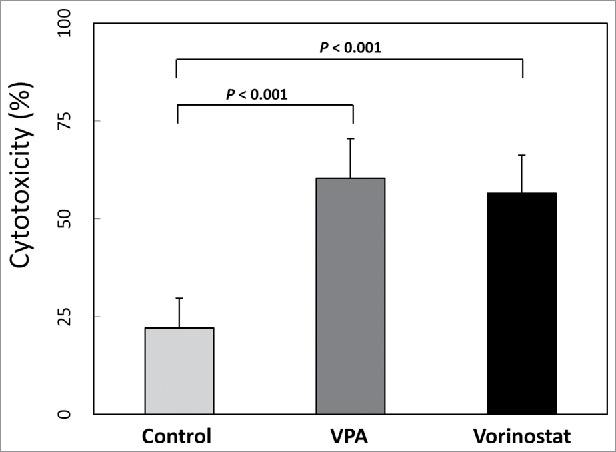
In vitro cytotoxicity assay. SNU-761 cells were incubated with VPA (5 mM) or vorinostat (2 μM) for 40 hours, and aliquots of 2 × 105 target cells/mL were prepared. Human CIK cells (effector cells) were obtained from mononuclear cells cultured with interleukin 2 and immobilized monoclonal antibody to CD3, and centrifuged cells were resuspended to 3×106 cells/mL in RPMI 1640 with FBS 1%. For the in vitro cytotoxicity assay, each well consisted of 50 μl of target cells and 100 μl of effector cells. After a 4-hour incubation period, treated cells were centrifuged, and the optical densities of supernatants were measured. Both VPA (5 mM) and vorinostat (2 μM) significantly increased CIK cell cytotoxicity against SNU-761 cells (control: 22.2 ± 7.6; VPA: 60.40 ± 10.16, P < 0.001; vorinostat: 56.54 ± 9.73, P < 0.001).
Determination of the appropriate VPA dose in vivo
We next determined the appropriate VPA dosage for examining effects that were synergistic but not cytotoxic when VPA treatment was combined with other agents in a mouse model of HCC. Based on previous studies, we estimated that dosage to be 10 or 30 mg/kg/day administered once daily for 7 d. After 7 d of VPA treatment, the changes in the tumor volume in the control group (saline) and in the group of mice receiving 10 mg/kg/day of VPA were not different (P = 0.100). However, the tumor volume was significantly reduced in the group receiving 30 mg/kg/day of VPA compared with that in the control group (P = 0.008). Therefore, the maximal VPA dose was determined to be 10 mg/kg/day because it did not have significant cytotoxicity (Fig. 4).
Figure 4.
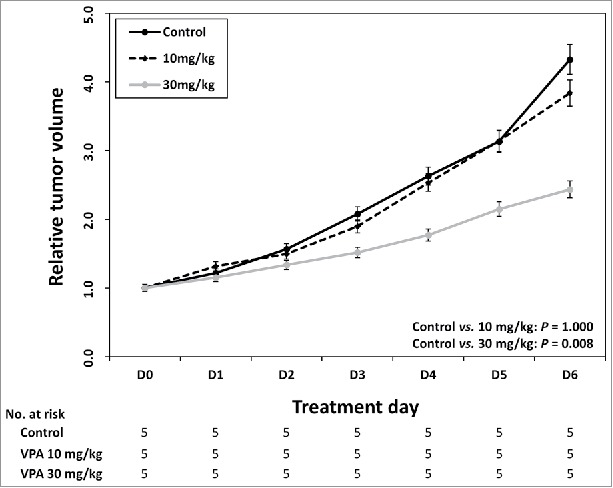
Identification of maximum VPA dose without cytotoxicity in vivo. After every 7 day of VPA application, control group and 10 mg/kg/day group were not different in the tumor volume change (P = 0.100). The tumor volume was significantly reduced in 30mg/kg/day VPA application group compared with control group (P = 0.008).
