

Abstract
Inversion of chromosome 16 (inv(16)) generates the CBFβ-SMMHC fusion protein and is found in nearly all patients with acute myeloid leukemia subtype M4 with Eosinophilia (M4Eo). Expression of CBFβ-SMMHC is causative for leukemia development, but the molecular mechanisms underlying its activity are unclear. Recently, there have been important advances in defining the role of CBFβ-SMMHC and its binding partners, the transcription factor RUNX1 and the histone deacetylase HDAC8. Importantly, initial trials demonstrate that small molecules targeting these binding partners are effective against CBFβ-SMMHC induced leukemia. This review will discuss recent advances in defining the mechanism of CBFβ-SMMHC activity, as well as efforts to develop new therapies for inv(16) AML.
Keywords: inv(16), CBFβ-SMMHC, CBFβ, RUNX1, AML
INTRODUCTION
Acute myeloid leukemia (AML) is often classified by the presence of specific, recurrent chromosomal abnormalities that generate characteristic fusion genes [1-3]. Expression of these fusion genes is likely the initiating event, although additional cooperating mutations are required for transformation to a frank leukemia. While the profile of cooperating mutations may vary in different leukemia sub-clones, the specific fusion protein involved is likely present in all leukemia cells of a given patient, making them attractive therapeutic targets [1, 4, 5]. However, many leukemia fusion proteins involve transcriptional regulators, which have historically been difficult to target. Recently, multiple inhibitors that target fusion proteins have been described, including 3 different drugs targeting the fusion protein expressed in inversion 16 AML [6-10].
Inversion of chromosome 16, inv(16)(p13q22) (inv(16)) and the related, but less common translocation t(16;16)(p13;q22), are associated with nearly all cases of French-American-British (FAB) classified AML subtype M4 with Eosinophilia (M4Eo) [11, 12]. Both of these rearrangements generate a fusion between the gene for Core Binding Factor β (CBFB) and the MYH11 gene, which encodes Smooth Muscle Myosin Heavy Chain (SMMHC) (Figure 1) Expression of CBFB-MYH11 is thought to be the initiating event in inv(16) AML [13, 14].
Figure 1. Schematic representation of the CBFβ-SMMHC fusion protein.
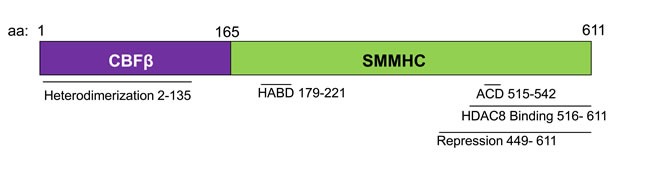
Diagram representing the indicated domains of the fusion protein, and the associated amino acid (aa) numbers. HABD: High Affinity Binding Domain. ACD: Assembly Competence Domain.
Inv(16) and t(16;16) also generate the reciprocal MYH11-CBFB fusion gene. However, this region is lost in some M4Eo AML patients, with no discernable clinical effect. Consequently, the MYH11-CBFB fusion is thought to be dispensable for leukemia development [15]. This is in contrast to other reciprocal chromosomal rearrangements, such as t15;17, which generates the PML-RARα and RARα-PML fusions, and t(4;11)(q21;q23), which generates the MLL-AF4 and AF4-MLL fusions. Both products of these chromosomal rearrangements are known to contribute to leukemogenesis [16-19].
Studies in mice indicate that expression of CBFB-MYH11, which encodes the protein CBFβ-SMMHC, is necessary, but not sufficient for AML development [20, 21]. CBFβ-SMMHC alone causes defects in hematopoietic differentiation, but additional cooperating mutations are required for transformation to frank leukemia [22-28].
Inv(16) is found in 8-10% of pediatric and adult AML cases [2, 29, 30]. Inv(16) is considered a good prognosis AML due to the high rate of response to current treatments [3, 31]. Nearly 90% of inv(16) AML patients achieve clinical remission under current chemotherapy protocols, although approximately half will eventually relapse [32-35]. In addition, current treatments are associated with significant morbidity and mortality [36-38].
CBFβ-SMMHC AND RUNX1
CBFβ is a member of the Core Binding Factor (CBF) family of transcription factors, and heterodimerizes with the DNA binding α subunits, RUNX1, RUNX2, and RUNX3 [39, 40]. CBFβ does not directly bind DNA, but stabilizes the interaction between the α subunits and DNA, and protects them from degradation [39, 41, 42].
RUNX1 is a key transcription factor during hematopoiesis. During embryogenesis, RUNX1 is required for the generation of hematopoietic stem cells (HSCs) from the hemogenic epithelium and for the proper differentiation of primitive erythrocytes [43-46]. During adult hematopoiesis, RUNX1 regulates HSC self-renewal and survival, as well as the differentiation of megakaryocytes, T-, and B-cells [47-51].
RUNX1 is also associated with a variety of hematological malignancies [52-58]. Point mutations and deletions of RUNX1 are frequently found in patients with myelodysplastic syndrome (MDS), chronic myelomonocytic leukemia (CMML) and FAB M0 AML [59-62]. Heterozygous germline mutations in RUNX1 cause Familial Platelet disorder with a predisposition to AML (FPD-AML) [63, 64]. Translocations involving RUNX1, t(8;21)(q22;q22) and t(12;21)(p13;q22), are associated with M2 AML and acute lymphoblastic leukemia (ALL), respectively [65-69].
Dominant negative model of CBFβ-SMMHC activity
CBFβ-SMMHC retains the ability to bind RUNX1 through the N-terminal half of the fusion protein (Figure 1) [42, 70]. In addition, there is a high-affinity binding domain (HABD) in the SMMHC tail. This allows the fusion protein to bind RUNX1 at two sites and outcompete wildtype CBFβ for RUNX1 binding [71]. Because of RUNX1′s established role in hematopoiesis and leukemogenesis, it has been proposed that CBFβ-SMMHC acts as a dominant repressor of RUNX1 [71-73].
Early studies in mice demonstrate that CBFβ-SMMHC dominantly represses RUNX1 in vivo. Knockin mice with a single copy of the fusion gene expressed from the endogenous Cbfb promoter (Cbfb+/MYH11) have a phenotype remarkably similar to mice homozygous for null alleles of Runx1 (Runx1−/−) or Cbfb (Cbfb−/−) [43, 44, 74-77]. Embryos of all three of these genotypes have a complete block in definitive hematopoiesis, central nervous system hemorrhaging, and embryonic lethality between embryonic day 12.5 and 13.5 (e12.5- e13.5). These findings indicate that CBFβ-SMMHC represses RUNX1 activity, consistent with a dominant negative model of CBFβ-SMMHC activity [43, 44, 74-77]. However, more recent work has demonstrated that the fusion protein has additional activities, as well.
CBFβ-SMMHC and RUNX1 independent activities
During primitive hematopoiesis, the initial wave of embryonic blood development that generates primarily nucleated erythrocytes, Cbfb+/MYH11 embryos have differentiation defects that are not seen in either Runx1−/− or Cbfb−/− embryos [74, 78]. At e10.5, Runx1−/− embryos have a subtle differentiation defect, resulting in a small population of circulating immature erythrocytes [46]. Cbfb+/MYH11 embryos, have a significantly more severe differentiation defect, with cells arrested at an earlier stage of differentiation and a larger population of circulating immature cells [78]. This indicates that Cbfb-MYH11 has RUNX1 repression-independent activities during primitive hematopoiesis.
