

Abstract
A controlled, reproducible, gram‐scale method is reported for the covalent functionalization of graphene sheets by a one‐pot nitrene [2+1] cycloaddition reaction under mild conditions. The reaction between commercially available 2,4,6‐trichloro‐1,3,5‐triazine and sodium azide with thermally reduced graphene oxide (TRGO) results in defined dichlorotriazine‐functionalized sheets. The different reactivities of the chlorine substituents on the functionalized graphene allow stepwise post‐modification by manipulating the temperature. This new method provides unique access to defined bifunctional 2D nanomaterials, as exemplified by chiral surfaces and multifunctional hybrid architectures.
Keywords: cycloaddition, graphene materials, nanostructures, polyglycerol, triazines
Graphene is a two‐dimensional (2D) carbon network with unusual properties, including extraordinary mechanical stiffness, strength, and elasticity, outstanding electrical and thermal conductivity, and many others.1, 2 Despite these unique properties, its low solubility, poor reactivity, and the limited accessibility of a well‐defined basal plane are major challenges that must be addressed for future applications. An ideal method to overcome these problems is the covalent attachment of functional molecules to its surface.3, 4, 5, 6, 7, 8, 9, 10, 11, 12 The nitrene [2+1] cycloaddition reaction is one of the most useful approaches to conjugate diverse functional groups to the surface of graphene.13 However, this approach requires high temperatures to functionalize carbon‐based nanomaterials efficiently.14, 15 Therefore, controlling the functionality and surface structure of the obtained 2D nanomaterials is challenging.16 The functionalization of graphene under ambient conditions by perfluoroazides as precursors of the highly reactive nitrenes has been explored to overcome this drawback.13 The mild reaction conditions and well‐defined functionality provide superior efficiency and simplicity compared to common methods for the functionalization of nitrenes.17, 18 In addition to efficiency and simplicity, a universal and affordable functionalization method should also involve readily available starting materials and provide reactive functional groups for flexible post‐modification. 2,4,6‐Trichloro‐1,3,5‐triazine is a cheap, commercially available compound and is highly reactive towards nucleophiles such as amines. The reaction between sodium azide and 2,4,6‐trichloro‐1,3,5‐triazine at low temperature results in 2‐azido‐4,6‐dichloro‐1,3,5‐triazine, which is a precursor to the highly reactive nitrene intermediates.19 Inspired by perfluoroazides,20, 21 the lifetime of these electron‐poor nitrenes could be long enough to conjugate to the π‐system of graphene through a [2+1] cycloaddition reaction.
Herein, dichlorotriazine‐functionalized TRGO (TRGO‐Trz) was successfully synthesized by the reaction of TRGO with 2‐azido‐4,6‐dichloro‐1,3,5‐triazine (Figures 1 and S1). The nitrogen content of TRGO‐Trz was determined by elemental analysis to calculate the degree of functionalization (DF; triazine/carbon atoms, see the Supporting Information, page S12). Since NMP interacts with the surface of graphene strongly,22 control experiments were performed to subtract any nitrogen content originating from the solvent (Supporting Information, page S7). To increase the DF of TRGO‐Trz, it was repeatedly functionalized up to three times by nitrene [2+1] cycloaddition reactions. TRGO functionalized once, twice, or three times are denoted as TRGO‐Trz1, TRGO‐Trz2, and TRGO‐Trz3, respectively (Table S1). Subsequent functionalizations dramatically increase the DF of TRGO‐Trz from 1:50 to 1:25 after three runs (Table S2). The DF values of TRGO‐Trz1, TRGO‐Trz2, and TRGO‐Trz3 that were calculated on the basis of mass loss in the thermogravimetric analysis (TGA) diagrams at 450 °C are in good agreement with the obtained elemental analysis results (Table S2 and Figure S2). The correlation between the DF and the number of reactions allows this method to be considered as a controlled covalent functionalization.
Figure 1.