CIK cell culture
CIK cells were generated according to the protocol described in Materials and Methods section. Murine splenocytes obtained by splenectomy for manufacturing the CIK cells were collected from the spleens of 5-week-old C3H mice. Mononuclear cells derived from the spleen were separated and cultured for 14 d with interleukin 2 and a monoclonal antibody to CD3. During cell culture, the absolute cell count was increased and the proportions of cells that were positive for the T-cell marker CD3 (D0, 3370.3 ± 763.4; D7, 7826.7 ± 930.8; D14, 8868.0 ± 446.5; P = 0.050, D7 vs. D0; P = 0.050, D14 vs. D0) and the cytotoxic T-cell marker CD8α (D0, 1452.3 ± 474.5; D7, 2539.0 ± 631.2; D14, 3738.3 ± 1178.7; P = 0.127, D7 vs. D0; P = 0.127, D14 vs. D0) were also increased. The proportion of cells positive for the natural killer cell marker CD161 was maintained during the 2 weeks of culture (D0, 499.3 ± 174.0; D7, 409.7 ± 91.5; D14, 477.3 ± 152.4; P = 0.127, D7 vs. D0; P = 0.827, D14 vs. D0). These results suggest that CIK cells, which have a mixed natural killer cell- and cytotoxic T cell-like phenotype, proliferated well compared with isotype IgG control (Fig. 5).
Figure 5.
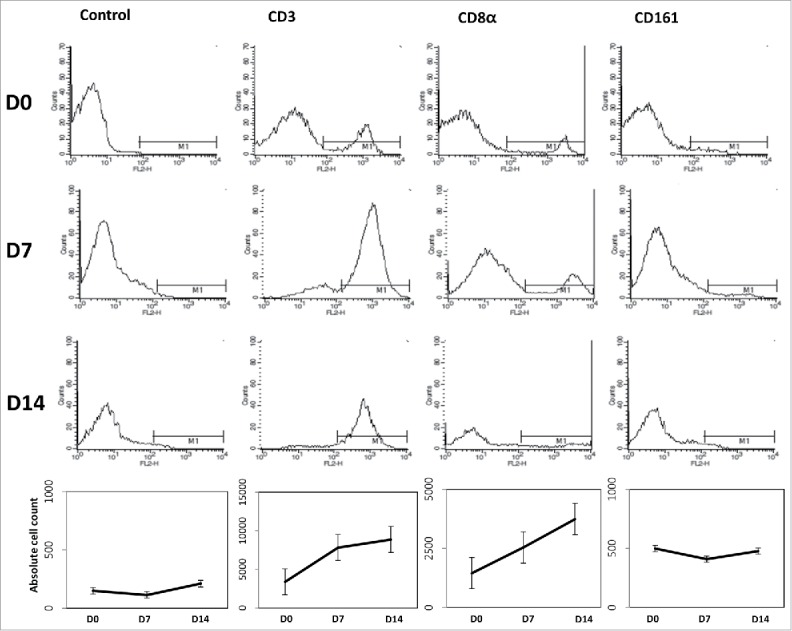
Generation of mouse CIK cells. Four spleens of 5-week-old C3H mice were acquired and incubated with interferon γ (100 U/ml) for 24 hours. After then, cell dishes were moved to CD3 monoclonal antibody coated dishes followed by interleukin 2 (500 U/ml). CD3, CD8α, and CD161 were checked to identify the CIK cell differentiation. CIK cells were proliferated well compared with isotype IgG control.
Synergistic effect of CIK with VPA
Compared with that in the control group, the relative tumor volume in the group treated with CIK cells alone was slightly but not significantly reduced (P = 0.597) and that in the group treated with VPA only was also unchanged (P = 0.754). This result confirms that this dose of VPA (10 mg/kg/day) did not have a cytotoxic effect. However, the combination therapy of CIK cells and VPA produced a significant synergistic effect. Following 7 d of combined CIK cells and VPA treatment, the relative tumor volume increased to 5.20 while that in the control group increased to 11.25 (P = 0.047; Fig. 6).
Figure 6.
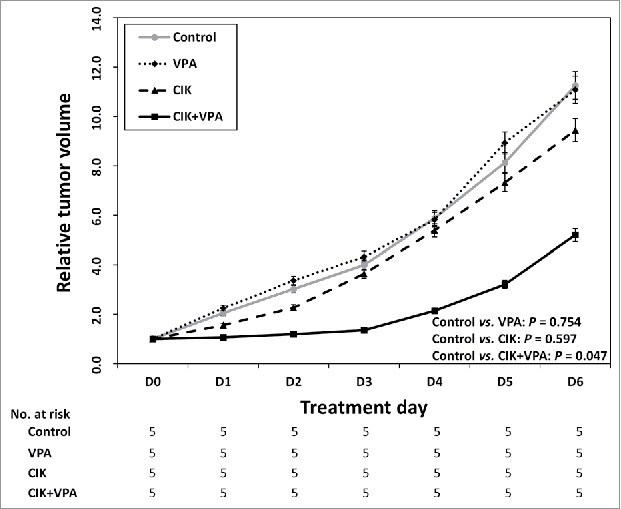
Synergistic effect of CIK with VPA in vivo. Relative tumor volume of VPA single or CIK single treatment group was not statistically different from that of control group (P = 0.754 and P = 0.597 respectively). The combination therapy of CIK cells + VPA produced a significant synergistic effect (P = 0.047). The relative tumor volume of combination therapy group increased to 5.20 while that in the control group increased to 11.25.
