
Figure 6. Localization of FYCO1 in hereditary myopathies with rimmed vacuoles and in idiopathic inflammatory myopathies.
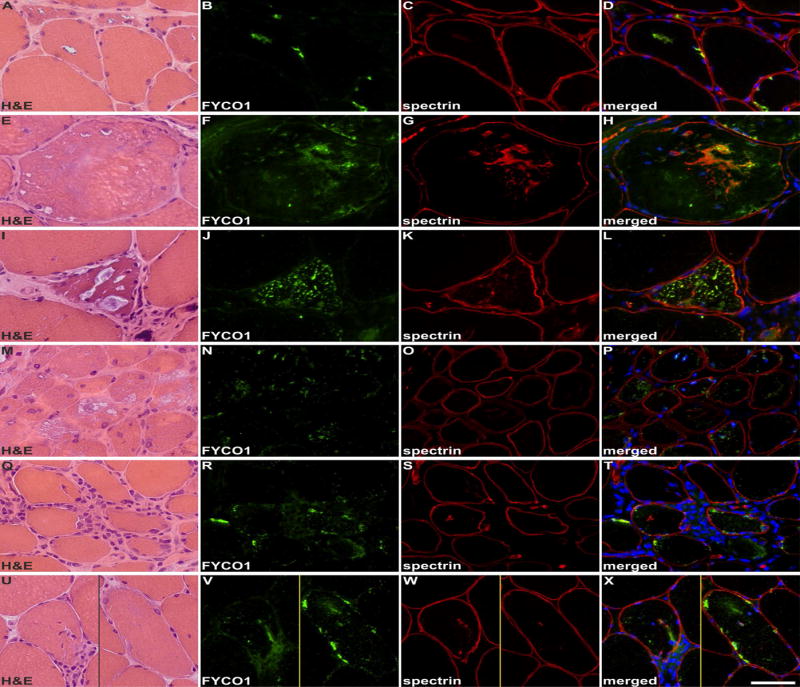
Shown are findings in patients with: GNE-related hereditary inclusion body myopathy (A-D), myofibrillar myopathy caused by FLNC mutation (E-H), glycogen storage disease type II (I-L), dermatomyositis (M-P), polymyositis (Q-T) and a morphological diagnosis of polymyositis but a typical sIBM clinical phenotype (U-X). Serial skeletal muscle sections were stained with H&E and double-immunostained with antibodies recognizing FYCO1 (green) and the constituent muscle protein spectrin (red). Nuclei were stained with DAPI (blue). RVs in hereditary inclusion body myopathy, myofibrillar myopathy and glycogen storage disease type II showed a strong immunoreactivity for FYCO1. In myofibrillar myopathy, FYCO1 was also located in cytoplasmic protein aggregates. In dermatomyositis, some perifascicular muscle fibers showed an increased immunoreactivity for FYCO1 but “punched-out” areas of myofibrillar loss were not rimmed or markedly filled with FYCO1. In polymositis and in sIBM with a morphological phenotype of polymyositis, muscle fibers surrounded by inflammatory cells displayed a diffuse/punctate immunoreactivity for FYCO1. In addition, some fibers showed FYCO1 accumulations in subsarcolemmal areas similar to that found in RV areas in sIBM. These areas were basophilic in H&E staining. Scale bar = 50 μm.