Cbfb+/MYH11 embryos also have changes in gene expression that are not observed in Cbfb−/− mice. Microarray analysis of peripheral blood from Cbfb+/MYH11 embryos identified deregulated expression of 658 genes, while the same analysis of Cbfb−/− embryos identified only 174 differentially expressed genes, with only 71 genes deregulated in both Cbfb+/MYH11 and Cbfb−/− embryos [78]. The majority of the genes deregulated in both Cbfb+/MYH11 and Cbfb−/− embryos showed increased expression (95% and 77%, respectively). Importantly, many of the genes that showed deregulated expression uniquely in Cbfb+/MYH11 embryos are also expressed in inv(16) patient samples [78]. This indicates that CBFβ-SMMHC has effects on gene expression that are not due to loss of the RUNX1 activity, and that RUNX1 repression-independent activities may be important for leukemia development.
Clinical data from inv(16) AML patients is also consistent with the CBFβ-SMMHC fusion protein having RUNX1 repression-independent activities. If dominant repression of RUNX1 were CBFβ-SMMHC's only activity, one would expect that loss of RUNX1 would result in leukemia with similar characteristics to those with inv(16). Instead, RUNX1 point mutations are associated with stem cell-like, M0 AML with poor prognosis, while expression of CBFβ-SMMHC is associated with a more differentiated, myelomonocytic M4 AML with relatively good prognosis [32-35, 59-61]. These differences in clinical presentation and outcome imply fundamental differences in the underlying leukemogenic process of these two AML subtypes.
Interactions between CBFβ-SMMHC and the other CBFα subunits, RUNX2 and RUNX3, are tempting explanations for the differences between RUNX1 mutated and inv(16) AML. Both RUNX2 and RUNX3 are expressed in adult hematopoietic stem and progenitor cells, and are predicted to heterodimerize with CBFβ-SMMHC [49]. Studies in mice show that decreased RUNX2 activity slows CBFβ-SMMHC induced leukemia, while increased RUNX2 expression accelerates it [79]. These findings imply that repression of RUNX2 by CBFβ-SMMHC is likely not the cause of the unique inv(16) AML phenotype. However, it is possible that the fusion protein alters RUNX2 activity in a way that contributes to the leukemogenesis.
How RUNX3 may contribute to the differences between the two leukemia subtypes is less well understood. RUNX3 is frequently silenced by hypermethylation in inv(16) patient samples, and re-expression of RUNX3 decreases their proliferation in vitro. This implies that RUNX3 activity inhibits CBFβ-SMMHC induced leukemogenesis. [80]. How RUNX3 becomes hypermethylated and when it occurs in the leukemic process is not known.
RUNX1 is necessary for CBFβ-SMMHC leukemogenesis
The observation that CBFβ-SMMHC has RUNX1 repression- independent activities raises the possibility that RUNX1 may be dispensable for CBFβ-SMMHC induced leukemogenesis. To test this possibility, mice expressing a conditional Cbfb-MYH11 allele (Cbfb56M) under the control of the Mx1-Cre Recombinase (Mx1-Cre+) transgene were crossed with mice expressing a semi-dominant negative allele of Runx1 in which the 3′ end of the gene is fused to the bacterial beta-galactosidase gene, lacZ (Runx1lz) [81]. The Runx1+/lz mice retain enough RUNX1 activity to bypass the embryonic lethality associated with nullizygous Runx1−/− mice, but have less RUNX1 activity than Runx1+/− mice. Mx1-Cre+; Cbfb+/56M; Runx1+/lz mice have a partial rescue of the differentiation and gene expression defects induced by the fusion gene [82]. In addition, Mx1-Cre+; Cbfb+/56M; Runx1+/lz mice show significantly delayed leukemia development as compared to Mx1-Cre+; Cbfb+/56M; Runx1+/+ mice [82]. These findings demonstrate that Runx1 is required for efficient CBFβ-SMMHC induced leukemia.
The fact that the majority of the Mx1-Cre+; Cbfb+/56M; Runx1+/lz mice eventually develop leukemia does not necessarily mean that the fusion protein's RUNX1-independent activities are sufficient for leukemia development, if given enough time. These mice have one wildtype Runx1 allele, so CBFβ-SMMHC:RUNX1 complexes can still form. Due to limited RUNX1, fewer of these complexes may assemble, and those that do form may have less transcriptional repression or activation activity. However, this residual RUNX1 activity may be enough to drive leukemia development.
Although the mechanism of RUNX1 activity is currently not understood, these results demonstrate a genetic requirement for Runx1 for efficient CBFβ-SMMHC induced leukemia in mice. A similar requirement for RUNX1 has been demonstrated in the inv(16) patient derived cell line, ME-1. When short hairpin RNAs are used to knockdown RUNX1, ME-1 cells show decreased proliferation and increased apoptosis [83]. This confirms the importance of RUNX1 in CBFβ-SMMHC-expressing leukemia cells.
A new model of CBFβ-SMMHC leukemogenesis
The finding that CBFβ-SMMHC has activities that don't involve RUNX1 repression, but that RUNX1 is required for leukemogensis has led to a new model of CBFβ-SMMHC activity. Rather than acting as a dominant repressor of RUNX1, it is possible that the fusion protein cooperates with RUNX1 to induce changes in gene expression [78, 82, 84]. In support of this model, immunohistochemistry of inv(16) patient samples demonstrate that CBFβ-SMMHC is localized to the nucleus [85]. In addition, chromatin immunoprecipitation (ChIP) experiments in ME-1 cells demonstrate that CBFβ-SMMHC and RUNX1 co-localize at genes whose expression is regulated by the fusion protein. When the fusion protein is knocked down in ME-1 cells, the majority of the genes bound by the fusion protein and RUNX1 show decreased expression, implying that the CBFβ-SMMHC:RUNX1 complex can act as a transcriptional activator [86]. Consistent with this observation, the majority of the genes deregulated in Cbfb+/MYH11 embryos show increased expression as compared to wildtype littermates [78].
These observations do not exclude the possibility that CBFβ-SMMHC also acts as a transcriptional repressor of some target genes [78, 86]. CBFβ-SMMHC binds the transcriptional co-repressor Sin3a via its C-terminal repression domain (Figure 1) [87]. Therefore, it is possible that, in concert with Sin3a, CBFβ-SMMHC:RUNX1 directly represses expression of some target genes. In addition, some of these repression targets may be transcriptional repressors themselves. Silencing of transcriptional repressors by CBFβ-SMMHC may indirectly contribute to the overall increased gene expression caused by the fusion protein.
Using a model in which CBFβ-SMMHC directly regulates target gene expression, one would predict that the fusion protein requires heterodimerization with RUNX1 for its activity, and that with the loss of RUNX1, the fusion protein is no longer functional. However, it is also possible that the genetic requirement for RUNX1 observed in CBFβ-SMMHC expressing cells is due to the activities of the wildtype CBFβ:RUNX1 heterodimer or to CBFβ-independent activities of RUNX1. Both the dominant negative Runx1-lz allele and shRNA knockdown of RUNX1 are predicted to affect all RUNX1 containing complexes [82, 83]. ChIP experiments in ME-1 cells identified genes associated with RUNX1 and wildtype CBFβ, as well as genes bound by RUNX1 alone. This implies that RUNX1 has transcriptional activities independent of the fusion protein in inv(16) leukemia cells [86].