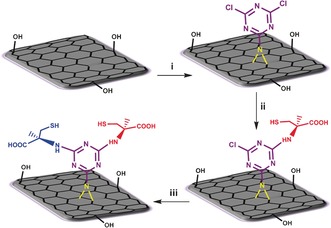
Functionalization of TRGO by a nitrene [2+1] cycloaddition reaction and controlled post‐modification of the product (TRGO‐Trz) by stepwise attachment of l‐ and d‐Cys to its triazine functional groups. i) 2,4,6‐Trichloro‐1,3,5‐triazine, NaN3, N‐methyl‐2‐pyrrolidone (NMP), TRGO, 0–60 °C, 24 h. ii) l‐Cysteine (l‐Cys), Et3N, DMF, 25 °C, 48 h. iii) d‐Cysteine (d‐Cys), Et3N, DMF, 60 °C, 48 h (further details about TRGO and its source can be found in the Supporting Information, page S3).
The binding energy (BE) range between 285 and 289 eV in the highly resolved C1s XPS spectrum is assigned to contributions of the new dichlorotriazine groups containing C−N and C−Cl bonds (Figures 2 a and S5).23 The stepwise increase in the intensity of these component peaks from TRGO‐Trz1 to TRGO‐Trz3 supports the hypothesis of an increase in the DF after repetitive runs. The C K‐edge NEXAFS spectrum for pristine TRGO surfaces is displayed in Figure 2 b (and Figure S3). The spectra arise from transitions from the C1s orbital to various empty, bound final states. At the low‐energy side, the NEXAFS spectrum shows a sharp resonance at a photon energy of 285.4 eV (d), which corresponds to a C1s→π* transition.22, 24, 25 After functionalization of TRGO, all the TRGO‐related resonances are found again in the C K‐edge spectrum. New resonances (e–g) related to the dichlorotriazine groups and their covalent bonds to TRGO occur in the “fingerprint” region at photon energies between 286 and 290 eV. The intensities of these resonances increase from TRGO‐Trz1 to TRGO‐Trz3, thus revealing an improved DF. At the same time, the intensity of the TRGO‐related π*‐resonance decreases. This can be explained by a consumption of sp2 carbon bonds during the reaction and the formation of other types of carbon bonds, which are represented by the new resonance features (e–g) in the “fingerprint” region.25
Figure 2.
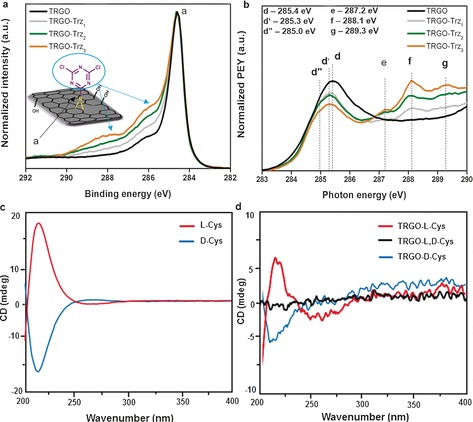
XPS spectra of TRGO‐Trz. a) Highly resolved C1s XPS spectra of TRGO, TRGO‐Trz1, TRGO‐Trz2, and TRGO‐Trz3. b) Expanded low‐energy section of the C K‐edge NEXAFS image of pristine TRGO, TRGO‐Trz1, TRGO‐Trz2, and TRGO‐Trz3 (for assignments and full spectra, see Supporting Information Figures S4 and S5, and Table S3 and S4). c) Circular dichroism (CD) spectra of l‐Cys and d‐Cys showing peaks at 210–220 nm. d) CD spectra of TRGO‐l‐Cys (red), TRGO‐l,d‐Cys (black), and TRGO‐d‐Cys (blue).
TRGO‐Trz can be used as a platform with a defined DF. Furthermore, it is expected to have a similar chemical reactivity as dichlorotriazine derivatives.26 Stepwise nucleophilic substitution of the dichlorotriazine groups results in the formation of well‐defined bifunctional 2D nanomaterials (Figure 1).