CIK cells infiltration and apoptosis in tumor tissue
CIK cell infiltrations were observed in tumor tissue of CIK cell treated mice and CIK cells combined with VPA treated mice. More CIK cell infiltrations and apoptosis were found in tumor tissue of combination treatment mice than that of CIK cell single treatment mice. Tumor tissue of VPA single treatment mice showed some apoptosis in TUNEL assay without lymphocyte infiltration in H and E stain. In TUNEL assay, ratio of stain positive cells to total stain cell are 24.31 ± 6.28% in control, 28.31 ± 2.30% in VPA treated mice, 28.77 ± 4.50% in CIK cell treated mice, and 38.30 ± 2.79% in combination treatment mice. Single treatment with VPA or CIK cells represented a mild increase of apoptosis in each tumor tissue (Fig. 7).
Figure 7.
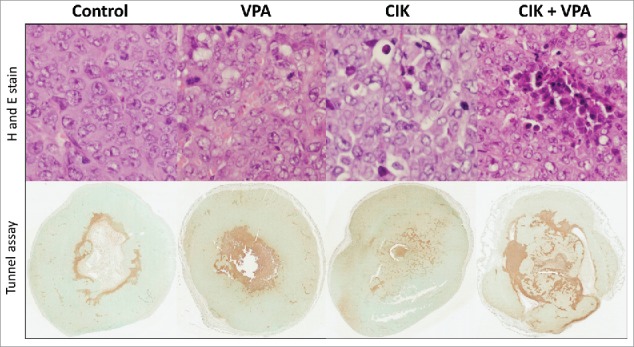
CIK cells infiltration and apoptosis in tumor tissue. More CIK cell infiltration and apoptosis were observed in CIK cells + VPA combination treatment group than CIK or VPA single treatment mice tissue. Apoptosis of each group was evaluated in TUNEL assay. Percent of positivity is 24.31 ± 6.28% in control, 28.31 ± 2.30% in VPA treated mice, 28.77 ± 4.50% in CIK cell treated mice, and 38.30 ± 2.79% in combination treatment mice.
Discussion
We attempted to prove the synergism of CIK cells and VPA in an HCC mouse model, and obtained results that supported our hypothesis. To determine whether VPA and/or vorinostat alters the expression levels of MIC-A, which is a stress-inducible surface glycoprotein for the NKG2D12 in HCC cells, we used FACS and found that the MIC-A expression level was increased in VPA- and vorinostat-treated compared with untreated HCC cell lines. We also found that VPA and vorinostat decreased the expression levels of PD-L1 and PD-L2 in HCC cells. As a next step, we investigated whether VPA treatment-induced expression of the NKG2D ligand on the surface of tumor cells enhances the therapeutic efficacy of CIK cells. In our study, the synergistic effect of CIK cells and VPA treatment was investigated in a subcutaneous HCC model. Treatment with either therapy alone was ineffective, whereas combination therapy with the same dosages of CIK cells and VPA showed significant cytotoxic effects. These results suggest that VPA has significant activity as a helper for CIK cells, and the molecular biology background for these molecules supports this suggestion.
Many previous studies have reported antitumor effects of CIK cells in diverse tumors. CIK cells show a potent MHC unrestricted tumor killing ability against various tumors, including HCC, leukemia, gastric, ovarian, renal, and gastric cancers, mainly through the use of the innate immune system.9,13-15 Those CIK cells are mixtures of T lymphocytes. Among them, the CD3+CD56+ subset is the main effectors against tumor cells. Unfortunately, this subset comprises less than 5% of fresh peripheral blood mononuclear cells, suggesting that the human immune system alone is insufficient to cope with tumor cells. However, this CD3+CD56+ subset of T lymphocytes can be expanded ex vivo through the use of molecular biology.5
CIK cells kill tumor cells by recognizing NKG2D ligands on the tumor cell surface.16 Although CIK cells show clear antitumor effects, these effects are insufficient for application in clinical treatment. A satisfactory antitumor effect of CIK cell therapy may be achieved once the ratio of CIK:tumor cells is greater than 30:1.13 However, this high level of CIK cells causes adverse effects. Thus, combinations with novel agents have been investigated in an attempt to reduce the CIK cell concentration to that which does not induce adverse toxicity. Moreover, owing to molecular biology development, the antitumor mechanisms of CIK cells are now better understood and indicate that CIK cell-based antitumor therapy is a promising novel strategy.