It is likely that RUNX1 acts through multiple different protein complexes in inv(16) AML cells, and that the balance of these different activities influences the growth and survival of leukemia cells. This idea has been proposed to explain the unexpected finding that CBFβ-SMMHC fusions with reduced RUNX1 binding retain the ability to cause leukemia. Knockin mice have been generated that express a deletion mutant of CBFβ-SMMHC lacking the HABD (CBFβ-SMMHC d179-221) and with reduced RUNX1 binding affinity (Figure 1) [88]. Heterozygous mice expressing CBFβ-SMMHC d179-221 are viable, indicating that the loss of the HABD eliminates the embryonic lethality caused by dominant repression of RUNX1. Surprisingly, mice expressing CBFβ-SMMHC d179-221 rapidly develop leukemia, with significantly reduced latency as compared to mice with full length CBFβ-SMMHC [88]. In addition, a small number of inv(16) patients express fusion genes lacking the HABD, referred to as a type I fusion [89, 90]. The blast morphology and clinical course of patients with type I fusions are indistinguishable from patients with the more common fusions that include the HABD. Together, these findings indicate that high affinity binding of RUNX1 is not required for CBFβ-SMMHC induced leukemia [89, 90].
Currently the mechanism of accelerated leukemia development caused by the CBFβ-SMMHC d179-221 mutant is not known. One possible explanation is that the fusion protein transcriptionally regulates target genes that have both pro- and anti-leukemic effects, but that control of these genes requires different levels of CBFβ-SMMHC activity. The CBFβ-SMMHC d179-221 deletion mutant may retain enough activation and/or repression function to regulate genes associated with a pro-leukemic effect, but not enough activity to regulate anti-leukemic target genes. This would result in accelerated leukemia development. A similar mechanism has been demonstrated in the case of the RUNX1-ETO (also known as AML1-ETO or RUNX1-RUNX1T1) fusion protein, which is the product of the t(8;21) translocation. A splice variant of this fusion gene, RUNX1-ETO9a, lacks c-terminal co-repressor binding domains, and has reduced transcriptional repression and cellular dysregulation activity as compared to full length RUNX1-ETO. However, RUNX1-ETO9a causes accelerated leukemia development in mice and its expression is correlated with worse survival in patients [67, 91].
A second possible explanation for the accelerated leukemia development by the CBFβ-SMMHC d179-221 mutant is that it alters the amount of RUNX1 available to non-fusion protein complexes. Deletion of CBFβ-SMMHC's HABD may allow more RUNX1 to bind wildtype CBFβ or to participate in complexes with other transcription factors. This may result in increased expression of pro-survival genes, and an expanded pool of pre-leukemic cells capable of acquiring cooperating mutations [82, 83, 88].
The idea that the balance of RUNX1 activities in various complexes impacts CBFβ-SMMHC activity may explain the observation that loss of wildtype CBFβ causes accelerated leukemogenesis. Mice with one inducible Cbfb-MYH11 knockin allele (Cbfb56M) were crossed with mice heterozygous for a null Cbfb allele (Cbfb−). The resulting Cbfb56M/− mice developed leukemia significantly faster than mice expressing the fusion protein in the presence of one wildtype Cbfb allele (Cbfb56M/+), indicating that wildtype CBFβ has activities that counter balance those of the fusion protein [21, 92]. Loss of wildtype CBFβ is expected to allow more RUNX1 to act in complexes with the fusion protein or with other transcription factors, and perhaps the activity of these other complexes accelerates the development of leukemia.
TARGETING THE CBFβ-SMMHC:RUNX1 INTERACTION
Due to the importance of RUNX1 for CBFβ-SMMHC activity, efforts to develop inhibitors of the fusion protein have focused on disrupting the CBFβ-SMMHC:RUNX1 interaction. To date, two different small molecule inhibitors of the fusion protein have been identified, both of which are able to kill CBFβ-SMMHC expressing leukemia cells [8, 9]. Both current CBFβ-SMMHC inhibitors are unlikely to be used in patients due to poor pharmacokinetics and high effective concentrations. Nonetheless, these studies provide important proof-of-principal support for targeting the CBFβ-SMMHC:RUNX1 interaction for the treatment of inv(16) patients.
Ro5-3335 is a benzodiazepine that was identified in a screen for inhibitors of the interaction between wildtype CBFβ and the runt homology domain (RHD) of RUNX1 (Figures 2 and 3) [8]. Ro5-3335 binds both RUNX1 and CBFβ, and inhibits RUNX1 activity in a cell based promoter assay and in zebrafish. Treatment with Ro5-3335 reduces the viability of ME-1 cells. In leukemic mice, Ro5-3335 reduces disease burden and increases survival [8]. Because Ro5-3335 binds both RUNX1 and CBFβ, it is predicted to inhibit RUNX1 complexes with either the fusion protein or wildtype CBFβ (Figure 3). Extended treatment of wildtype mice with Ro5-3335 causes minor changes in platelet and red blood cell counts, and differences in white blood cell differentials. However, no obvious illness was observed, indicating that Ro5-3335 is relatively well tolerated in mice [8].
Figure 2. Small molecules targeting CBFβ-SMMHC.
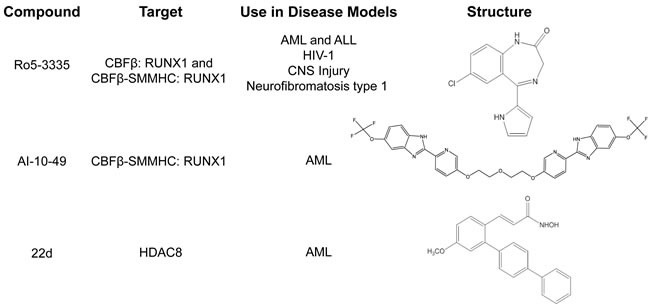
Table listing current small molecule inhibitors targeting CBFβ-SMMHC, and other disease models each drug has been tested in. AML: Acute Myeloid Leukemia. ALL: Acute Lymphoid Leukemia. CNS: Central Nervous System.
Figure 3. Proposed mechanism of CBFβ-SMMHC inhibitors.
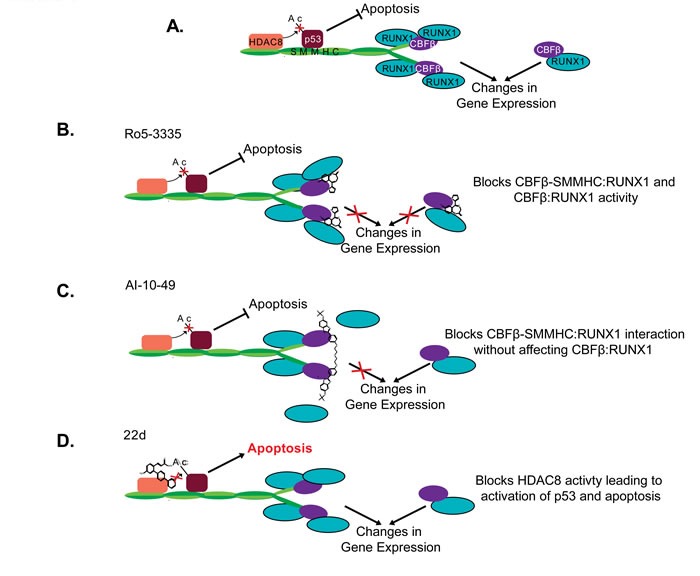
Schematic representation of proposed CBFβ-SMMHC activities A. in the absence of inhibitors, or in the presence of B. Ro5-3335, C. AI-10-49, or D. 22d. Proteins are represented by the colored shapes labeled in (A). Ac: Acetylation group.
Currently, how Ro5-3335 inhibits RUNX1 activity is not known. In vitro assays with purified proteins show that Ro5-3335 blocks the interaction between CBFβ and the RHD. However, experiments with cellular extracts indicate that the drug does not prevent dimerization between full length RUNX1 and CBFβ, or binding to DNA. It is possible that Ro5-3335 causes a confirmation change in either RUNX1 or CBFβ which doesn't block heterodimerization, but alters the complex's DNA binding specificity or it's ability to activate and/or repress transcription [8].