To prove the controlled bifunctionalization, l‐ and d‐cysteine (l‐Cys and d‐Cys) were conjugated selectively and in a stepwise manner to TRGO‐Trz1 by tuning the temperature of the nucleophilic reactions (Figure 1). Based on the XPS results and elemental analysis, the ratio of cysteine molecules to TRGO carbon atoms for TRGO‐l‐Cys and TRGO‐l,d‐Cys was approximately 1:47 and 1:22, respectively (Figures S6 and S7, and Table S5). This clearly indicates a quantitative substitution of the first and second chlorine atoms of TRGO‐Trz1 by controlled post‐modification (Supporting Information, page S32 and Figure S13). Since they are enantiomers, substitution of the first chlorine atom of the dichlorotriazine groups by l‐Cys should produce chiral 2D nanomaterials.27 Meanwhile, substitution of the second chlorine atom would racemize the system. Accordingly, the controlled and stepwise modification of TRGO‐Trz1 by l‐ and d‐Cys is also demonstrated by the chiroptical properties of the synthesized 2D stereoisomers. The circular dichroism (CD) analysis shows characteristic peaks of l‐ and d‐Cys at 210–220 nm (Figure 2 c) for TRGO‐l‐Cys and TRGO‐d‐Cys, thus confirming successful conjugation of those amino acids to the TRGO‐Trz1 under ambient conditions (Figure 2 d). However, conjugation of the next enantiomer to those 2D stereoisomers results in a racemized 2D nanomaterial without any special absorption in the CD spectra (Figure 2 d).
The utility of our method is further demonstrated by stepwise attachment of differently functionalized macromolecules, that is, azido‐polyglycerol (hPG‐N3) and polyglycerol amine (hPG‐NH2) or amino‐β‐cyclodextrin (β‐CD‐NH2) to TRGO‐Trz. To minimize the steric hindrance between the macromolecules, we synthesized a TRGO‐Trz with a low DF (TRGO‐TrzL). The synthesis and characterization of TRGO‐TrzL is explained in the Supporting Information (page S9, Tables S2 and S3, and Figure S8). Nucleophilic substitution of the first chlorine atom of TRGO‐TrzL by propargylamine at 25 °C resulted in a TRGO derivative suitable for subsequent click reactions (Figure 3 a). Accordingly, the click reaction between hPG‐N3 and propargylamine‐functionalized TRGO led to water‐dispersible 2D nanomaterials with low polymer coverage (TRGO‐hPGL, Figure 3 a). A subsequent nucleophilic reaction between TRGO‐hPGL and β‐CD‐NH2 or hPG‐NH2 at 60 °C afforded 2D nanomaterials with hPG/β‐CD dual functionality (TRGO‐hPGL‐CD) or fully coated by polyglycerol derivatives (TRGO‐hPGH), respectively (Figures 3 a, S14, and S12). The two‐step post‐modification of TRGO‐Trz by macromolecules has been evaluated by different thermal and spectroscopic methods (see Supporting Information pages 36–42). After a click reaction, the ratio of the XPS peak areas of C1s components related to polyglycerol and TRGO (ChPG/CTRGO) was 0.46:1, thereby suggesting that TRGO‐hPGL contains about 31 wt % polyglycerol and about 69 wt % TRGO carbon atoms (Figure S11). This result agrees with TGA data showing a 30 wt % polyglycerol content for TRGO‐hPGL (Figure S15b). In the XPS spectrum, the peak area ratio of C1s at 286.6 eV increased upon conjugation of β‐CD‐NH2 to TRGO‐hPGL. Since this peak is assigned to components with C−O bonds, it proves the successful attachment of cyclodextrin to graphene sheets in the second step of post‐modification (Figures 3 b and S12, and Table S3).
Figure 3.