VPA has been used as an antipsychotic agent and in the treatment of epilepsy, bipolar disorder, migraine, and other disorders. Even though VPA has been widely used for decades and its antitumor effects have been gradually identified, its diverse antitumor mechanisms have not yet been completely elucidated. However, the HDAC inhibitory mechanism of VPA is relatively well known.8-10 HDAC is reported to be aberrantly expressed in diverse cancers. HDAC inhibition can increase the expression of several genes by accelerating their transcription, which can enhance the innate immune system and lead to suppression of tumor growth.9,17
VPA alters the expression of various genes involved in the interaction with the cellular immune system, which is known as NKG2D ligand in tumor cells. The observed NKG2D-mediated activation of cytotoxic lymphocytes critically relies on NKG2D ligand expression levels at the tumor cell surface.18 Among NKG2D ligands, MIC-A and MIC-B are constitutively expressed at low levels compared with transcript levels of a housekeeping gene in the HCC cell line. Treatment with VPA increases MIC-A and MIC-B mRNA without significantly altering the signal for the housekeeping gene.19 These results suggest that VPA as an HDAC inhibitor may induce HCC cell death through cytotoxic NK cells by increasing NKG2D ligand expression. The mechanism of tumor immune-escape is achieved by degradation of NKG2D ligand on the tumor cell surface,20 reducing the host's innate immunity against tumor cells. As already mentioned, VPA increases mRNA levels of NKG2D ligand, which can activate the innate immune system through cytotoxic NK cells.19 This cytotoxicity through the innate immune system has been shown to be critically dependent on the NKG2D/NKG2D ligands interaction, because it is blocked by addition of anti-NKG2D.20 NKG2D ligands make tumor cells susceptible to the innate immune system. Like cytotoxic NK cells, CIK cells can enhance antitumor effect under NKG2D ligand overexpression condition which can be induced by VPA in innate immune system.
Although CIK cell therapy has shown efficacy as an adjuvant therapy for HCC recurrence after curative treatment, it has limited applicability in primary HCC therapy. Here, we showed that the antitumor effect of CIK cells could be enhanced by co-treatment with VPA. These results suggest that combination therapy of CIK cells and VPA may be applied in primary HCC or more severe conditions, such as advanced stage HCC or metastatic HCC. The adverse effects of CIK cell therapy can be decreased by using a lower dosage in conjunction with treatment of VPA, which is readily available, inexpensive, and tolerable.
In conclusion, this study determined that a VPA-induced increase in NKG2D ligand expression enhanced the effects of CIK cell therapy in the treatment of HCC. We also determined the appropriate VPA concentration for enhancing MIC-A expression without causing cytotoxicity for use in future studies using this mouse model of HCC.
Financial Support: Funding: This study was supported by grants from Ministry for Trade, Industry, and Energy, Republic of Korea and from the Liver Research Foundation of Korea.
Materials and methods
Chemicals
VPA and vorinostat were purchased from Sigma-Aldrich Co., LLC. (Seoul, South Korea) and were dissolved in distilled water before each experiment. All other chemicals and reagents were from standard commercial sources and of the highest purity available.
Cell lines, cell culture, and reagents
The following 4 HCC cell lines were used in this study: MH-134 (a mouse HCC cell line), Huh-7 (a well-differentiated HCC cell line), SNU-475 (a poorly differentiated HCC cell line), and SNU-761 (a poorly differentiated HCC cell line). MH-134, SNU-475, and SNU-761 cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 100,000 U/L penicillin and 100 mg/L streptomycin.21 Huh-7 cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS, 100,000 U/L penicillin, and 100 mg/L streptomycin, with or without 100 nM insulin. In all experiments performed in this study, cells were serum-starved overnight before the experiments to avoid the effects of serum-induced signaling. The cells were incubated under standard culture conditions (20% O2 and 5% CO2, at 37°C).