Because Ro5-3335 inhibits RUNX1 and not just the CBFβ-SMMHC fusion protein, it may also be useful for the treatment of other leukemia subtypes that require RUNX1 activity (Figure 2). In fact, treatment with Ro5-3335 reduced viability in cells expressing other RUNX1 fusion proteins (RUNX1-ETO, and ETV6-RUNX1, also known as TEL-AML1), or the fusion protein MLL-AF9, which was recently shown to require RUNX1 for its leukemogenic activity [8, 93]. In addition, there are potential applications for Ro5-3335 in other diseases involving RUNX1, including Human Immunodeficiency Virus (HIV) infection, injuries involving the central nervous system, and Neurofibromatosis, type 1 [94-98].
A second small molecular drug targeting the CBFβ-SMMHC: RUNX1 interaction is called AI-10-49. In contrast to Ro5-3335, AI-10-49 specifically inhibits only the fusion protein (Figures 2 and 3) [9]. The parent compound for this inhibitor was identified in a screen for inhibitors of the CBFβ:RUNX1 interaction, and then subsequently shown to bind CBFβ and block its interaction with RUNX1. In order to generate an inhibitor that would preferentially interact with the fusion protein, a linker was used to generate a bivalent molecule. This strategy takes advantage of the observation that CBFβ-SMMHC exists as a multimer, while wildtype CBFβ is monomeric [71, 73, 99-101]. The bivalent inhibitor, AI-10-49, has a higher binding affinity for CBFβ-SMMHC than wildtype CBFβ. Treatment with AI-10-49 induces cell death in ME-1 cells, but in contrast to Ro5-3335, did not affect the growth of Kasumi-1 cells which express the RUNX1-ETO fusion protein. This indicates that AI-10-49 specifically inhibits the CBFβ-SMMHC:RUNX1 complex, and not the wildtype CBFβ:RUNX1 complex. Similarly, AI-10-49 did not significantly affect the survival or colony forming ability in culture of normal human hematopoietic cells in vitro. Importantly, treatment with AI-10-49 increased survival in mice with CBFβ-SMMHC induced leukemia, and decreased viability of primary inv(16) AML patient samples [9]. These results demonstrate that the bivalent inhibitor approach has potential for the treatment of inv(16) AML.
CBFβ-SMMHC's C-TERMINUS
Initially, examination of CBFβ-SMMHC's activity focused on its interaction with RUNX1. More recently, domains in the C-terminus of the fusion protein have been shown to be relevant for leukemogenesis.
The C-terminus of CBFβ-SMMHC is composed of a series of α-helical repeats [71]. Work in cell lines has demonstrated that within this region, there is a multimerization or assembly competence domain (ACD) that allows for the self-dimerization of the fusion protein, and a repression domain that interacts with transcriptional co-repressors such as Sin3a and HDAC8 (Figure 1) [72, 73, 87, 100-102].
Studies in mice show the importance of CBFβ-SMMHC's C-terminus. Knockin mice expressing a CBFβ-SMMHC deletion mutant lacking the final 95 amino acids (CBFβ-SMMHCΔC95) show an almost complete rescue of the full length fusion protein's activity (Figure 1). In contrast to knockin mice expressing full length CBFβ-SMMHC, mice heterozygous for the CBFβ-SMMHCΔC95 allele (Cbfb+/ΔC95) are viable, with no obvious defects in primitive or definitive hematopoiesis [103]. Importantly, Cbfb+/ΔC95 mice do not develop leukemia, even after treatment with the mutagen N-Ethyl-N-Nitrosourea (ENU). A small percentage of aged Cbfb+/ΔC95 mice develop a non-transplantable myeloprolifeative disease (MPD). In addition, homozygous CbfbΔC95/ ΔC95 embryos have similar differentiation defects during primitive hematopoiesis as described in Cbfb+/MYH11 embryos [103]. As neither the MPD nor the primitive hematopoietic defect have been observed in mice with the null allele of Cbfb, these results indicate that the CBFβ-SMMHCΔC95 mutant retains some of the fusion protein's activity, but that the C-terminus is required for leukemogenesis. In more recent work, knockin mice expressing a CBFβ-SMMHC point mutant that specifically disrupts multimerization (CbfbmDE) were generated. Cbfb+/mDE recapitulate many of the features of the Cbfb+/ΔC95 including the failure to develop leukemia [101, 104]. This finding indicates that multimerization is required for CBFβ-SMMHC induced leukemia.
Another important CBFβ-SMMHC activity lost with the deletion of the terminal 95 amino acids is the interaction with the histone deacetylase, HDAC8 (Figure 2) [10, 87]. HDAC8 is an enzyme that catalyzes the removal of acetyl groups from lysine residues [105, 106]. Recent works demonstrates that the interaction between CBFβ-SMMHC and HDAC8 leads to the deacetylation and subsequent inactivation of the transcription factor p53, which also binds CBFβ-SMMHC (Figure 3) [10]. Genetic experiments show that Hdac8 is required for CBFβ-SMMHC activity. Mice expressing the fusion protein but lacking HDAC8 show significantly delayed leukemia development. Importantly, treatment with the HDAC8 inhibitor 22d, an ortho-Aryl-N-hydroxycinnamide, selectively induced apoptosis in mouse and human CBFβ-SMMHC expressing leukemia cells. 22d had only minimal effect on the survival of non-inv(16) leukemia cells or CD34+ cells from healthy individuals [10, 107]. The precise mechanism of action for 22d is not known, but molecular modeling suggests that the drug interacts with a hydrophobic pocket at HDAC8′s active site, preventing the enzyme's interaction with substrates such as p53. In inv(16) leukemia cells, treatment with 22d prevents de-acetylation and subsequent inactivation of p53, allowing the transcription factor to remain in its active state and capable of inducing apoptosis [10, 107]. These results indicate that inhibition of HDAC8 is a potential strategy for the treatment of inv(16) AML.
CONCLUSIONS
Although expression of CBFβ-SMMHC is known to be causative in inv(16) AML, the mechanism of its activity has been unclear. Recent advances demonstrate that the fusion protein's activity depends on at least two different interacting partners, RUNX1 and HDAC8 [10, 82, 83]. Importantly, pharmacological inhibition of either factor is effective at specifically targeting CBFβ-SMMHC expressing leukemia cells, with little effect on normal hematopoietic cells [8-10]. In addition, recent studies have pointed to additional targets for drug development. The importance of CBFβ-SMMHC multimerization and its inhibition of p53 are only beginning to be appreciated. Early studies indicate that both activities are critical for inv(16) AML and are intriguing avenues for future drug development [10, 104]. While much work remains, these recent advances indicate a promising future for the treatment of patients with inv(16) AML.