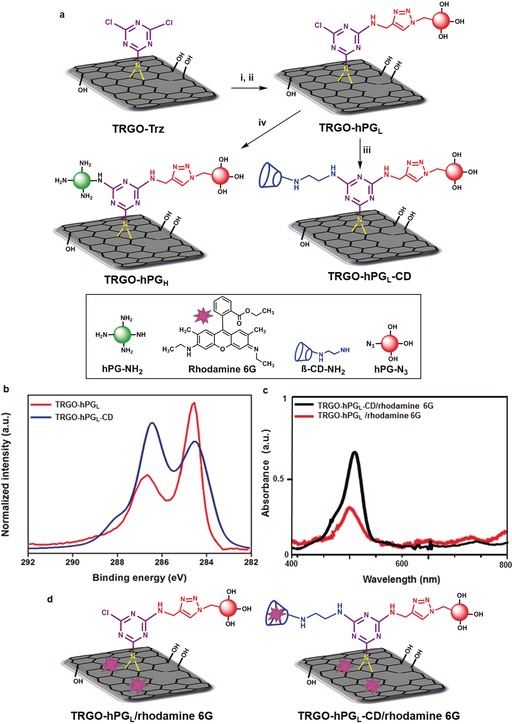
a) Stepwise attachment of polyglycerol and cyclodextrin to TRGO‐Trz. i) Propargylamine, DMF, Et3N, 25 °C, 24 h; ii) polyglycerol azide 4 %, sodium ascorbate, CuSO4⋅5 H2O, DMF, 50 °C, 24 h; iii) β‐CD‐NH2, Et3N, DMF, 60 °C, 12 h; iv) polyglycerol amine 50 %, Et3N, NMP, 60 °C, 48 h. b) Highly resolved C1s spectra of TRGO‐hPGL and TRGO‐hPGL‐CD (for assignments and full spectra see Figures S8–S11, and Tables S3 and S4). c) UV spectra of TRGO‐hPGL/rhodamine 6G and TRGO‐hPGL‐CD/rhodamine 6G. d) Host–guest interaction of TRGO‐hPGL/rhodamine 6G and TRGO‐hPGL‐CD/rhodamine 6G, Milli‐Q water, rhodamine 6G, 30 min incubation (see page S28 in the Supporting Information for loading capacity).
Furthermore, UV measurements showed that the loading capacity of TRGO‐hPGL‐CD with rhodamine 6G is 1.6 times higher than TRGO‐hPGL (Figure 3 c; Supporting Information page 28). As a consequence of the presence of the conjugated cyclodextrins, TRGO‐hPGL‐CD can deliver suitable guest molecules not only by π‐π staking but also through host–guest interactions (Figure 3 d).
In a second example of post‐modification, the nucleophilic reaction between TRGO‐hPGL and hPG‐NH2 results in a positively charged 2D nanomaterial (TRGO‐hPGH). In the XPS spectrum of TRGO‐hPGH, the ratio of the peak areas of C1s components related to polyglycerol and TRGO (ChPG/CTRGO) increased from 0.46:1 to 1.23:1 (Figure S11, Table S3). This clearly proves efficient conjugation of hPG‐NH2 to the carbon surface. The C K‐edge spectrum (Figure S10) shows that the intensity of the TRGO‐related π*‐resonance (j) decreases with increasing polymer coverage. This is due to the increased thickness of the TRGO with polyglycerol, which attenuates the electron signal originating from the TRGO substrate. The other prominent change in the C K‐edge NEXAFS is the occurrence of the resonance (n) at 289.4 eV, which is assigned to C−H* resonances.28 This is taken as proof of the successful synthesis of TRGO‐hPGL and TRGO‐hPGH. Scanning force microscopy (SFM) in tapping mode (TM) and quantitative nanomechanical mapping mode (QNM) were used to investigate the TRGO functionalization (Figure S16). SFM height images of the TRGO‐hPGL show spherical objects on the TRGO surface (Figures 3 a and 4 a). Both TRGO‐hPGL and TRGO‐hPGH have a height of (11±2) nm at 35–40 % (ambient) relative humidity (Figures 4 b,c). However, in the case of TRGO‐hPGH, the polyglycerol branches were tightly packed (Figure S18). SFM suggests an integrated polyglycerol layer with a height of 4–5 nm on both sides of the functionalized TRGO‐Trz, thereby leading to an overall thickness of 11–12 nm. Furthermore, sample deformation images obtained from QNM‐SFM provides complementary information that demonstrates the attachment of hPG to TRGO‐Trz. The softer regions are positions where spherical objects are observed. Regions with deformations of 1–2.5 nm under applied forces of 0.3–1 nn by the SFM tip are attributed in the case of TRGO‐hPGL to single or a few polyglycerol molecules (Figures S19a,b). The more deformable objects observed on TRGO‐hPGH are attributed to the tightly packed polyglycerol branches (Figures S19c,d).