Estimation of RAE-1 or MIC-A expression in HCC cell lines treated with VPA in vitro
MH-134, Huh-7, SNU-475, and SNU-761 cell lines were detached from culture dishes using 0.25% Trypsin/EDTA and incubated with VPA (5 mM). Cell line was harvested 40 hours after drug treatment, as well as at 8 hours intervals after the medium was replaced with a drug-free medium. After being incubated with a specific monoclonal antibody for 30 min at 4°C, the cells were washed 3 times with cold phosphate-buffered saline (PBS). Finally, FACS was conducted and the results analyzed. SFI of RAE-1 labeling in MH-134 cells and MIC-A labeling in Huh-7, SNU-475, and SNU-761 cells were calculated by dividing the median fluorescence level that was obtained with the specific monoclonal antibody by the corresponding isotype control.
Real-time PCR analysis
The mRNA expression of PD-L1 and PD-L2 were evaluated by a real-time reverse-transcriptase polymerase chain reaction (PCR) assay. Total RNA was extracted by Trizol Reagent (Invitrogen; Carlsbad, CA, USA). cDNA (cDNA) templates were synthesized using oligo-dT random primers and Moloney murine leukemia virus reverse transcriptase. The primer sequences were as follows: PD-L1, forward: 5′-aaatggaacctggcgaaagc;-3′ and reverse: 5′-gatgagcccctcaggcattt-3′; PD-L2, forward: 5′- gtcttgggagccagggtgac;-3′ and reverse: 5′- tgaaaagtgcaaatggcaagc;-3′.22 After the reverse transcription reaction, the cDNA template was amplified by Taq polymerase (Invitrogen; Carlsbad, CA, USA). Relative quantification was calculated using the 2−ΔΔCT method, with GAPDH as the internal control. All PCR experiments were performed in triplicate.
Immunoblot analysis
Antibodies against PD-L1 and PD-L2 were purchased from Invitrogen (Carlsbad, CA, USA). Cells were lysed for 20 min on ice in lysis buffer (50 mM Tris-HCl, pH 7.4; 1% Nonidet P-40, 0.25% sodium deoxycholate; 150 mM NaCl; 1 mmol/L EDTA; 1 mM phenylmethylsulfonyl-fluoride; 1 mM Na3VO4; 1 mM NaF; and 1 μg/mL each of aprotinin, pepstatin, and leupeptin) and then centrifuged at 14,000 g for 15 min at 4°C. Samples were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, blotted with appropriate primary antibodies, and incubated with peroxidase-conjugated secondary antibodies (Biosource International; Camarillo, CA, USA). Bound antibodies were visualized by a chemiluminescent substrate (ECL; Amersham, Arlington Heights, IL, USA) and exposed to Kodak X-OMAT film.
In vitro cytotoxicity assay
SNU-761 cells were incubated with VPA (5 mM) or vorinostat (2 μM) for 40 hours. The cells were then mixed with washing media (RPMI 1640 with FBS 1%), centrifuged (300 g, 4 minutes), and the cell pellet was resuspended at concentrations of 2 × 105 cell/mL in RPMI 1640 with FBS 1%. Following IRB approval (Seoul National University Hospital IRB No. 1605–143–765) and informed consents, human CIK cells were obtained from human mononuclear cells cultured at 37°C for 12–21 d with interleukin-2 and immobilized monoclonal antibody to CD3, according to a modified protocol.23,24 Washing medium was added to separated CIK cells (effecter cells) to a final concentration of 3 × 106 cells/mL. Each well consisted of 50 μL of target cells and 100 μL of CIK cells. After a 4-hour incubation period, treated cells were centrifuged (300 g, 4 minutes), and 75 μL of supernatant were added to wells of a Black plate (SPL Life Sciences, Cat No. 31496). Optical density was estimated at 485 ± 9 nm.
Identification of maximum VPA dose without cytotoxicity in vivo
Animal experiments were conducted to determine the maximum dose of VPA that did not affect HCC cell growth. MH-134 cells were cultured for 2 weeks to a density of 2.5 × 106/mL. MH-134 cells (2.5 × 106/mL in 100 µL of RPMI-1640) were subcutaneously injected into 15 male C3H mice (6 weeks old). In previous studies, 10 mg/kg and 30 mg/kg were selected as minimum VPA doses.25,26 VPA treatment was started when tumors had grown to a palpable size (0.2 cm3). VPA was dissolved in distilled water and injected into the peritoneum of 10 mice once daily at a dose of 10 (n = 5) or 30 (n = 5) mg/kg/day for 7 d. The control group was injected with solvent (n = 5). The tumor size was measured with calipers. Tumor volumes were calculated by standard formula: π/6 × (length) × (width2).27 Relative tumor volume (relative to the volume of first treatment day) was also calculated. Body weights were recorded continuously to determine the maximum dose of VPA that did not inhibit the growth of the HCC cells.