Acknowledgments
This work was supported by funding from the National Institutes of Health, grant R00CA148963 (RKH), and from the Biochemistry and Molecular Biology Department and the Fred and Pamela Buffet Cancer Center, University of Nebraska Medical Center.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–64. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 2.Byrd JC, Mrozek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, Pettenati MJ, Patil SR, Rao KW, Watson MS, Koduru PR, Moore JO, Stone RM, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–36. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 3.Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136–52. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 4.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–42. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 5.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 6.Stein EM, Tallman MS. Mixed lineage rearranged leukaemia: pathogenesis and targeting DOT1L. Curr Opin Hematol. 2015;22:92–6. doi: 10.1097/MOH.0000000000000123. [DOI] [PubMed] [Google Scholar]
- 7.Senter T, Gogliotti R, Han C, Locuson CW, II, Morrison R, Daniels JS, Cierpicki T, Grembecka J, Lindsley CW, Stauffer SR. Progress towards small molecule inhibitors of the Menin-Mixed Lineage Leukemia (MLL) interaction with in vivo utility. Probe Reports from the NIH Molecular Libraries Program; Bethesda (MD): 2010. [PubMed] [Google Scholar]
- 8.Cunningham L, Finckbeiner S, Hyde RK, Southall N, Marugan J, Yedavalli VR, Dehdashti SJ, Reinhold WC, Alemu L, Zhao L, Yeh JR, Sood R, Pommier Y, et al. Identification of benzodiazepine Ro5-3335 as an inhibitor of CBF leukemia through quantitative high throughput screen against RUNX1-CBFbeta interaction. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14592–7. doi: 10.1073/pnas.1200037109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Illendula A, Pulikkan JA, Zong H, Grembecka J, Xue L, Sen S, Zhou Y, Boulton A, Kuntimaddi A, Gao Y, Rajewski RA, Guzman ML, Castilla LH, et al. Chemical biology. A small-molecule inhibitor of the aberrant transcription factor CBFbeta-SMMHC delays leukemia in mice. Science. 2015;347:779–84. doi: 10.1126/science.aaa0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi J, Singh S, Hua WK, Cai Q, Chao SW, Li L, Liu H, Ho Y, McDonald T, Lin A, Marcucci G, Bhatia R, Huang WJ, et al. HDAC8 Inhibition Specifically Targets Inv(16) Acute Myeloid Leukemic Stem Cells by Restoring p53 Acetylation. Cell Stem Cell. 2015 doi: 10.1016/j.stem.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Beau MM, Larson RA, Bitter MA, Vardiman JW, Golomb HM, Rowley JD. Association of an inversion of chromosome 16 with abnormal marrow eosinophils in acute myelomonocytic leukemia. A unique cytogenetic-clinicopathological association. N Engl J Med. 1983;309:630–6. doi: 10.1056/NEJM198309153091103. [DOI] [PubMed] [Google Scholar]
- 12.Bloomfield CD, Ruppert AS, Mrozek K, Kolitz JE, Moore JO, Mayer RJ, Edwards CG, Sterling LJ, Vardiman JW, Carroll AJ, Pettenati MJ, Stamberg J, Byrd JC, et al. Core binding factor acute myeloid leukemia. Cancer and Leukemia Group B (CALGB) Study 8461. Ann Hematol. 2004;83(Suppl 1):S84–5. doi: 10.1007/s00277-004-0850-2. [DOI] [PubMed] [Google Scholar]
- 13.Liu P, Tarle SA, Hajra A, Claxton DF, Marlton P, Freedman M, Siciliano MJ, Collins FS. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261:1041–4. doi: 10.1126/science.8351518. [DOI] [PubMed] [Google Scholar]
- 14.Liu P, Seidel N, Bodine D, Speck N, Tarle S, Collins FS. Acute myeloid leukemia with Inv (16) produces a chimeric transcription factor with a myosin heavy chain tail. Cold Spring Harb Symp Quant Biol. 1994;59:547–53. doi: 10.1101/sqb.1994.059.01.061. [DOI] [PubMed] [Google Scholar]
- 15.Marlton P, Claxton DF, Liu P, Estey EH, Beran M, LeBeau M, Testa JR, Collins FS, Rowley JD, Siciliano MJ. Molecular characterization of 16p deletions associated with inversion 16 defines the critical fusion for leukemogenesis. Blood. 1995;85:772–9. [PubMed] [Google Scholar]
- 16.Pandolfi PP. Oncogenes and tumor suppressors in the molecular pathogenesis of acute promyelocytic leukemia. Hum Mol Genet. 2001;10:769–75. doi: 10.1093/hmg/10.7.769. [DOI] [PubMed] [Google Scholar]
- 17.Bursen A, Moritz S, Gaussmann A, Moritz S, Dingermann T, Marschalek R. Interaction of AF4 wild-type and AF4. MLL fusion protein with SIAH proteins: indication for t(4;11) pathobiology? Oncogene. 2004;23:6237–49. doi: 10.1038/sj.onc.1207837. [DOI] [PubMed] [Google Scholar]
- 18.Bursen A, Schwabe K, Ruster B, Henschler R, Ruthardt M, Dingermann T, Marschalek R. The AF4.MLL fusion protein is capable of inducing ALL in mice without requirement of MLL.AF4. Blood. 2010;115:3570–9. doi: 10.1182/blood-2009-06-229542. [DOI] [PubMed] [Google Scholar]
- 19.Wilkinson AC, Ballabio E, Geng H, North P, Tapia M, Kerry J, Biswas D, Roeder RG, Allis CD, Melnick A, de Bruijn MF, Milne TA. RUNX1 is a key target in t(4;11) leukemias that contributes to gene activation through an AF4-MLL complex interaction. Cell Rep. 2013;3:116–27. doi: 10.1016/j.celrep.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castilla LH, Garrett L, Adya N, Orlic D, Dutra A, Anderson S, Owens J, Eckhaus M, Bodine D, Liu PP. The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia [letter] Nat Genet. 1999;23:144–6. doi: 10.1038/13776. [DOI] [PubMed] [Google Scholar]
- 21.Kuo YH, Landrette SF, Heilman SA, Perrat PN, Garrett L, Liu PP, Le Beau MM, Kogan SC, Castilla LH. Cbf beta-SMMHC induces distinct abnormal myeloid progenitors able to develop acute myeloid leukemia. Cancer Cell. 2006;9:57–68. doi: 10.1016/j.ccr.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 22.Castilla LH, Perrat P, Martinez NJ, Landrette SF, Keys R, Oikemus S, Flanegan J, Heilman S, Garrett L, Dutra A, Anderson S, Pihan GA, Wolff L, et al. Identification of genes that synergize with Cbfb-MYH11 in the pathogenesis of acute myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4924–9. doi: 10.1073/pnas.0400930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landrette SF, Kuo YH, Hensen K, Barjesteh van Waalwijk van Doorn-Khosrovani S, Perrat PN, Van de Ven WJ, Delwel R, Castilla LH. Plag1 and Plagl2 are oncogenes that induce acute myeloid leukemia in cooperation with Cbfb-MYH11. Blood. 2005 Dec 7;105:2900–7. doi: 10.1182/blood-2004-09-3630. Epub 004. [DOI] [PubMed] [Google Scholar]
- 24.Xue L, Pulikkan JA, Valk PJ, Castilla LH. NrasG12D oncoprotein inhibits apoptosis of preleukemic cells expressing Cbfbeta-SMMHC via activation of MEK/ERK axis. Blood. 2014;124:426–36. doi: 10.1182/blood-2013-12-541730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao L, Melenhorst JJ, Alemu L, Kirby M, Anderson S, Kench M, Hoogstraten-Miller S, Brinster L, Kamikubo Y, Gilliland DG, Liu PP. KIT with D816 mutations cooperates with CBFB-MYH11 for leukemogenesis in mice. Blood. 2012;119:1511–21. doi: 10.1182/blood-2011-02-338210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno-Miralles I, Pan L, Keates-Baleeiro J, Durst-Goodwin K, Yang C, Kim HG, Thompson MA, Klug CA, Cleveland JL, Hiebert SW. The inv(16) cooperates with ARF haploinsufficiency to induce acute myeloid leukemia. J Biol Chem. 2005;280:40097–103. doi: 10.1074/jbc.M506855200. [DOI] [PubMed] [Google Scholar]
- 27.Carella C, Bonten J, Sirma S, Kranenburg TA, Terranova S, Klein-Geltink R, Shurtleff S, Downing JR, Zwarthoff EC, Liu PP, Grosveld GC. MN1 overexpression is an important step in the development of inv(16) AML. Leukemia. 2007;21:1679–90. doi: 10.1038/sj.leu.2404778. [DOI] [PubMed] [Google Scholar]
- 28.Kim HG, Kojima K, Swindle CS, Cotta CV, Huo Y, Reddy V, Klug CA. FLT3-ITD cooperates with inv(16) to promote progression to acute myeloid leukemia. Blood. 2008;111:1567–74. doi: 10.1182/blood-2006-06-030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A, Paietta E, Willman CL, Head DR, Rowe JM, Forman SJ, Appelbaum FR. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96:4075–83. [PubMed] [Google Scholar]
- 30.Kihara R, Nagata Y, Kiyoi H, Kato T, Yamamoto E, Suzuki K, Chen F, Asou N, Ohtake S, Miyawaki S, Miyazaki Y, Sakura T, Ozawa Y, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014;28:1586–95. doi: 10.1038/leu.2014.55. [DOI] [PubMed] [Google Scholar]
- 31.Haferlach T, Schnittger S, Kern W, Hiddemann W, Schoch C. Genetic classification of acute myeloid leukemia (AML) Ann Hematol. 2004;83(Suppl 1):S97–100. doi: 10.1007/s00277-004-0850-2. [DOI] [PubMed] [Google Scholar]
- 32.Marcucci G, Mrozek K, Ruppert AS, Maharry K, Kolitz JE, Moore JO, Mayer RJ, Pettenati MJ, Powell BL, Edwards CG, Sterling LJ, Vardiman JW, Schiffer CA, et al. Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): a Cancer and Leukemia Group B study. Journal of clinical oncology. 2005;23:5705–17. doi: 10.1200/JCO.2005.15.610. [DOI] [PubMed] [Google Scholar]
- 33.Appelbaum FR, Kopecky KJ, Tallman MS, Slovak ML, Gundacker HM, Kim HT, Dewald GW, Kantarjian HM, Pierce SR, Estey EH. The clinical spectrum of adult acute myeloid leukaemia associated with core binding factor translocations. British journal of haematology. 2006;135:165–73. doi: 10.1111/j.1365-2141.2006.06276.x. [DOI] [PubMed] [Google Scholar]
- 34.Hospital MA, Prebet T, Bertoli S, Thomas X, Tavernier E, Braun T, Pautas C, Perrot A, Lioure B, Rousselot P, Tamburini J, Cluzeau T, Konopacki J, et al. Core-binding factor acute myeloid leukemia in first relapse: a retrospective study from the French AML Intergroup. Blood. 2014 doi: 10.1182/blood-2014-01-549212. [DOI] [PubMed] [Google Scholar]
- 35.Ustun C, Marcucci G. Emerging diagnostic and therapeutic approaches in core binding factor acute myeloid leukaemia. Curr Opin Hematol. 2015;22:85–91. doi: 10.1097/MOH.0000000000000124. [DOI] [PubMed] [Google Scholar]
- 36.Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–74. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 37.Creutzig U, van den Heuvel-Eibrink MM, Gibson B, Dworzak MN, Adachi S, de Bont E, Harbott J, Hasle H, Johnston D, Kinoshita A, Lehrnbecher T, Leverger G, Mejstrikova E, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood. 2012;120:3187–205. doi: 10.1182/blood-2012-03-362608. [DOI] [PubMed] [Google Scholar]
- 38.Paschka P, Dohner K. Core-binding factor acute myeloid leukemia: can we improve on HiDAC consolidation? Hematology Am Soc Hematol Educ Program. 2013;2013:209–19. doi: 10.1182/asheducation-2013.1.209. [DOI] [PubMed] [Google Scholar]
- 39.Adya N, Castilla LH, Liu PP. Function of CBFbeta/Bro proteins. Semin Cell Dev Biol. 2000;11:361–8. doi: 10.1006/scdb.2000.0189. [DOI] [PubMed] [Google Scholar]
- 40.van Wijnen AJ, Stein GS, Gergen JP, Groner Y, Hiebert SW, Ito Y, Liu P, Neil JC, Ohki M, Speck N. Nomenclature for Runt-related (RUNX) proteins. Oncogene. 2004;23:4209–10. doi: 10.1038/sj.onc.1207758. [DOI] [PubMed] [Google Scholar]
- 41.Tang Y, Huang X, Berardi M, Yan J, Shi J, Davis A, Speck N, Bushweller J. Structural insights into the mechanism by which the core-binding factor beta subunit (CBFbeta) enhances DNA-binding by the CBFalpha subunit. Blood. 1999;94:683a. [Google Scholar]
- 42.Tang YY, Shi J, Zhang L, Davis A, Bravo J, Warren AJ, Speck NA, Bushweller JH. Energetic and functional contribution of residues in the core binding factor beta (CBFbeta ) subunit to heterodimerization with CBFalpha. J Biol Chem. 2000;275:39579–88. doi: 10.1074/jbc.M007350200. [DOI] [PubMed] [Google Scholar]
- 43.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–30. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 44.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci U S A. 1996;93:3444–9. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature. 2009;457:887–91. doi: 10.1038/nature07619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yokomizo T, Hasegawa K, Ishitobi H, Osato M, Ema M, Ito Y, Yamamoto M, Takahashi S. Runx1 is involved in primitive erythropoiesis in the mouse. Blood. 2008;111:4075–80. doi: 10.1182/blood-2007-05-091637. [DOI] [PubMed] [Google Scholar]
- 47.Cai X, Gaudet JJ, Mangan JK, Chen MJ, De Obaldia ME, Oo Z, Ernst P, Speck NA. Runx1 loss minimally impacts long-term hematopoietic stem cells. PLoS One. 2011;6:e28430. doi: 10.1371/journal.pone.0028430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, Curley DP, Kutok JL, Akashi K, Williams IR, Speck NA, Gilliland DG. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106:494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Bruijn MFTR, Speck NA. Core-binding factors in hematopoiesis and immune function. Oncogene. 2004;23:4238–48. doi: 10.1038/sj.onc.1207763. [DOI] [PubMed] [Google Scholar]
- 50.Okuda T, Nishimura M, Nakao M, Fujita Y. RUNX1/AML1: a central player in hematopoiesis. Int J Hematol. 2001;74:252–7. doi: 10.1007/BF02982057. [DOI] [PubMed] [Google Scholar]
- 51.Imperato MR, Cauchy P, Obier N, Bonifer C. The RUNX1-PU.1 axis in the control of hematopoiesis. Int J Hematol. 2015;101:319–29. doi: 10.1007/s12185-015-1762-8. [DOI] [PubMed] [Google Scholar]
- 52.Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002;2:502–13. doi: 10.1038/nrc840nrc840. [DOI] [PubMed] [Google Scholar]
- 53.Harada H, Harada Y. Recent advances in myelodysplastic syndromes: Molecular pathogenesis and its implications for targeted therapies. Cancer Sci. 2015;106:329–36. doi: 10.1111/cas.12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jhanwar SC. Genetic and epigenetic pathways in myelodysplastic syndromes: A brief overview. Adv Biol Regul. 2015;58:28–37. doi: 10.1016/j.jbior.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 55.Tang JL, Hou HA, Chen CY, Liu CY, Chou WC, Tseng MH, Huang CF, Lee FY, Liu MC, Yao M, Huang SY, Ko BS, Hsu SC, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114:5352–61. doi: 10.1182/blood-2009-05-223784. [DOI] [PubMed] [Google Scholar]
- 56.Schnittger S, Dicker F, Kern W, Wendland N, Sundermann J, Alpermann T, Haferlach C, Haferlach T. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011;117:2348–57. doi: 10.1182/blood-2009-11-255976. [DOI] [PubMed] [Google Scholar]
- 57.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brettingham-Moore KH, Taberlay PC, Holloway AF. Interplay between Transcription Factors and the Epigenome: Insight from the Role of RUNX1 in Leukemia. Front Immunol. 2015;6:499. doi: 10.3389/fimmu.2015.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roumier C, Fenaux P, Lafage M, Imbert M, Eclache V, Preudhomme C. New mechanisms of AML1 gene alteration in hematological malignancies. Leukemia. 2003;17:9–16. doi: 10.1038/sj.leu.24027662402766. [DOI] [PubMed] [Google Scholar]
- 60.Osato M, Asou N, Abdalla E, Hoshino K, Yamasaki H, Okubo T, Suzushima H, Takatsuki K, Kanno T, Shigesada K, Ito Y. Biallelic and Heterozygous Point Mutations in the Runt Domain of the AML1/PEBP2alphaB Gene Associated With Myeloblastic Leukemias. Blood. 1999;93:1817–24. [PubMed] [Google Scholar]
- 61.Osato M. Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene. 2004;23:4284–96. doi: 10.1038/sj.onc.12077791207779. [DOI] [PubMed] [Google Scholar]
- 62.Kuo MC, Liang DC, Huang CF, Shih YS, Wu JH, Lin TL, Shih LY. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23:1426–31. doi: 10.1038/leu.2009.48. [DOI] [PubMed] [Google Scholar]
- 63.Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, Ratajczak J, Resende IC, Haworth C, Hock R, Loh M, Felix C, Roy DC, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23:166–75. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- 64.Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia--a review. Br J Haematol. 2008;140:123–32. doi: 10.1111/j.1365-2141.2007.06909.x. [DOI] [PubMed] [Google Scholar]
- 65.Erickson P, Gao J, Chang KS, Look T, Whisenant E, Raimondi S, Lasher R, Trujillo J, Rowley J, Drabkin H. Identification of breakpoints in t(8;21) acute myelogenous leukemia and isolation of a fusion transcript, AML1/ETO, with similarity to Drosophila segmentation gene, runt. Blood. 1992;80:1825–31. [PubMed] [Google Scholar]
- 66.Golub TR, Barker GF, Bohlander SK, Hiebert SW, Ward DC, Bray-Ward P, Morgan E, Raimondi SC, Rowley JD, Gilliland DG. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 1995;92:4917–21. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lam K, Zhang DE. RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Front Biosci (Landmark Ed) 2012;17:1120–39. doi: 10.2741/3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O'Neil J, Look AT. Mechanisms of transcription factor deregulation in lymphoid cell transformation. Oncogene. 2007;26:6838–49. doi: 10.1038/sj.onc.1210766. [DOI] [PubMed] [Google Scholar]
- 69.Harrison CJ. Targeting signaling pathways in acute lymphoblastic leukemia: new insights. Hematology Am Soc Hematol Educ Program. 2013;2013:118–25. doi: 10.1182/asheducation-2013.1.118. [DOI] [PubMed] [Google Scholar]
- 70.Warren AJ, Bravo J, Williams RL, Rabbitts TH. Structural basis for the heterodimeric interaction between the acute leukaemia-associated transcription factors AML1 and CBFbeta. Embo J. 2000;19:3004–15. doi: 10.1093/emboj/19.12.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lukasik SM, Zhang L, Corpora T, Tomanicek S, Li Y, Kundu M, Hartman K, Liu PP, Laue TM, Biltonen RL, Speck NA, Bushweller JH. Altered affinity of CBF beta-SMMHC for Runx1 explains its role in leukemogenesis. Nat Struct Biol. 2002;9:674–9. doi: 10.1038/nsb831. [DOI] [PubMed] [Google Scholar]
- 72.Lutterbach B, Hou Y, Durst KL, Hiebert SW. The inv(16) encodes an acute myeloid leukemia 1 transcriptional corepressor. Proc Natl Acad Sci U S A. 1999;96:12822–7. doi: 10.1073/pnas.96.22.12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adya N, Stacy T, Speck NA, Liu PP. The leukemic protein core binding factor beta (CBFbeta)-smooth-muscle myosin heavy chain sequesters CBFalpha2 into cytoskeletal filaments and aggregates. Mol Cell Biol. 1998;18:7432–43. doi: 10.1128/mcb.18.12.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Castilla LH, Wijmenga C, Wang Q, Stacy T, Speck NA, Eckhaus M, Marin-Padilla M, Collins FS, Wynshaw-Boris A, Liu PP. Failure of embryonic hematopoiesis and lethal hemorrhages in mouse embryos heterozygous for a knocked-in leukemia gene CBFB-MYH11. Cell. 1996;87:687–96. doi: 10.1016/s0092-8674(00)81388-4. [DOI] [PubMed] [Google Scholar]
- 75.Sasaki K, Yagi H, Bronson RT, Tominaga K, Matsunashi T, Deguchi K, Tani Y, Kishimoto T, Komori T. Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc Natl Acad Sci U S A. 1996;93:12359–63. doi: 10.1073/pnas.93.22.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Q, Stacy T, Miller JD, Lewis AF, Gu TL, Huang X, Bushweller JH, Bories JC, Alt FW, Ryan G, Liu PP, Wynshaw-Boris A, Binder M, et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell. 1996;87:697–708. doi: 10.1016/s0092-8674(00)81389-6. [DOI] [PubMed] [Google Scholar]
- 77.Niki M, Okada H, Takano H, Kuno J, Tani K, Hibino H, Asano S, Ito Y, Satake M, Noda T. Hematopoiesis in the fetal liver is impaired by targeted mutagenesis of a gene encoding a non-DNA binding subunit of the transcription factor, polyomavirus enhancer binding protein 2/core binding factor. Proc Natl Acad Sci U S A. 1997;94:5697–702. doi: 10.1073/pnas.94.11.5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hyde RK, Kamikubo Y, Anderson S, Kirby M, Alemu L, Zhao L, Liu PP. Cbfb/Runx1 repression-independent blockage of differentiation and accumulation of Csf2rb-expressing cells by Cbfb-MYH11. Blood. 2010;115:1433–43. doi: 10.1182/blood-2009-06-227413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kuo YH, Zaidi SK, Gornostaeva S, Komori T, Stein GS, Castilla LH. Runx2 induces acute myeloid leukemia in cooperation with Cbfbeta-SMMHC in mice. Blood. 2009;113:3323–32. doi: 10.1182/blood-2008-06-162248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Estecio MR, Maddipoti S, Bueso-Ramos C, DiNardo CD, Yang H, Wei Y, Kondo K, Fang Z, Stevenson W, Chang KS, Pierce SA, Bohannan Z, Borthakur G, et al. RUNX3 promoter hypermethylation is frequent in leukaemia cell lines and associated with acute myeloid leukaemia inv(16) subtype. Br J Haematol. 2015;169:344–51. doi: 10.1111/bjh.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.North T, Gu TL, Stacy T, Wang Q, Howard L, Binder M, Marin-Padilla M, Speck NA. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development. 1999;126:2563–75. doi: 10.1242/dev.126.11.2563. [DOI] [PubMed] [Google Scholar]
- 82.Hyde RK, Zhao L, Alemu L, Liu PP. Runx1 is required for hematopoietic defects and leukemogenesis in Cbfb-MYH11 knock-in mice. Leukemia. 2015;29:1771–8. doi: 10.1038/leu.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ben-Ami O, Friedman D, Leshkowitz D, Goldenberg D, Orlovsky K, Pencovich N, Lotem J, Tanay A, Groner Y. Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell Rep. 2013;4:1131–43. doi: 10.1016/j.celrep.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 84.Hyde RK, Liu PP. RUNX1 repression-independent mechanisms of leukemogenesis by fusion genes CBFB-MYH11 and AML1-ETO (RUNX1-RUNX1T1) J Cell Biochem. 2010;110:1039–45. doi: 10.1002/jcb.22596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhao W, Claxton DF, Medeiros LJ, Lu D, Vadhan-Raj S, Kantarjian HM, Nguyen MH, Bueso-Ramos CE. Immunohistochemical analysis of CBFbeta-SMMHC protein reveals a unique nuclear localization in acute myeloid leukemia with inv(16)(p13q22) Am J Surg Pathol. 2006;30:1436–44. doi: 10.1097/01.pas.0000213301.19273.66. [DOI] [PubMed] [Google Scholar]
- 86.Mandoli A, Singh AA, Jansen PW, Wierenga AT, Riahi H, Franci G, Prange K, Saeed S, Vellenga E, Vermeulen M, Stunnenberg HG, Martens JH. CBFB-MYH11/RUNX1 together with a compendium of hematopoietic regulators, chromatin modifiers and basal transcription factors occupies self-renewal genes in inv(16) acute myeloid leukemia. Leukemia. 2013 doi: 10.1038/leu.2013.257. [DOI] [PubMed] [Google Scholar]
- 87.Durst KL, Lutterbach B, Kummalue T, Friedman AD, Hiebert SW. The inv(16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol Cell Biol. 2003;23:607–19. doi: 10.1128/MCB.23.2.607-619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kamikubo Y, Zhao L, Wunderlich M, Corpora T, Hyde RK, Paul TA, Kundu M, Garrett L, Compton S, Huang G, Wolff L, Ito Y, Bushweller J, et al. Accelerated leukemogenesis by truncated CBF beta-SMMHC defective in high-affinity binding with RUNX1. Cancer Cell. 2010;17:455–68. doi: 10.1016/j.ccr.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dissing M, Le Beau MM, Pedersen-Bjergaard J. Inversion of chromosome 16 and uncommon rearrangements of the CBFB and MYH11 genes in therapy-related acute myeloid leukemia: rare events related to DNA-topoisomerase II inhibitors? J Clin Oncol. 1998;16:1890–6. doi: 10.1200/JCO.1998.16.5.1890. [DOI] [PubMed] [Google Scholar]
- 90.Van der Reijden BA, de Wit L, van der Poel S, Luiten EB, Lafage-Pochitaloff M, Dastugue N, Gabert J, Lowenberg B, Jansen JH. Identification of a novel CBFB-MYH11 transcript: implications for RT-PCR diagnosis. Hematol J. 2001;2:206–9. doi: 10.1038/sj.thj.6200103. [DOI] [PubMed] [Google Scholar]
- 91.DeKelver RC, Yan M, Ahn EY, Shia WJ, Speck NA, Zhang DE. Attenuation of AML1-ETO cellular dysregulation correlates with increased leukemogenic potential. Blood. 2013;121:3714–7. doi: 10.1182/blood-2012-11-465641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Heilman SA, Kuo YH, Goudswaard CS, Valk PJ, Castilla LH. Cbfbeta reduces Cbfbeta-SMMHC-associated acute myeloid leukemia in mice. Cancer Res. 2006;66:11214–8. doi: 10.1158/0008-5472.CAN-06-0959. [DOI] [PubMed] [Google Scholar]
- 93.Goyama S, Schibler J, Cunningham L, Zhang Y, Rao Y, Nishimoto N, Nakagawa M, Olsson A, Wunderlich M, Link KA, Mizukawa B, Grimes HL, Kurokawa M, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013;123:3876–88. doi: 10.1172/JCI68557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Klase Z, Yedavalli VS, Houzet L, Perkins M, Maldarelli F, Brenchley J, Strebel K, Liu P, Jeang KT. Activation of HIV-1 from latent infection via synergy of RUNX1 inhibitor Ro5-3335 and SAHA. PLoS Pathog. 2014;10:e1003997. doi: 10.1371/journal.ppat.1003997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Witvrouw M, Pauwels R, Vandamme AM, Schols D, Reymen D, Yamamoto N, Desmyter J, De Clercq E. Cell type-specific anti-human immunodeficiency virus type 1 activity of the transactivation inhibitor Ro5-3335. Antimicrob Agents Chemother. 1992;36:2628–33. doi: 10.1128/aac.36.12.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kira T, Hashimoto K, Baba M, Okamoto T, Shigeta S. 2-Glycineamide-5-chlorophenyl 2-pyrryl ketone, a non-benzodiazepin Tat antagonist, is effective against acute and chronic HIV-1 infections in vitro. Antiviral Res. 1996;32:55–62. doi: 10.1016/0166-3542(95)00980-9. [DOI] [PubMed] [Google Scholar]
- 97.Li H, Zhao X, Yan X, Jessen WJ, Kim MO, Dombi E, Liu PP, Huang G, Wu J. Runx1 contributes to neurofibromatosis type 1 neurofibroma formation. Oncogene. 2015 doi: 10.1038/onc.2015.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Logan TT, Rusnak M, Symes AJ. Runx1 promotes proliferation and neuronal differentiation in adult mouse neurosphere cultures. Stem Cell Res. 2015;15:554–64. doi: 10.1016/j.scr.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 99.Huang X, Peng JW, Speck NA, Bushweller JH. Solution structure of core binding factor beta and map of the CBF alpha binding site. Nat Struct Biol. 1999;6:624–7. doi: 10.1038/10670. [DOI] [PubMed] [Google Scholar]
- 100.Kummalue T, Lou J, Friedman AD. Multimerization via its myosin domain facilitates nuclear localization and inhibition of core binding factor (CBF) activities by the CBFbeta-smooth muscle myosin heavy chain myeloid leukemia oncoprotein. Mol Cell Biol. 2002;22:8278–91. doi: 10.1128/MCB.22.23.8278-8291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang L, D'Costa J, Kummalue T, Civin CI, Friedman AD. Identification of a region on the outer surface of the CBFbeta-SMMHC myeloid oncoprotein assembly competence domain critical for multimerization. Oncogene. 2006;25:7289–96. doi: 10.1038/sj.onc.1209725. [DOI] [PubMed] [Google Scholar]
- 102.D'Costa J, Chaudhuri S, Civin CI, Friedman AD. CBFbeta-SMMHC slows proliferation of primary murine and human myeloid progenitors. Leukemia. 2005;19:921–9. doi: 10.1038/sj.leu.2403755. [DOI] [PubMed] [Google Scholar]
- 103.Kamikubo Y, Hyde RK, Zhao L, Alemu L, Rivas C, Garrett LJ, Liu PP. The C-terminus of CBFbeta-SMMHC is required to induce embryonic hematopoietic defects and leukemogenesis. Blood. 2013;121:638–42. doi: 10.1182/blood-2012-06-434688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao LA, L, Cheng J, Zhen T, Friedman AD, Liu P.P. The Multimerization Domain of Cbfb-SMMHC is Required for Leukemogenesis. Blood. 2015:126. [Google Scholar]
- 105.Bertrand P. Inside HDAC with HDAC inhibitors. Eur J Med Chem. 2010;45:2095–116. doi: 10.1016/j.ejmech.2010.02.030. [DOI] [PubMed] [Google Scholar]
- 106.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–9. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 107.Huang WJ, Wang YC, Chao SW, Yang CY, Chen LC, Lin MH, Hou WC, Chen MY, Lee TL, Yang P, Chang CI. Synthesis and biological evaluation of ortho-aryl N-hydroxycinnamides as potent histone deacetylase (HDAC) 8 isoform-selective inhibitors. ChemMedChem. 2012;7:1815–24. doi: 10.1002/cmdc.201200300. [DOI] [PubMed] [Google Scholar]