Figure 4.
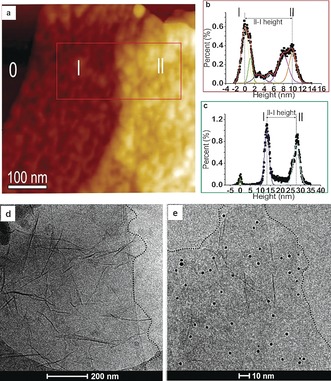
a) TM‐SFM height image of mica (0), covered with single (l) and double (ll) layers of TRGO‐hPGL. b) Height histogram of single (l) and double (ll) layered TRGO‐hPGL. c) Height histogram of single (l) and double (ll) layered TRGO‐hPGH (Figure S18). d) Cryo‐TEM image of TRGO‐hPGL and e) TRGO‐hPGH after incubation with gold nanoparticles (sheet boundaries are highlighted by dotted lines).
Finally, the ability of TRGO‐hPGH to function as a multivalent platform to bind gold nanoparticles was investigated. Although no specific interactions between negatively charged gold nanoparticles (AuNPs) and TRGO‐hPGL were observed in phosphate buffered saline (PBS buffer, Figure 4 d), the immobilized AuNPs (ca. 5 nm) can be detected on the surface of TRGO‐hPGH in stereoscopic cryo‐TEM image pairs (Figures 4 e and S21). This is due to the electrostatic interactions between AuNPs and positively charged polyglycerolamine branches that are attached to the surface of TRGO‐hPGH (Figure S15a).
In conclusion, we have developed a simple and mild method for the controlled covalent functionalization of TRGO by an in situ nitrene cycloaddition reaction using commercially available reagents. The functionalized graphene sheets can be transformed into defined 2D surfaces by stepwise post‐modification in high yields. The advantage of this method is the construction of bifunctional carbon nanomaterials with controlled ligand densities, which even allows the conjugation of sensitive bio(macro)molecules at room temperature.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the German Science Foundation (DFG) for financial support within the grants SFB 658 and SFB 765 as well as the Cluster of Excellence “Image Knowledge Gestaltung”. We would like to acknowledge Dieter Treu for operating the XPS instrument at BAM and Guy Guday for careful proof‐reading of the manuscript. We also acknowledge support by the team at the BESSY II synchrotron radiation facility as well as Dr. A. Nefedov (Karlsruhe Institute of Technology, KIT) from the HE‐SGM Collaborate Research Group.
A. Faghani, I. S. Donskyi, M. Fardin Gholami, B. Ziem, A. Lippitz, W. E. S. Unger, C. Böttcher, J. P. Rabe, R. Haag, M. Adeli, Angew. Chem. Int. Ed. 2017, 56, 2675.