Generation of mouse CIK cells
We acquired 4 spleens from 5-week-old C3H mice and maintained the splenocytes in RPMI at a density of 5 × 106 cells/ml. These cells were incubated with interferon gamma (1000 U/mL) for 24 h. After 24 h, the cells were transferred to CD3 monoclonal antibody-coated dishes and exposed to interleukin 2 (750 U/mL). When the cell density reached over 2 × 106 cells/mL, the cells were split and transferred to new CD3-free culture dishes with additional interleukin 2 exposure (750 U/mL). To identify the differentiation to CIK cells, flow cytometry was performed using CD3, CD8, CD161 markers (Fig. 8).
Figure 8.
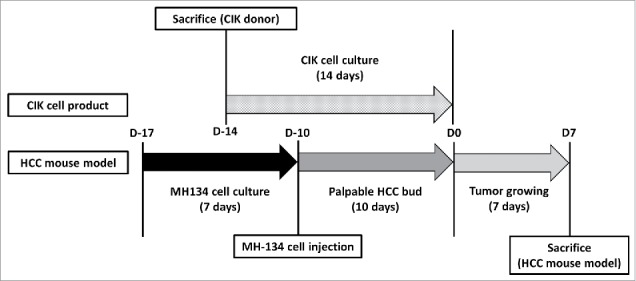
Schedule of CIK cells differentiation and a HCC mouse model generation. Schedule of CIK cells differentiation and a HCC mouse model generation. MH-134 cells were cultured for 7 d. 4 d before MH-134 cell injection to C3H mice, splenocytes were obtained by sacrificing other 5-week-old C3H mice. With these splenocytes, CIK cells were cultured for 14 d. Interventions (control, VPA, CIK cells, CIK cells + VPA) were performed at D0. After then, each tumor volume was observed for 7 d. All mice were killed at D7 to acquire their tumors for analysis.
Synergistic effect of CIK and VPA in vivo
MH-134 cells (2.5 × 106/mL in 100 µL of RPMI-1640) were injected subcutaneously into the back side of 6-week-old C3H mice (n = 20). Once the implanted MH-134 tumor bud grew to 0.2 cm3 in more than 80% of mice, the mice were divided into the following 4 treatment groups with 5 mice each group: control, VPA, CIK cells, CIK cells + VPA. The mice in the VPA group were injected with 10 mg/kg of VPA intraperitoneally once daily between D0 and D6 for 7 d. The mice in the CIK cell group were injected once at D0 with 1 × 107 CIK cells around the tumor bud. The mice in the CIK cell + VPA group were treated with both the VPA and CIK cell injections as described above. All mice were killed on day 7 to acquire their tumors for analysis at D7 (Fig. 8). Tumor volumes and relative tumor volumes were calculated by standard formula: π/6 × (length) × (width2).27 The pathologic analysis was performed by using Aperio image analysis (Aperio Technologies, Inc., Vista, CA, USA). Staining positivity was defined as strong positive and positive results in Aperio image analysis.
Statistical analysis
Results from at least 3 independent in vitro experiments and at least 5 mice in vivo experiments are expressed as the mean ± SEM and analyzed using Spearman's rank order correlation analysis, Mann Whitney U-test, and student T test. P values <0.05 are considered statistically significant.
Ethics statement
All animal experiment protocols were approved by the Seoul National University Institutional Animal Care and Use Committee (approval number, 12–2014–00–22).
Disclosure of potential conflicts of interest
These authors disclose the following: Su Jong Yu has received a lecture fee from Bayer HealthCare Pharmaceuticals; Yoon Jun Kim has received research grants from Bristol-Myers Squibb, Roche, JW Creagene, Bukwang Pharmaceuticals, Handok Pharmaceuticals, Hanmi Pharmaceuticals, Yuhan Pharmaceuticals, and Pharmaking, and has received lecture fees from Bayer HealthCare Pharmaceuticals, Gilead Science, MSD Korea, Yuhan Pharmaceuticals, Samil Pharmaceuticals, CJ Pharmaceuticals, Bukwang Pharmaceuticals, and Handok Pharmaceuticals; and Jung-Hwan Yoon has received a research grant from Bayer HealthCare Pharmaceuticals. The remaining authors disclose no conflicts.
Funding
This study was supported by grants from Seoul National University Hospital Research Fund (03–2016–0380) and from the Liver Research Foundation of Korea (Bio Future Strategies Research Project).
References
- 1.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012; 142:1264-73.e1; PMID:22537432; http://dx.doi.org/ 10.1053/j.gastro.2011.12.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El-Serag HB, Kanwal F. Epidemiology of hepatocellular carcinoma in the United States: where are we? Where do we go? Hepatology 2014; 60:1767-75; PMID:24839253; http://dx.doi.org/ 10.1002/hep.27222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen W-T, Chau G-Y, Lui W-Y, Tsay S-H, King K-L, Loong C-C, Wu CW. Recurrent hepatocellular carcinoma after hepatic resection: prognostic factors and long-term outcome. Eur J Surg Oncol (EJSO) 2004; 30:414-20; PMID:15063895; http://dx.doi.org/ 10.1016/j.ejso.2004.01.013 [DOI] [PubMed] [Google Scholar]
- 4.Lee JH, Lee J-H, Lim Y-S, Yeon JE, Song T-J, Yu SJ, Gwak GY, Kim KM, Kim YJ, Lee JW, et al.. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015; 148:1383-91. e6; PMID:25747273; http://dx.doi.org/ 10.1053/j.gastro.2015.02.055 [DOI] [PubMed] [Google Scholar]
- 5.Sangiolo D. Cytokine induced killer cells as promising immunotherapy for solid tumors. J Cancer 2011; 2:363-8; PMID:21716717; http://dx.doi.org/ 10.7150/jca.2.363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Lin J, Guo Z-Q, Lin W-S, Zhou Z-F, Huang C-Z, Chen Q, Ye YB. MHC I-related chain a expression in gastric carcinoma and the efficacy of immunotherapy with cytokine-induced killer cells. Am J Cancer Res 2015; 5:3221; PMID:26693072 [PMC free article] [PubMed] [Google Scholar]
- 7.Blaheta RA, Michaelis M, Driever PH, Cinatl J. Evolving anticancer drug valproic acid: insights into the mechanism and clinical studies. Med Res Rev 2005; 25:383-97; PMID:15637697; http://dx.doi.org/ 10.1002/med.20027 [DOI] [PubMed] [Google Scholar]
- 8.Li J, Bonifati S, Hristov G, Marttila T, Valmary‐Degano S, Stanzel S, Schnölzer M, Mougin C, Aprahamian M, Grekova SP, et al.. Synergistic combination of valproic acid and oncolytic parvovirus H‐1PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol Med 2013; 5:1537-55; PMID:24092664; http://dx.doi.org/ 10.1002/emmm.201302796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Zhu Y, He H, Lou L, Ye W, Chen Y, Wang J. Synergistically killing activity of aspirin and histone deacetylase inhibitor valproic acid (VPA) on hepatocellular cancer cells. Biochem Biophys Res Commun 2013; 436:259-64; PMID:23726914; http://dx.doi.org/ 10.1016/j.bbrc.2013.05.088 [DOI] [PubMed] [Google Scholar]
- 10.Shirsath N, Rathos M, Chaudhari U, Sivaramakrishnan H, Joshi K. Potentiation of anticancer effect of valproic acid, an antiepileptic agent with histone deacetylase inhibitory activity, by the cyclin-dependent kinase inhibitor P276-00 in human non-small-cell lung cancer cell lines. Lung Cancer 2013; 82:214-21; PMID:24051085; http://dx.doi.org/ 10.1016/j.lungcan.2013.08.010 [DOI] [PubMed] [Google Scholar]
- 11.Wu X, Tao Y, Hou J, Meng X, Shi J. Valproic acid upregulates NKG2D ligand expression through an ERK-dependent mechanism and potentially enhances NK cell-mediated lysis of myeloma. Neoplasia 2012; 14:1178-89; PMID:23308050; http://dx.doi.org/ 10.1593/neo.121236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999; 285:727-9; PMID:10426993; http://dx.doi.org/ 10.1126/science.285.5428.727 [DOI] [PubMed] [Google Scholar]
- 13.Kim HM, Lim J, Park S-K, Kang JS, Lee K, Lee CW, Lee KH, Yun MJ, Yang KH, Han G, et al.. Antitumor activity of cytokine-induced killer cells against human lung cancer. Int Immunopharmacol 2007; 7:1802-7; PMID:17996691; http://dx.doi.org/ 10.1016/j.intimp.2007.08.016 [DOI] [PubMed] [Google Scholar]
- 14.Rettinger E, S KuçI, Naumann I, Becker P, Kreyenberg H, Anzaghe M, Willasch A, Koehl U, Bug G, Ruthardt M, et al.. The cytotoxic potential of interleukin-15-stimulated cytokine-induced killer cells against leukemia cells. Cytotherapy 2012; 14:91-103; PMID:21973023; http://dx.doi.org/ 10.3109/14653249.2011.613931 [DOI] [PubMed] [Google Scholar]
- 15.Kim HM, Kang JS, Lim J, Park S-K, Lee K, Yoon YD, Lee CW, Lee KH, Han G, Yang KH, et al.. Inhibition of human ovarian tumor growth by cytokine-induced killer cells. Arch Pharm Res 2007; 30:1464-70; PMID:18087816; http://dx.doi.org/ 10.1007/bf02977372 [DOI] [PubMed] [Google Scholar]
- 16.Kim YJ, Lim J, Kang JS, Kim HM, Lee HK, Ryu HS, Kim JY, Hong JT, Kim Y, Han SB. Adoptive immunotherapy of human gastric cancer with ex vivo expanded T cells. Arch Pharm Res 2010; 33:1789-95; PMID:21116782; http://dx.doi.org/ 10.1007/s12272-010-1111-7 [DOI] [PubMed] [Google Scholar]
- 17.Tsai C, Leslie JS, Franko-Tobin LG, Prasnal MC, Yang T, Mackey LV, Fuselier JA, Coy DH, Liu M, Yu C, et al.. Valproic acid suppresses cervical cancer tumor progression possibly via activating Notch1 signaling and enhances receptor-targeted cancer chemotherapeutic via activating somatostatin receptor type II. Arch Gynecol Obstet 2013; 288:393-400; PMID: 23435724; http://dx.doi.org/ 10.1007/s00404-013-2762-7 [DOI] [PubMed] [Google Scholar]
- 18.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001; 413:165-71; PMID:11557981; http://dx.doi.org/ 10.1038/35093109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, et al.. Natural killer cell–mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res 2005; 65:6321-9; PMID:16024634; http://dx.doi.org/ 10.1158/0008-5472.can-04-4252 [DOI] [PubMed] [Google Scholar]
- 20.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002; 419:734-8; PMID:12384702; http://dx.doi.org/ 10.1038/nature01112 [DOI] [PubMed] [Google Scholar]
- 21.Park JG, Lee JH, Kang MS, Park KJ, Jeon YM, Lee HJ, Kwon HS, Park HS, Yeo KS, Lee KU, et al.. Characterization of cell lines established from human hepatocellular carcinoma. Int J Cancer 1995; 62:276-82; PMID:7543080 [DOI] [PubMed] [Google Scholar]
- 22.Hassan SS, Akram M, King EC, Dockrell HM, Cliff JM. PD-1, PD-L1 and PD-L2 gene expression on T-cells and natural killer cells declines in conjunction with a reduction in PD-1 protein during the intensive phase of tuberculosis treatment. PloS One 2015; 10:e0137646; PMID:26359860 ; http://dx.doi.org/ 10.1371/journal.pone.0137646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ochoa A, Gromo G, Alter B, Sondel P, Bach F. Long-term growth of lymphokine-activated killer (LAK) cells: role of anti-CD3, beta-IL 1, interferon-gamma and-beta. J Immunol 1987; 138:2728-33; PMID:2435804 [PubMed] [Google Scholar]
- 24.Anderson PM, Bach FH, Ochoa AC. Augmentation of cell number and LAK activity in peripheral blood mononuclear cells activated with anti-CD3 and interleukin-2. Cancer Immunol, Immunother 1988; 27:82-8; PMID:3260824; http://dx.doi.org/23372654 10.1007/bf00205763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juengel E, Makarević J, Tsaur I, Bartsch G, Nelson K, Haferkamp A, Blaheta RA. Resistance after chronic application of the HDAC-inhibitor valproic acid is associated with elevated Akt activation in renal cell carcinoma in vivo. PloS One 2013; 8:e53100; PMID:23372654; 10.1371/journal.pone.0053100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackenzie GG, Huang L, Alston N, Ouyang N, Vrankova K, Mattheolabakis G, Constantinides PP, Rigas B. Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. PLoS One 2013; 8:e61532; PMID:23650499; http://dx.doi.org/ 10.1371/journal.pone.0061532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou H, Lariviere JP, Demidenko E, Gladstone D, Swartz H, Khan N. Repeated tumor pO 2 measurements by multi-site EPR oximetry as a prognostic marker for enhanced therapeutic efficacy of fractionated radiotherapy. Radiother Oncol 2009; 91:126-31; PMID:19013657; http://dx.doi.org/ 10.1016/j.radonc.2008.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]