Contributor Information
Prof. Dr. Rainer Haag, Email: haag@chemie.fu-berlin.de
Prof. Dr. Mohsen Adeli, Email: aadeli@fu-berlin.de
References
- 1. Geim A. K., Novoselov K. S., Nat. Mater. 2007, 6, 183–191. [DOI] [PubMed] [Google Scholar]
- 2. Geim A. K., Science 2009, 324, 1530–1534. [DOI] [PubMed] [Google Scholar]
- 3. Zheng Z., Nottbohm C. T., Turchanin A., Muzik H., Beyer A., Heinemann M., Sauer M., Golzhauser A., Angew. Chem. Int. Ed. 2010, 49, 8493–8497; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8671–8675. [Google Scholar]
- 4. Toth P. S., Ramasse Q. M., Velický M., Dryfe R. A. W., Chem. Sci. 2015, 6, 1316–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chua C. K., Sofer Z., Luxa J., Pumera M., Chem. Eur. J. 2015, 21, 8090–8095. [DOI] [PubMed] [Google Scholar]
- 6. Criado A., Melchionna M., Marchesm S., Prato M., Angew. Chem. Int. Ed. 2015, 54, 10734–10750; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10882–10900. [Google Scholar]
- 7. Eigler S., Hirsch A., Angew. Chem. Int. Ed. 2014, 53, 7720–7738; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7852–7872. [Google Scholar]
- 8. Li J., Li M., Zhou L. L., Lang S. Y., Lu H. Y., Wang D., Chen C. F., Wan L. J., J. Am. Chem. Soc. 2016, 138, 7448–7451. [DOI] [PubMed] [Google Scholar]
- 9. Knirsch K. C., Schafer R. A., Hauke F., Hirsch A., Angew. Chem. Int. Ed. 2016, 55, 5861–5864; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5956–5960. [Google Scholar]
- 10. Paulus G. L. C., Wang Q. H., Strano M. S., Acc. Chem. Res. 2013, 46, 160–170. [DOI] [PubMed] [Google Scholar]
- 11. Hirsch A., Englert J. M., Hauke F., Acc. Chem. Res. 2013, 46, 87–96. [DOI] [PubMed] [Google Scholar]
- 12. Vadukumpully S., Gupta J., Zhang Y., Xu G. Q., Valiyaveettil S., Nanoscale 2011, 3, 303–308. [DOI] [PubMed] [Google Scholar]
- 13. Park J., Yan M., Acc. Chem. Res. 2013, 46, 181–189. [DOI] [PubMed] [Google Scholar]
- 14. Gao C., He H., Zhou L., Zheng X., Zhang Y., Chem. Mater. 2009, 21, 360–370. [Google Scholar]
- 15. He H., Gao C., J. Am. Chem. Soc. 2010, 132, 5054–5064.20307064 [Google Scholar]
- 16. Han J., Gao C., Nano-Micro Lett. 2010, 2, 213–226. [Google Scholar]
- 17. Liu L. H., Yan M., Nano Lett. 2009, 9, 3375–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu L. H., Lerner M. M., Yan M., Nano Lett. 2010, 10, 3754–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bucher G., Siegler F., Wolff J. J., Chem. Commun. 1999, 2113–2114. [Google Scholar]
- 20. Platz M. S., Acc. Chem. Res. 1995, 28, 487–492. [Google Scholar]
- 21. Park J., Jayawardene H. S. N., Chen X., Jayawardana K. W., Sundhoro M., Ada E., Yan M., Chem. Commun. 2015, 51, 2882–2885. [DOI] [PubMed] [Google Scholar]
- 22. Dubin S., Gilje S., Wang K., Tung V. C., Cha K., Hall A. S., Farrar J., Varshneya R., Yang Y., Kaner R. B., ACS Nano 2010, 4, 3845–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yuan B., Bao C., Song L., Hong N., Liew K. M., Hu Y., Chem. Eng. J. 2014, 237, 411–420. [Google Scholar]
- 24. Batson P. E., Phys. Rev. B 1993, 48, 2608–2610. [DOI] [PubMed] [Google Scholar]
- 25. Ehlert C., Unger W. E. S., Saalfrank P., Phys. Chem. Chem. Phys. 2014, 16, 14083–14095. [DOI] [PubMed] [Google Scholar]
- 26. Apen E., Hitchcock A. P., Gland J. L. J., Phys. Chem. 1993, 97, 6859–6866. [Google Scholar]
- 27. Suzuki N., Wang Y., Elvati P., Qu Z. B., Kim K., Jiang S., Baumeister E., Lee J., Yeom B., Bahng J. H., Lee J., Violi A., Kotov N. A., ACS Nano 2016, 10, 1744–1755. [DOI] [PubMed] [Google Scholar]
- 28. Principles S. J., Techniques, and Instrumentation of NEXAFS, Springer, Berlin, 1992. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary