

Summary
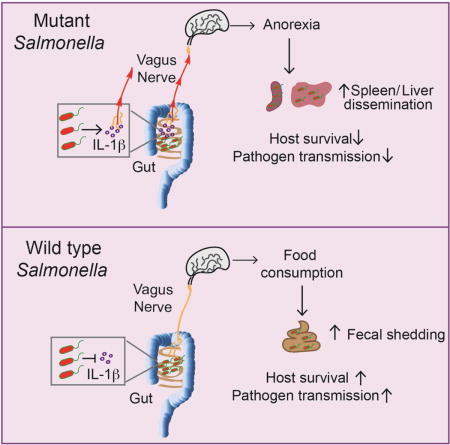
Sickness-induced anorexia is a conserved behavior induced during infections. Here, we report that an intestinal pathogen, Salmonella Typhimurium, inhibits anorexia by manipulating the gut-brain axis. Inhibition of inflammasome activation by the S. Typhimurium effector, SlrP, prevented anorexia caused by IL-1β mediated signaling to the hypothalamus via the vagus nerve. Rather than compromising host defenses, pathogen-mediated inhibition of anorexia increased host survival. SlrP-mediated inhibition of anorexia prevented invasion and systemic infection by wild-type S. Typhimurium, reducing virulence while increasing transmission to new hosts, suggesting there are trade-offs between transmission and virulence. These results clarify the complex and contextual role of anorexia in host defense and suggest that microbes have evolved mechanisms to modulate sickness-induced behaviors to promote health of their host and their transmission at the expense of virulence.
Keywords: sickness-induced anorexia, pathogen transmission, virulence, Salmonella, inflammasome, IL-1β, vagus nerve
Graphical abstract
Introduction
Infections trigger stereotypical behavioral changes in the host including anorexia, fever, sleep disturbances, social withdrawal and changes in grooming, collectively referred to as “sickness behaviors” (Dantzer, 2009; Hart, 1988). These behaviors have been proposed to be adaptive strategies to increase the chance of survival from acute illness (Exton, 1997; Hart, 1988; Kyriazakis et al., 1998). Animal studies have suggested that any benefits conferred by anorexia caused by infection are context dependent. In mice systemically infected with Listeria monocytogenes, animals that were force fed succumbed to infection more rapidly than animals that were allowed to develop the anorexic response (Murray and Murray, 1979). However, in Drosophila, anorexia was maladaptive for surviving an L. monocytogenes systemic infection (Ayres and Schneider, 2009). Because many biomedical practices interfere with sickness-induced behaviors, it is important to understand the mechanisms that lead to the induction of these behaviors and the contexts in which they are beneficial or detrimental for the host.
Whether sickness-induced anorexia is beneficial for the host is largely dependent on how the fasted state functionally influences host resistance and tolerance defenses (Medzhitov et al., 2012; Schneider and Ayres, 2008). Acute starvation and diet restriction studies have primarily focused on how nutrition influences resistance mechanisms (Bedoyan et al., 1992; Dunn et al., 1994). In contrast to expectations, food restriction had a negative impact on the outcome of infection for the host (Burger et al., 2007; Kristan, 2007; Libert et al., 2008; Ritz et al., 2008; Sun et al., 2001). However, in a fruit fly model, Drosophila that developed anorexia or were diet restricted during systemic Salmonella infection lived significantly longer despite equivalent pathogen levels compared to infected flies fed a normal diet (Ayres and Schneider, 2009). More recently, anorexia promoted tolerance in mice infected with L. monocytogenes, indicating that in these contexts, the fasted state promoted tolerance defenses (Wang et al., 2016).
Pathogens and the microbiota are dependent on energy intake of their hosts during infection. Whether anorexia will have a beneficial effect on host outcome will also likely be determined by how microbial virulence is affected under fasted states. The effects of anorexia on pathogen virulence have been most theorized as creating a less hospitable niche, starving pathogens from essential nutrients required for replication (Hart, 1988). Although anorexia may have some positive and undefined effects on inhibiting pathogen growth, it is equally plausible that the fasted state may trigger increased virulence by altering microbial behavior independent of any effects on microbial growth. Microbes vary in their metabolic capacity and foraging strategies to adjust to situations in which nutrients are scarce (Ayres, 2016). Acute starvation in humans is associated with an increased risk of invasive bacterial infections (Page et al., 2013). While this increased risk has been assumed to be dependent on the poor immune status of the patient due to malnutrition (Gordon et al., 2012), it is possible that the increased invasiveness and virulence may potentially reflect an adaptive strategy of certain microbes under nutrient limiting conditions. Thus, the discrepancies shown in previous studies regarding any benefits afforded by sickness-induced anorexia for the host may in part be due to microbe-induced behavioral changes caused by food restricted conditions that lead to increased virulence of infecting pathogens, the resident microbiota or both.
Salmonella enterica serovar Typhimurium (ST) is a gram-negative bacterium that causes enteric and systemic typhoid-like diseases in diverse animal models and humans. Upon oral infection in mice, ST penetrates the gut epithelial layer typically by invasion of specialized epithelial cells called M cells (Monack et al., 2004). Once in the lamina propria (LP), bacteria are engulfed by innate immune cells including neutrophils and macrophages, and infection induces the infiltration of T and B cells (Monack et al., 2004). The SPI-I type 3 secretion system (T3SS) and associated effectors are important for the gut stage of ST infection (Galan, 1996). In adapted salmonellosis such as typhoid fever, with the aid of a second T3SS, SPI-2 and associated effectors (Hensel et al., 1998), the bacteria disseminate via the lymphatics and bloodstream to systemic organs, including the spleen and liver. The gut phase of salmonellosis in mice induces an anorexic response in the host (Schieber et al., 2015), and this pathogen can be transmitted to new mice via the fecal-oral route. As ST has evolved mechanisms to manipulate diverse physiological aspects of the mouse, a natural host of ST, it is an excellent pathogen to dissect the relationship among anorexia, host health, pathogen virulence and transmission.
Here we examined how sickness-induced anorexia affected infection-induced lethality in a transmissible model of infection. We found that the ST effector, Salmonella leucine rich repeat protein (SlrP) negatively regulated virulence and promoted survival of the host by inhibiting the infection-induced anorexic response. We found that SlrP inhibited inflammasome activation and IL-1β maturation in the small intestine (SI), preventing the anorexic feeding program in the hypothalamus that is dependent on the vagus nerve. Failure to inhibit IL-1β and the anorexic response resulted in increased extra-intestinal dissemination and increased virulence of the pathogen but at the expense of pathogen transmission to new hosts. Our study provides mechanistic aspects for how local tissue response to microbes can signal to the brain to induce anorexia and demonstrates that microbes have evolved anti-virulence strategies that inhibit sickness-induced anorexia to promote host survival and pathogen transmission.
Results
A Salmonella effector promotes survival of its host
In ongoing efforts to identify microbial factors that promote host health, we tested the importance of the ST effector, SlrP, in regulating host health during oral infection. SlrP is a member of the novel E3 ubiquitin ligases (NEL) class, a class of ubiquitin ligases that are encoded by some bacteria (Maculins et al., 2016; Miao et al., 1999; Tsolis et al., 1999). The function of NELs and other prokaryotic ubiquitin ligases is traditionally thought to promote virulence during infection (Maculins et al., 2016). However because commensal bacteria and microbes that can colonize the host asymptomatically, including Salmonella species (Boyer and Lemichez, 2004), also encode mechanisms to interact with the host ubiquitin system, we hypothesized that SlrP may be important for negatively regulating virulence. Specific pathogen free (SPF) C57Bl/6 (B6) mice orally infected with a ST SL1344 strain deficient in slrP (ΔslrP) exhibited faster death kinetics than mice infected with the parental ST SL1344 strain (wildtype, wt) (Figure 1A). ΔslrP-infected animals exhibited a median time to death of seven days post-infection, while ~70% of mice infected with the parental strain were alive at day 10 post-infection (Figure 1A). The increased death kinetics of ΔslrP-infected mice was associated with increased weight loss (Figure 1B, C). We previously showed that ST infection in B6 mice causes wasting of lean and fat stores (Schieber et al., 2015). We found that while ΔslrP-infected mice had slightly higher lean body mass (Figure 1D), they exhibited significantly more wasting of adipose tissue stores compared to wt-infected mice (Figure 1E). These data suggest that SlrP function is necessary for controlling ST virulence and sustaining health of the host during infection in vivo of B6 mice.
Figure 1. A Salmonella effector promotes survival of its host.
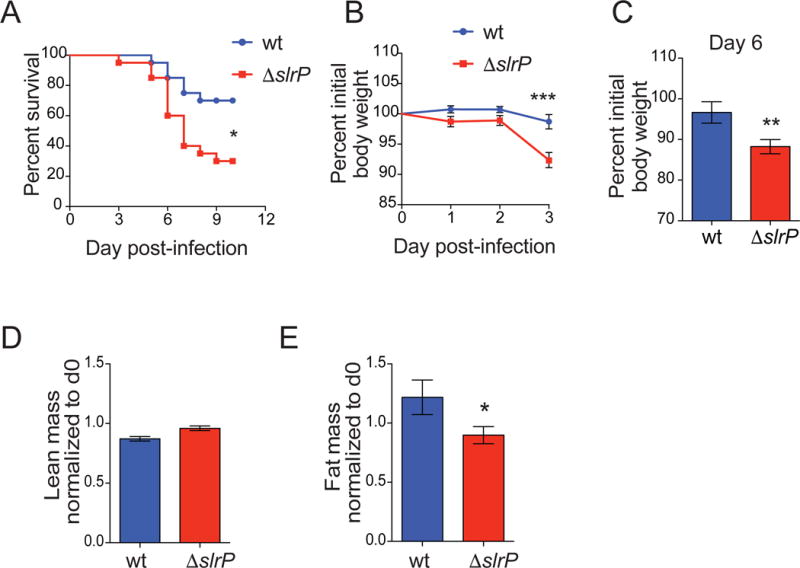
(A) Survival of B6 mice orally infected with wt (n=20) or ΔslrP (n=20) ST. Data represent 2 independent experiments combined.
(B–C) Weight loss of B6 mice orally infected with wt or ΔslrP ST on days 1–3 (B) and day 6 (C) post-infection. n=18/group. Data represent 3 independent experiments combined
(B). n=12 wt-infected and n=17 ΔslrP-infected mice (C).
(D–E) MRI analysis of lean (D) and fat mass (E) of B6 mice orally infected with wt (n=9) or ΔslrP (n=10) ST for 72hr. Measurements at 72hr are normalized to those taken on day 0 for each mouse. Data represent 2 independent experiments combined.
*** indicates p < 0.001, ** indicates p < 0.01, * indicates p<0.05 by unpaired student’s t test or Log rank analysis (survival). Error bars +/− SEM.
Inhibition of the sickness-induced anorexia response controls virulence
We performed an in-depth metabolic characterization of infected animals using the Comprehensive Laboratory Animal Monitoring System (CLAMS) and found that mice infected with the ΔslrP strain exhibited differences in the rate of oxygen consumption (VO2, Figure S1A) and CO2 production (VCO2, Figure S1B), resulting in a decreased respiratory exchange ratio (RER) compared to mice infected with wt ST (Figure 2A). In agreement with increased wasting of adipose tissue, lower RER suggests that ΔslrP-infected mice preferentially utilize fat as an energy substrate resulting in increased lipid oxidationat and depletion of fat stores, while wt-infected mice with higher RER use carbohydrates as the preferential fuel source (Schmidt-Nielsen, 1997).
Figure 2. Inhibition of the sickness-induced anorexia response controls virulence.
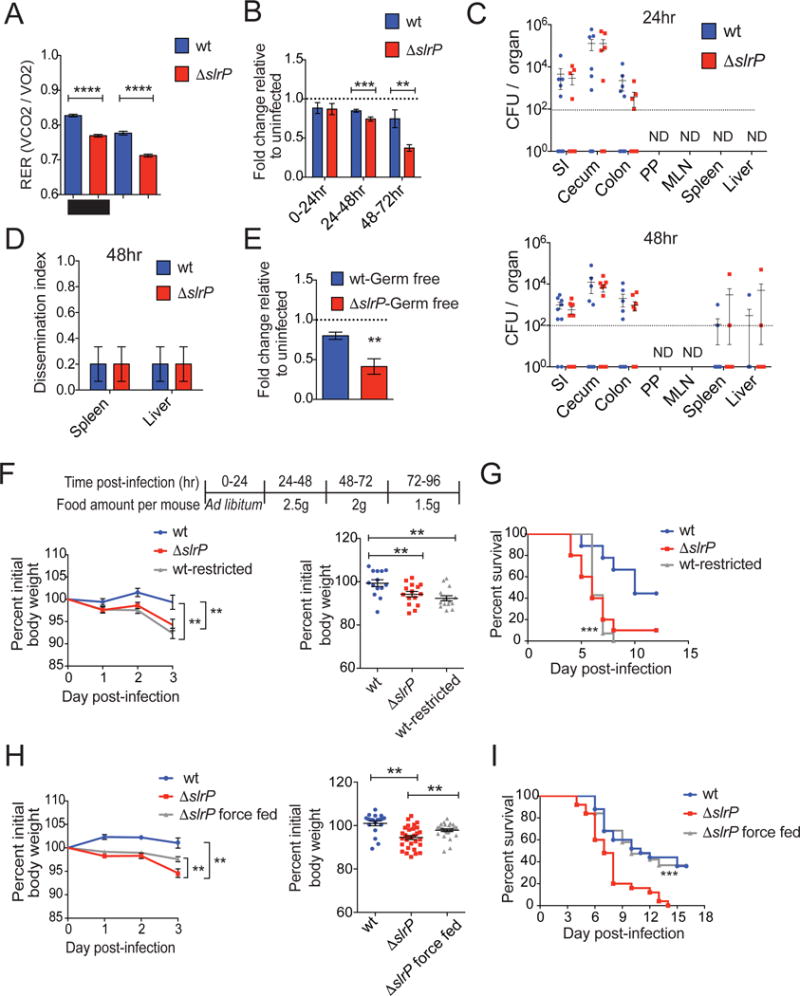
(A) RER at 72hr post-infection of B6 mice orally infected with wt or ΔslrP ST. Black bar indicates dark/night cycle, and white bar indicates light/day cycle. n=5/group.
(B) Food consumption of B6 mice orally infected with wt or ΔslrP ST. Quantification is expressed as a ratio of grams of food per animal at each timepoint to that of vehicle (PBS)-gavaged animals. 0–24hr: n=30 mice/group; 24–48hr: n=30 mice/group; 48–72hr: n=15 mice/group. Data represent 3 experiments combined.
(C) CFU in indicated tissues of B6 mice orally infected with wt or ΔslrP ST for 24hr (top) and 48hr (bottom). n=10–16 mice/group. Data represent 2 independent experiments combined. Dotted line indicates limit of detection and mice with no CFU detected are indicated by symbols below this line. ND indicates none detected.
(D) Animals from (C) were assigned a dissemination score of “1” (at least 1 CFU at the level of detection in indicated systemic organ) or “0” (no CFU detected in indicated systemic organ) 48hr post-infection. n=10/group
(E) Food consumption of germ-free B6 mice orally infected with wt (n=12) or ΔslrP (n=14) ST for 24hr. Grams of food per animal normalized to uninfected consumption value of germ-free mice. Data combined from 2 independent experiments.
(F) Weight loss from days 1–3 of wt ST infection (n=14), ΔslrP ST infection (n=15), and wt ST infection during which food consumption was restricted (n=15) as indicated in table. Right graph shows weight loss on day 3 post-infection from left graph. Data represent 2 independent experiments combined.
(G) Survival of mice from (F). ***indicates p=0.0006 between wt-infected and wt-infected food-restricted mice by Log rank analysis.
(H) Weight loss from days 1–3 of wt ST infection (n=19), ΔslrP ST infection (n=30) and ΔslrP ST infection during which mice were force fed twice daily in addition to ad libitum feeding (n=29). Right graph shows weight loss on day 3 post-infection from left graph. Data represent 3 independent experiments combined.
(I) Survival of mice from (H). ***indicates p=0.0012 between ΔslrP-infected mice and ΔslrP-infected force fed mice by Log rank analysis.
****p<0.0005, ***p<0.01, **p<0.05 by unpaired student’s t test. Error bars indicate +/−SEM. See Figure S1.
We hypothesized that nutrient availability may be compromised in ΔslrP-infected mice due to the development of a more severe anorexic response in the absence of SlrP function. Consistent with our previous findings (Schieber et al., 2015), we found that mice infected with wt ST exhibited an anorexic response (Figure 2B). However, we found that ΔslrP-infected mice exhibited a more severe anorexic response compared to wt-infected mice that was apparent within the first 24hrs post-infection and became more severe by 24–48hrs post-infection (Figure 2B). Between 0–24hrs post-infection, ΔslrP-infected mice consumed ~6% less food compared to wt infected animals and this difference increased to ~20% less food by 24–48hrs post-infection (Figure 2B). Expression of a wt copy of slrP under the native promoter in the ΔslrP mutant protected from infection induced anorexia (Figure S1C–D). These data raise the possibility that the more severe anorexic response results from increased pathogen burden, indicating the ΔslrP strain may grow faster and/or is cleared less efficiently than the parental strain. However, we found that at both 0–24 and 24–48hrs post-infection, the timeframes in which we observe the onset of the more severe anorexic response in ΔslrP-infected mice (Figure 2B), that the pathogen burdens in wt- and ΔslrP-infected mice were comparable in all target tissues of this pathogen (SI, cecum, colon, Peyer’s patches [PP], MLN, liver and spleen) (Figure 2C). We further found at 48hrs post-infection the percentage of hosts with an extraintestinal dissemination event to be identical between ΔslrP- and wt-infected B6 mice (Figure 2D). Another possibility is that ΔslrP infection induces perturbations in the intestinal microbiota, resulting in a more pathogenic microbiota that leads to increased anorexia. Similar to our findings with SPF mice, germ-free mice monoinfected orally with ΔslrP exhibited a more severe anorexic response compared to germ-free mice that were monoinfected orally with the parental strain (Figure 2E), consuming approximately 50% less food within the first 24hrs post infection. Thus the more severe anorexic response observed in ΔslrP-infected mice is not due to increased pathogen burdens, differences in pathogen tissue tropism, dissemination or differential responses of the intestinal microbiota.
To determine if the host nutrient status was related to pathogen virulence, we compared infection of food-restricted wt-infected animals to that of wt-infected mice that were fed ad libitum. Food-restricted wt-infected mice exhibited increased wasting that was associated with increased death kinetics compared to wt-infected mice that were fed ad libitum (Figure 2F, G). This was specific to infection, as uninfected food-restricted animals exhibited no mortality (Figure S1E–F). We next tested whether force feeding could dampen ΔslrP virulence in B6 mice. ΔslrP-infected mice force fed a liquefied diet during infection were protected from weight loss and had increased survival comparable to that of wt-infected B6 mice fed ad libitum (Figure 2H, I). Taken together, our data suggest that reduced nutrient intake leads to increased virulence of ST oral infection. Since the protection against anorexia mediated by SlrP was neither due to regulation of pathogen burdens nor regulation of the pathogenicity of the intestinal microbiota, we hypothesized that SlrP interacts directly with host physiologies that negatively regulate the sickness-induced anorexic response and in doing so, regulates ST virulence.
SlrP prevents anorexia and regulates virulence through the inhibition of IL-1β
Protection from anorexia without a change in pathogen infection levels suggests that SlrP can negatively regulate ST virulence through 1) the inhibition of host processes that promote the anorexic response or 2) through the induction of a disease tolerance mechanism that promotes feeding. To distinguish between these two possibilities, we asked how the absence of SlrP function during ST infection influences the canonical mediators of sickness-induced anorexia. Using the CLAMS monitoring system, we found equivalent activity levels between ΔslrP- and wt-infected mice indicating that ΔslrP-infected mice are not more anorexic because they are less mobile (Figure 3A). Although we found no difference in the systemic levels of pathogen between mice infected with either strain at the onset of the more severe anorexic response (0–24 and 24–48hrs), we considered the hypothesis that the absence of SlrP rendered ST more stimulatory of an anorexic response. However, systemic challenge of B6 mice with either ΔslrP or wt ST resulted in an equivalent degree of anorexia (Figure 3B), indicating that ΔslrP is not more stimulatory of an anorexic response when systemic. Consistent with this and our burden analyses (Figure 2C), we found no significant difference in the extent of tissue pathology in ST target tissues liver, spleen and SI, between mice infected with wt or ΔslrP ST (Figure 3C and S2A). Thus SlrP does not inhibit anorexia by alleviating tissue damage in target organs of this pathogen. SlrP has been shown in in vitro assays to inhibit dendritic cell (DC) migration (McLaughlin et al., 2014). We found no difference in the levels of DCs or other immune cells associated with ST infection or intestinal inflammation in the LP, MLNs, liver or spleen between 0–24 and 24–48hrs post-infection, indicating that SlrP does not inhibit anorexia by limiting immune cell infiltration (Figure 3D, E and S2B–H). Additionally, levels of TNFα and IL-6, cytokines that can modulate sickness-induced behavioral changes, (Dantzer, 2009), in ST target tissues were not higher in ΔslrP-infected mice at 24 and 48hrs post-infection (Figure 3F, G, S2I and data not shown). Taken together, these data suggest that SlrP does not inhibit the anorexic response by alleviating the primary cause of disease (tissue damage), inhibit the induction of TNFα or IL-6, nor inhibit immune cell infiltration during infection.
Figure 3. Canonical mediators of sickness-induced anorexia are equivalent between wt and ΔslrP ST.
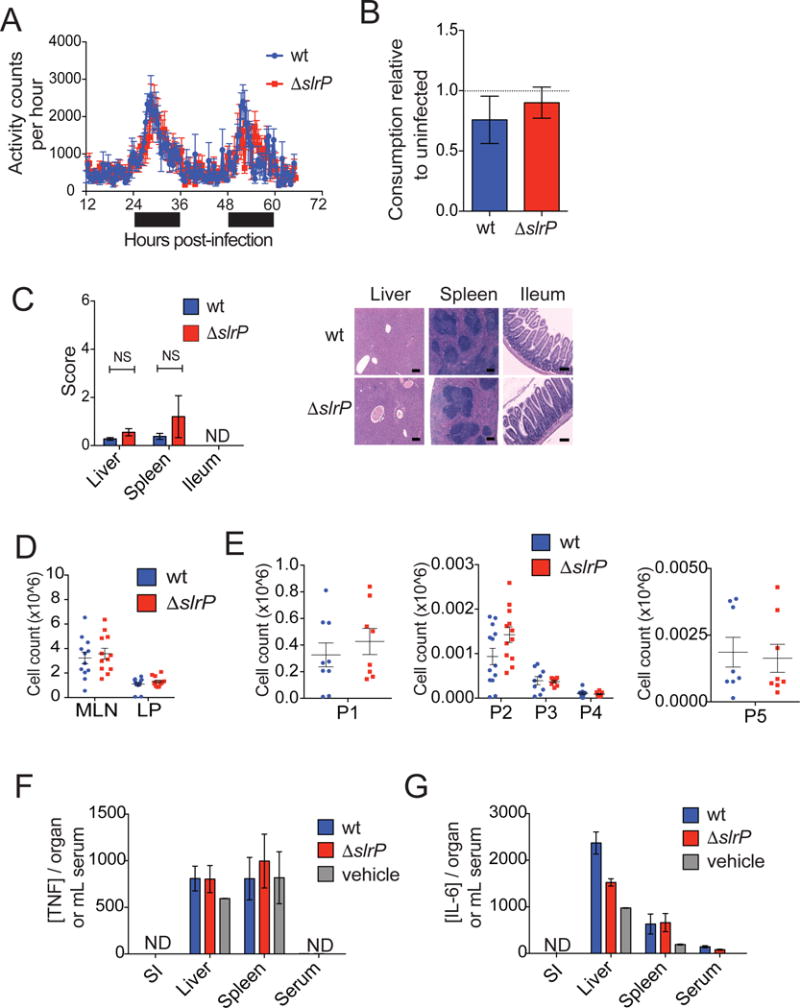
(A) Activity of B6 mice orally infected with wt or ΔslrP ST. Black bar indicates dark/night cycle, and white bar indicates light/day cycle. n=6 mice/group.
(B) B6 mice were infected i.p. with wt or ΔslrP ST, and food consumption was measured every 24hrs post-infection and normalized to consumption of uninfected animals. Grams of food per animal consumed within the first four days of infection shown. n=6/group.
(C) H&E staining and histological scoring of spleen, liver, and ileum 24hr after oral infection of B6 mice with wt or ΔslrP ST. ND indicates pathology above limit of detection not detected. NS indicates not significant. Scale bar indicates 200μm. n=5/group.
(D) Total cellularity of MLN and SI LP 48hr after oral infection of B6 mice with wt or ΔslrP ST. n=12–13/group. Data represent 3 independent experiments combined.
(E) Numbers of inflammatory cell populations 48hr after oral infection of B6 mice with wt or ΔslrP ST. LP: T/B lymphocytes (P1, n=8–9/group), neutrophils (P2, n=13/group), phagocytic macrophages (P3, n=8–9/group), inflammatory monocytes (P4, n=8–9/group); MLN: migratory DCs (P5, n=8/group). Data represent 3 independent experiments combined.
(F–G) ELISA measurements of TNFα (F) and IL-6 (G) in indicated tissues 48hr after oral infection of B6 mice with wt or ΔslrP ST. n=4–10/group. ND denotes Not Detected. Vehicle indicates PBS. Error bars indicate +/− SEM. See Figure S2.
Systemic IL-1β is believed to be the predominant mediator of sickness-induced anorexia (Dantzer, 2009). We did not detect any differences in the levels of serum, liver or spleen IL-1β between ΔslrP- and wt-infected mice within the first 48hrs of infection (Figure 4A, S3A). We did, however, find significantly increased levels of IL-1β in the SI of ΔslrP-infected mice compared to wt-infected mice within the first 48hrs of infection, which correlates with the onset of the more severe anorexic response observed in these mice (Figure 4A). This increased IL-1β was specific to the SI because IL-1β levels of other tissues of the digestive tract including the stomach, cecum, colon, pancreas, PP and MLN were not significantly different between wt and ΔslrP-infected mice (Figure S3B and data not shown). Expression of a wt copy of slrP under the native promoter in the ΔslrP mutant prevented increased levels of IL-1β in the SI of infected mice (Figure S1D and S3C).
Figure 4. ST SlrP prevents anorexia and regulates virulence through the inhibition of IL-1β.
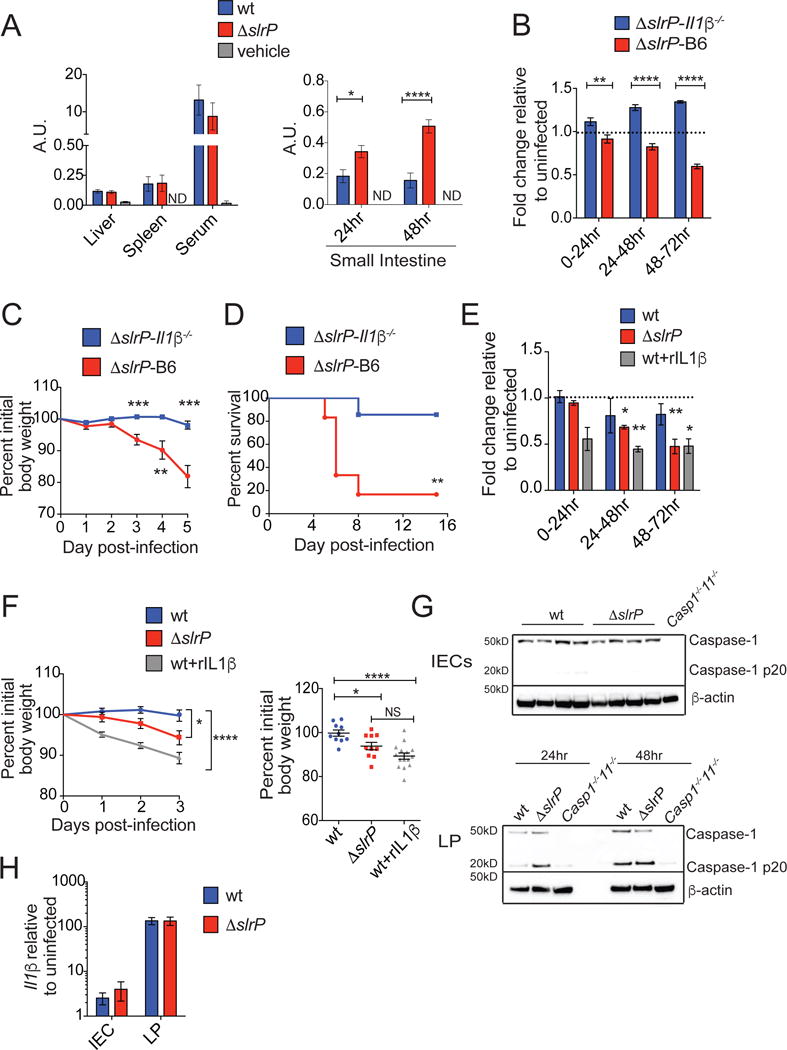
(A) Levels of mature IL-1β were determined by IL-1R reporter assay of whole organ homogenates in liver, spleen and serum of B6 mice 48hr post-infection (left, n=4–10/group) and in SI (right; n=7 vehicle-gavaged, n= 24 wt-infected and n= 24 ΔslrP-infected mice). Graph shown depicts reporter fluorescence (arbitrary units), indicative of active IL-1β. Data represent 2 experiments combined.
(B–D) Food consumption (B), weight loss (C), and survival (D) during ΔslrP infection of B6 (n=6–20) and Il1β−/− (n=7–15) mice. (B) Grams of food per animal was normalized to consumption of uninfected animals of appropriate genotype. Data represent 2 experiments combined.
(E–F) B6 mice were injected with PBS (n=10–20 wt-infected mice; n=10–20 ΔslrP-infected mice) or rIL-1β (0.05μg/gram mouse, n=15 wt-infected mice). Food consumption (E) and weight loss (F) were measured. (E) Data represent 2–3 experiments combined.
(G) Western blot analysis of CASPASE-1 cleavage (p20) in IECs (top) and LP leukocytes (bottom) isolated from infected mice. Leukocytes were combined from 5 wt-infected mice and from 5 ΔslrP-infected mice at 24hr and 48hr post-infection.
(H) Quantitative PCR analysis of Il1β expression in IECs and LP leukocytes 48hr after oral infection of B6 mice with wt or ΔslrP ST. n=5/group.
****p<0.0001, ***p<0.001, ** p<0.01, *p<0.05 by unpaired student’s t test or Log rank analysis for survival. NS indicates not significant by unpaired student’s t-test. ND indicates none detected above limit of detection. Error bars indicate +/− SEM. See Figure S3.
Testing the importance of IL-1β in mediating the anorexic response and its relationship to ST virulence, we found that Il1β/− mice orally infected with ΔslrP were protected from developing anorexia compared to B6 mice infected with ΔslrP ST (Figure 4B). This protection from anorexia was associated with reduced wasting and increased survival (Figure 4C, D). Protection was independent of the microbiota as both Il1β−/− mice that were co-housed with B6 mice or left separately housed were protected from anorexia and the associated morbidity and mortality when orally infected with ΔslrP ST (Figure 4B–D and S3D–F). Infection of Il1β−/− mice with wt ST resulted in similar mortality as wt-infected B6 mice (Figure S3G, H). Injection of wt-infected B6 mice with rIL-1β during infection resulted in anorexia and increased morbidity compared to vehicle-injected/wt-infected B6 mice (Figure 4E, F), demonstrating that IL-1β was sufficient to induce anorexia in these mice. Together, these findings suggest that SlrP antagonizes sickness-induced anorexia through the inhibition of IL-1β in the SI, resulting in reduced virulence. While our data strongly support that slrP inhibition of IL-1β in the SI prevents anorexia, inhibition of the anorexic signal in another tissue remains formally possible.
Salmonella SlrP regulates IL-1β levels by inhibiting inflammasome activation
Our data support a model in which IL-1β levels, rather than the response to IL-1β, are regulated by SlrP. Upon oral infection, ST invades intestinal epithelial cells (IECs). Once the bacteria cross the epithelial barrier, ST infects leukocytes residing in the LP (Ruby et al., 2012). Infection in both cell types induces the expression of pro-IL-1β. Upon activation of the inflammasome, multiprotein cytosolic complexes that lead to the cleavage and activation of Caspase-1, pro-IL-1β is cleaved into its active form, IL-1β (von Moltke et al., 2013). Yersinia pestis encodes a SlrP homolog (YopM) that has been shown to antagonize Caspase-1 activity and IL-1β secretion (LaRock and Cookson, 2012). We found that the increased level of IL-1β in the SI within the first 48hrs of infection was associated with increased Caspase-1 cleavage in LP leukocytes from ΔslrP-infected mice (Figure 4G). By contrast, we detected no Caspase-1 cleavage in IECs from mice infected with either strain of ST (Figure 4G and S3I, J). We found no difference in pro-IL-1β expression in LP leukocytes or IECs from mice infected with either strain of ST (Figure 4H), suggesting that SlrP regulates IL-1β levels by impacting Caspase-1 activation. Consistent with these observations, in vitro ΔslrP infection of myeloid cells resulted in increased secretion of IL-1β compared to wt-infected cells, which was dependent on Caspase-1. Furthermore, in vitro ΔslrP infection of viable B6 and Il1β−/− myeloid cells resulted in increased Caspase-1 cleavage compared to wt-infected cells (Figure S3K–N). Thus, the regulation of SI IL-1β levels was associated with regulation of inflammasome activation (either directly or indirectly by SlrP) in LP myeloid cells.
Salmonella regulates the anorexic response and virulence via the gut-brain axis
We hypothesized that the increased IL-1β in the SI of ΔslrP-infected mice directly regulates the anorexic response. We determined how food restriction affected ΔslrP virulence in Il1β−/− mice, which did not develop anorexia and were less susceptible to ΔslrP infection (Figure 4B–D and S3D–F). Under food restricted conditions, ΔslrP ST was more virulent in Il1β−/− mice compared to ΔslrP infection in Il1β−/− mice that were fed ad libitum (Figure 5A, B, and S4A). The increased morbidity and mortality of the food-restricted animals was specific to the infected state as food-restricted, uninfected Il1β−/− mice exhibited less weight loss and no mortality (Figure S4B–C) Since food restriction in the absence of IL-1β is sufficient to increase virulence of the ΔslrP strain, it suggests that ST virulence is regulated by the direct actions of IL-1β on modulating feeding behavior of the host.
Figure 5. Salmonella regulates the anorexic response and virulence via the gut-brain axis.
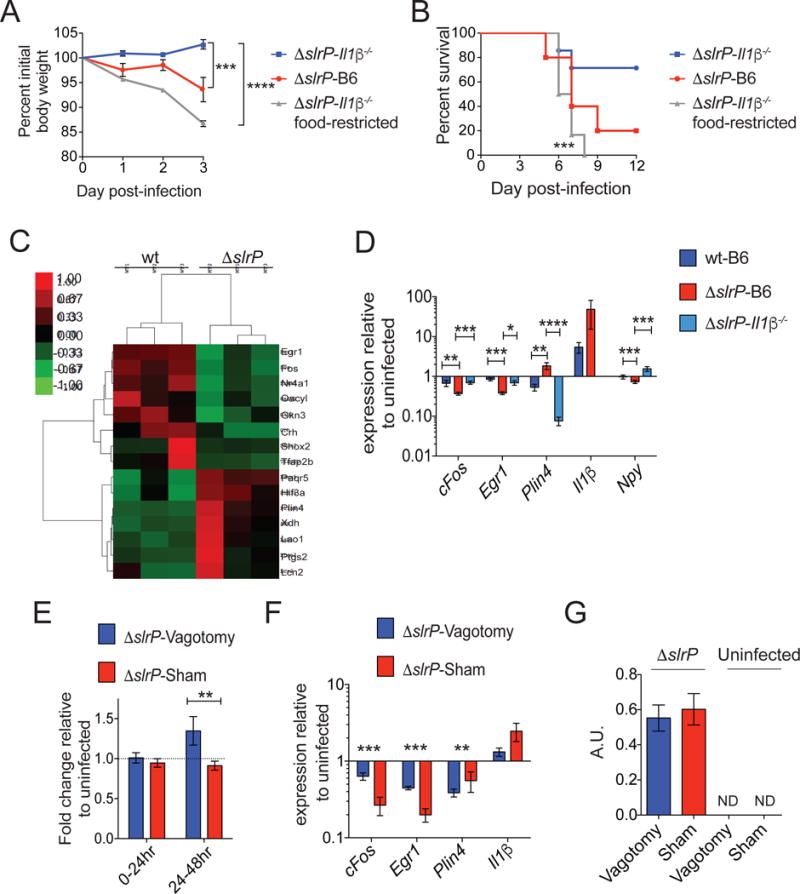
(A–B) Il1β−/− mice orally infected with ΔslrP were fed ad libitum (n=6) or given restricted amounts of food (n=6). ΔslrP-infected B6 mice (n=5) were fed ad libitum. Weight loss (A) and survival (B) were measured.
(C) Heat map of differentially expressed genes in the hypothalamus of wt (n=3) and ΔslrP (n=3) infected B6 mice 48hr post-infection.
(D) Quantitative PCR analysis of genes identified in (C) in the hypothalamus of B6 mice infected with wt (n=4) or ΔslrP (n=4) and Il1β−/− (n=10) mice infected with ΔslrP ST for 48hr.
(E) Feeding of ΔslrP-infected vagotomized or sham B6 mice. Values normalized to feeding amounts of uninfected mice. n=9–10/group. Data represent 2 independent experiments combined.
(F) Quantitative PCR analysis of genes identified in (C) in the hypothalamus of ΔslrP-infected vagotomized or sham B6 mice at 48hrs post-infection. n=4–5/group.
(G) Levels of mature IL-1β were determined by IL-1R reporter assay 48hr post-infection in SI of ΔslrP-infected vagotomized or sham B6 mice. Graph shown depicts reporter fluorescence (arbitrary units), indicative of active IL-1β. n=4–5/group.
****p<0.0001, ***<0.01, **p<0.05, *p=0.05. Unpaired student t-test, one-sample t test, or Log rank analysis for survival. Error bars indicate +/− SEM. Il1β for (D) is from hypothalamus from 48hr and 72hr. See Figure S4 and S5.
To understand the mechanism by which IL-1β regulates the anorexic response during ST oral infection, we considered that the behavioral changes that are triggered by IL-1β are mediated by its actions on the central nervous system (Pavlov and Tracey, 2012). Gene expression analysis of the hypothalamus revealed a cohort of genes involved in feeding and metabolism that were differentially regulated by the two different strains in B6 mice (Figure 5C–D, S4D, Table S1). Analysis of gene expression patterns in the hypothalamus of ΔslrP-infected Il1β−/− mice revealed that a subset of these genes are regulated by IL-1β during infection including Npy, Il1β, Egr1, Plin4 and cFos, all of which are involved in appetite regulation or neuronal activation (Figure 5D and S4D). These data indicate that mice infected with ΔslrP ST have dysregulation of the hypothalamic feeding and satiety network in an IL-1β dependent manner.
Sickness behaviors and anorexia induced by peripheral administration of LPS or IL-1β are abolished in mice in which the visceral afferent nerves have been severed (Goehler et al., 1999; Hansen et al., 2001; Pavlov and Tracey, 2012). Visceral afferent vagus nerve endings are likely necessary for transmitting information about distal inflammatory states to the brain when local levels of cytokines are present at low levels, but this has not been experimentally shown (Hansen et al., 2001; Pavlov and Tracey, 2004). Because we only observed IL-1β differences in the SI of ΔslrP-infected mice compared to wt-infected mice, we hypothesized that the vagus nerve was required for mediating the anorexic response and associated gene expression in the hypothalamus of ΔslrP-infected mice. To test our hypothesis, we infected vagotomized mice in which afferent nerves in the abdominal cavity were severed and surgical sham mice with ΔslrP and measured the anorexic response. Hepatic vagotomy results in vagal deinnervation of the SI, distal stomach, pancreas and liver, leaving the cecum and colon innervation intact. Feeding was equivalent between the vagotomized and sham mice under homeostatic conditions (Figure S5A). However, despite infection with ΔslrP ST, mice with a severed vagus nerve did not develop an anorexic response, while the infected sham mice did (Figure 5E). The IL-1β-dependent gene expression of the hypothalamus exhibited by ΔslrP-infected mice (specifically Il1β, Egr1, Plin4 and cFos) was also dependent on the vagus nerve (Figure 5F and S5B). Vagotomy did not however affect SI IL-1β activation during infection (Figure 5G). As we only detected IL-1β differences in the SI of ΔslrP-infected mice and not in other tissues affected by vagotomy (Figure 4A, S3A, B), our findings strongly suggest that the anorexic response and associated anorexic gene expression program in the hypothalamus requires IL-1β-dependent signaling via the vagus nerve from the gut to the brain during ΔslrP infection.
Anorexia creates trade-offs between pathogen virulence and transmission
In humans, complications of severe acute malnutrition (SAM) are invasive bacterial infections and sepsis (Page et al., 2013). Salmonella species are commonly isolated from the blood of SAM patients with invasive bacterial infections (Page et al., 2013). We hypothesized that the increased virulence resulting from anorexia and food restriction in our infection models was due to increased invasion and extra-intestinal dissemination of ST. We found that the percentage of hosts with a disseminated infection in liver, spleen, PP and MLN was significantly higher during ΔslrP infection compared to wt infection of B6 mice (Figure 6A, C, D and S6B–G). Ruling out that ΔslrP replicates more efficiently when systemic, we found that i.p. infection with wt or ΔslrP ST resulted in similar burden in the spleen and liver, indicating that ΔslrP disseminates to systemic sites more during oral infection (Figure S6A). The increased dissemination began at 72hrs post-infection (Figure 2C, D, 6A and S6B–G), indicating that dissemination occurred secondary to the more severe anorexic response seen in ΔslrP-infected mice, suggesting that anorexia leads to increased pathogen dissemination. In support of this, food restriction of wt-infected B6 mice increased the incident of disseminated infection in liver, spleen, PP and MLN, compared to wt-infected mice fed ad libitum (Figure 6A and S6B–G). Conversely, force feeding of ΔslrP-infected B6 mice decreased the incident of dissemination to systemic sites (Figure 6A and S6B–G). Like the anorexic response, dissemination was also dependent on IL-1β because administration of rIL-1β was associated with increased dissemination to systemic sites in wt-infected B6 mice (Figure 6A and S6B–G). Furthermore, the increased morbidity and mortality of food-restriction was associated with increased dissemination incident in ΔslrP-infected Il1β−/− mice (Figure S6H, I). Analyses of gut pathology and markers of barrier integrity showed that the increased dissemination was not due to a leaky gut, suggesting that the behavior of ST changes under food restricted conditions (Figure 3C, S6J and S2A). Thus, ST negatively regulates virulence by inhibiting IL-1β induced anorexia to prevent extra-intestinal dissemination.
Figure 6. Anorexia creates tradeoffs between virulence and transmission.
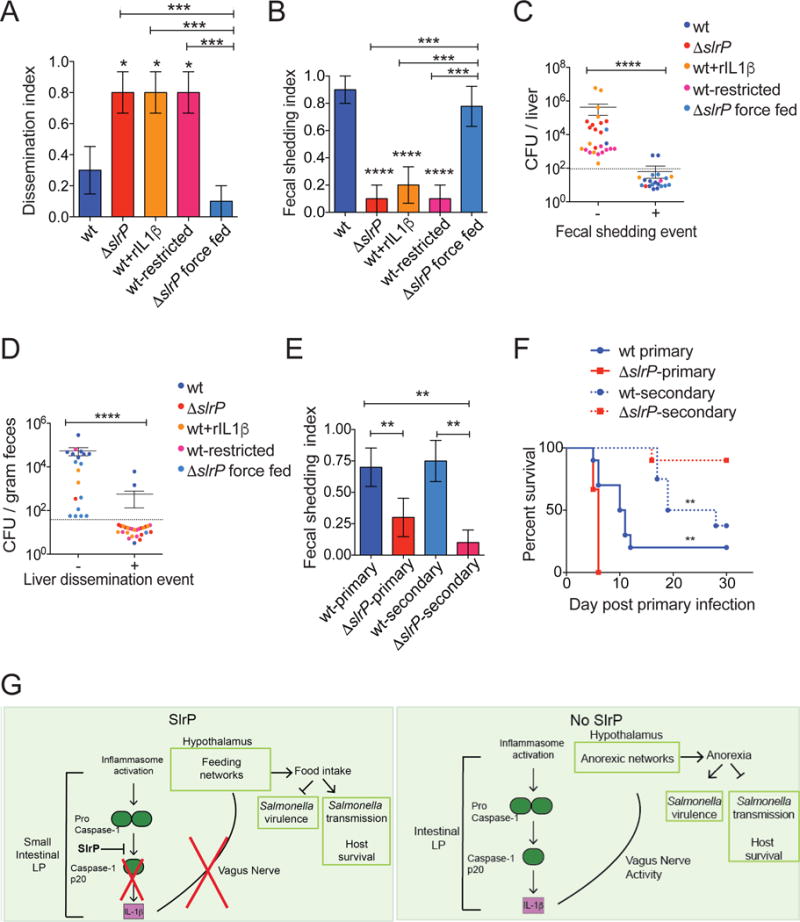
(A) Animals were assigned a liver dissemination score of “1” or “0” at 72hr post-infection. n=10/group
(B) Animals from (A) were assigned a score of “1” or “0” to determine fecal shedding index at 72hr post-infection.
(C) CFU in liver of mice from (A) was plotted against fecal shedding index (B) for each individual mouse. “−“ denotes mice with no fecal shedding, and “+” denotes mice with fecal burden.
(D) CFU detected in feces of mice from (A) was plotted against liver dissemination index (C) for each individual mouse. “−“ denotes mice with no liver dissemination, and “+” denotes mice with liver burden.
(E) B6 mice were orally infected with wt or ΔslrP ST (wt-primary or ΔslrP-primary) and co-housed with an uninfected B6 mouse (wt-secondary or ΔslrP-secondary). Fecal samples were collected every 24hrs post-infection from primary and secondary hosts for CFU analysis. Mice were assigned a score of “1” or “0” n=8–15 mice/group. Data combined from 2 experiments.
(F) Survival of mice from (E). 8–10 mice/group. ** represent statistical significance between both primary groups or both secondary groups.
(G) Model of how wt and ΔslrP ST regulate anorexia, virulence and transmission.
****p<0.0001, ***p<0.01, **p<0.05. unpaired student t-test, one way ANOVA with tukey post-test or Log rank analysis. Dotted line indicates limit of detection, and mice with no CFU detected in indicated tissue are indicated by symbols below this line. See Figure S6.
A fundamental question is why ST would maintain a trait that negatively regulates virulence through the inhibition of the sickness-induced anorexic response? Evolutionary theory predicts that the selection pressures on pathogens to maximize their transmission determines their optimal host exploitation strategies and thus their virulence. While the primary perspective in evolutionary medicine is that virulence increases with horizontal transmission (Alexander, 1981; Ewald, 1996), we reasoned that the absence of SlrP function and the resulting host anorexic response and increased pathogen virulence may come at a cost to ST transmission. Upon examination of all our experimental groups, we found that the fraction of hosts that shed at least one CFU of pathogen within the first 72hrs of infection was higher in experimental groups that had a lower fraction of hosts with a dissemination event (wt-infected B6 and force fed ΔslrP-infected B6, Figure 6A, B, S6B, E and data not shown). Additionally, individuals with at least one shedding event at 72hrs post-infection had lower levels of pathogen burden at extraintestinal tissues. These data also show that mice with a dissemination event had lower levels of shedding compared to individuals without dissemination (Figure 6C, D, S6C, D, F, G).
We next employed a horizontal transmission model in which we co-housed an infected mouse challenged with wt or ΔslrP ST (primary host) at the time of infection with an uninfected mouse (secondary host) and monitored shedding, wasting and mortality of the secondary host over the course of the infection. We detected that approximately 75% of the wt-secondary hosts shed pathogen in their feces, while approximately 10% of the ΔslrP-secondary hosts showed evidence of fecal shedding over the course of the experiment (Figure 6E). Interestingly, 60% of wt-secondary hosts succumbed to infection within one month after exposure to the wt-infected primary host. By contrast only 10% of ΔslrP-secondary hosts died within this same time frame (Figure 6F). Thus, the inhibition of anorexia afforded by SlrP function is associated with increased fecal shedding of ST and transmission to new hosts, creating a trade-off between virulence (extra-intestinal dissemination) and transmission (Figure 6G).
Discussion
The purpose of a physiological response during infection is often confounded by the complexities of counter-adaptation strategies by microbes. The purpose of the current study was to determine if there is a physiological role of sickness-induced anorexia for host defenses, how this response regulates pathogen virulence using a transmissible model of infection and the mechanism by which localized tissue responses to microbes can induce anorexia. Our finding that the ST effector SlrP promotes host health and transmission during infection by inhibiting anorexia via the gut-brain axis permits several conclusions: (1) that anorexia may have evolved as a mechanism to protect the host population rather than the individual host, (2) anorexia can increase pathogen virulence, (3) where increased virulence compromises transmission there will be tradeoffs, (4) local inflammatory responses in the intestine can signal to the brain via the vagus nerve to induce anorexia and (5) microbes can inhibit sickness-induced anorexia and directly modulate host feeding behavior.
NELs encoded by bacteria are believed to be important for virulence by mediating antagonistic manipulation of host processes by the microbe (Maculins et al., 2016). We discovered that the NEL SlrP, encoded by ST is important for dampening virulence during interactions with the host in vivo by directly antagonizing a host response that induces sickness-induced anorexia. In Balb/c mice, infection with a slrP mutant strain on the IR715 background had a higher lethal dose 50 (LD50) compared to infection with a parental strain (Tsolis et al., 1999). In an in vivo competition assay, this slrP mutant exhibited a colonization defect in PP but not in spleen (Tsolis et al., 1999). The phenotypic differences between this previous study and the one reported here may be explained by the different strains of ST in which SlrP function was tested, microbiota differences, housing and diet differences of the mice or the strain of mice used.
We showed that the ability of SlrP to regulate IL-1β in the SI was associated with inhibition of Caspase-1 cleavage. Several studies have shown a protective role for Caspase-1 during oral infection of mice with ST. However, if SlrP inhibits inflammasome function, why are mice infected with ΔslrP not more resistant to this strain compared to mice infected with a functional SlrP? By focusing on later stages of infection (Broz et al., 2010; Lara-Tejero et al., 2006; Muller et al., 2009; Raupach et al., 2006), prior studies identified an important role for the inflammasome for providing resistance in later stages of infection at extraintestinal sites but did not provide insight as to how ST-inflammasome interactions within specific populations of the gut in the early stages of infection influence the anorexic response, pathogen loads or host health. Additionally, our study focused on the function of SlrP inhibition of inflammasome activation within the intestine, and understanding how SlrP functions at systemic sites remains to be determined. A recent study showed that the NAIP/NLRC4 inflammasome is required for mediating resistance against ST in IECs in a murine model of enterocolitis (Sellin et al., 2014). It is worth noting, however, that we did not employ an enterocolitis model, and we observed no differences in the intestinal levels of wt or ΔslrP ST in our system. Nevertheless, these observations suggest that studies in inflammasome-deficient animals might be difficult to interpret. These considerations prompted our use of Il1β−/− mice.
It has been proposed that the vagus nerve is responsible for sensing local production of inflammatory cytokines in organs to relay messages to the brain to induce sickness behaviors (Pavlov and Tracey, 2012). However, we were unable to identify studies that provide evidence for these claims. Here our data strongly suggest that IL-1β from myeloid cells within the SI signals to the brain via the vagus nerve to induce the anorexic response during infection and that a microbe has evolved a trait to manipulate this pathway. As of yet, we do not know specifically which changes in the hypothalamus trigger the anorexic response, but we have identified likely candidates. We found SlrP to regulate cFos, Il1β, Egr1 and Plin4 expression levels in the hypothalamus in both an IL-1β and vagus nerve dependent manner. Distinct neuronal populations express cFos in response to hunger and satiety signals (Johnstone et al., 2006). IL-1β in the brain is sufficient to induce anorexia in rodent models (Pavlov and Tracey, 2012). Both Egr1 and perilipins (Plin4) have complex roles in regulating feeding and metabolic homeostasis (de Lartigue et al., 2010; Martinez-Botas et al., 2000). It is likely that SlrP-mediated regulation of all these genes alters the feeding networks to prevent anorexia during infection. Further, our studies show that the inflammasome contributes to sickness-induced behavior, adding to the evidence that this sensor is important for the control of homeostasis, tissue repair, and tolerance defenses (Ayres, 2013).
Sickness-induced behaviors have been theorized as evolved host behavioral strategies to promote survival upon challenge with infectious diseases (Dantzer, 2009; Exton, 1997; Hart, 1988). With the exception of social withdrawal, any benefits afforded by sickness-induced behaviors including anorexia have been viewed from the perspective of the individual host level, rather than that of the host population. By using a transmissible model and a loss-of-function mutant of ST, we determined how the host anorexic response influences individual host health and infection susceptibility at the population level. We found that while anorexia was maladaptive for the individual host in the context of a ST infection, the development of the anorexic response in a primary host spared the population from contracting the infection. While we found no difference in pathogen levels between hosts that developed the anorexic response and those that did not, at the population level, sickness-induced anorexia confers a negative impact on microbial fitness resulting in decreased transmission. Many infections induce anorexia in the host, and it is unlikely that the metabolic alterations that occur during this fasted state will be compatible for host defense against all infection types. Given our findings, we do suggest however, that in prior studies in which fasting increased morbidity during infection, the effects on transmission at the host population level would be of interest.
Microbes and their hosts share a wide range of resources that are required to support normal growth and metabolism. In the mouse, ST depends on a wide array of nutrients that are available in scarce amounts in host tissues (Steeb et al., 2013). In our study we show that reduced feeding of the host triggers virulence of a pathogen by promoting its invasion of extra-intestinal tissues. Our findings are supported by a recent study demonstrating that under specific conditions, LPS-induced anorexia can trigger pathogenicity of the microbiota (Pickard et al., 2014). How a reduction in food consumption changes the behavior of ST to become invasive is unknown. One possibility is that in an extra-intestinal niche, ST faces less competition than when competing for resources with other microbes inhabiting that particular niche in the intestine. It will be interesting to determine how invasion and virulence differ between wt and ΔslrP strains in gnotobitoic models.
Increased virulence has been proposed to benefit microbes by promoting transmission and, thus this benefit would be expected to drive the selection of traits in microbial populations that inhibit tolerance defenses or promote virulence (Alexander, 1981). (Ewald, 1996; Wickham et al., 2007). We found that there was an inverse correlation between ST virulence and transmission, suggesting that the selective pressures host anorexia places on pathogen transmission create trade-offs between these two traits. Trade-offs between virulence and transmission have been reported in animal models of malaria in which transmission success was inversely correlated with host mortality (Mackinnon et al., 2008; Paul et al., 2004). It has been proposed that microbes will evolve mechanisms to promote tolerance in their host so long as transmission of the microbe is not negatively affected by the increased tolerance (Ayres, 2013, 2016). Indeed, an intestinal microbe inhibited infection-induced muscle wasting through the induction of a tolerance mechanism in mice (Schieber et al., 2015). Anti-virulence strategies that inhibit pathological responses by the host without negatively affecting microbial fitness are phenotypically and evolutionarily analogous to such tolerance mechanisms. We provide evidence that there is also a selection of traits in microbial populations that act as anti-virulence strategies. Such relationships in the context of infection-induced anorexia have not been previously revealed as most studies rely on models in which transmission is not considered such as the systemic injection of immune elicitors that cause anorexia.
Anorexia and catabolism of energy stores are common co-morbidities of acute and chronic illnesses (Fearon et al., 2011). In these diseases, decreased energy intake and increased energy expenditure result in depletion of muscle and fat stores, or wasting (cachexia). Efforts to correct the nutritional deficits with force feeding have been met with little success clinically. Thus, therapeutic interventions that can modulate a patient’s physiology to promote appetite would be most desirable in combating these morbidities in critically ill patients. In patients with anorexia nervosa, efforts have been focused on treating the illness from a psychological perspective. Our work suggests that modulating local responses within the intestine by microbes may provide a therapeutic means to such illnesses.
STAR Methods
Contact for Reagent and Resource Sharing
Further information or requests for reagents may be directed to the corresponding author Janelle S. Ayres (jayres@salk.edu).
Experimental Model and Subject Details
Mice
Male mice were used for all experiments. C57Bl/6 (B6) mice were obtained from room 15 at Jackson Laboratories at 5 weeks of age and housed in our AALAC-certified vivarium. The whole body Il1b−/− knock-out mice were provided by Russell Vance (UC Berkeley) and were bred and maintained in our barrier facility. Vagotomized and sham B6 mice were obtained from Charles River Laboratories. Mice were specific pathogen free, maintained under a 12-hour light/dark cycle and given standard chow diet ad libitum unless otherwise stated. Mice were used for experimentation at 6–8 weeks of age. For germ-free mice, B6 mice were bred and maintained in our germ free mouse facility in sterile semi-flexible isolators and screened for bacterial, fungal, and viral contamination (Schieber et al., 2015). For shared bedding experiments, bedding from cages of B6 and Il1b−/− mice were mixed three times per week for two weeks prior to experimentation to control for floral differences. All animal experiments were done in accordance with The Salk Institute Animal Care and Use Committee.
Salmonella strains
The ΔslrP mutant was generated on the Salmonella enterica serovar Typhimurium SL1344 background (Brandt et al., 2004). Gene deletion was confirmed by PCR and sequencing using primers listed in Table S2 that have been previously reported (McLaughlin et al., 2014). The parental SL1344 strain was used for wildtype (wt) control for all of our experiments described. For preparation of Salmonella stocks for infection, single colonies of ΔslrP or wt ST that were struck out on LB agar plates supplemented with 100μg/mL streptomycin were grown overnight at 37 degrees C in 5mL liquid broth containing 100μg/mL streptomycin. After overnight growth, 1mL of liquid cultures was diluted in 49mL liquid broth containing streptomycin. Cultures were incubated in a 37-degree shaker for approximately 1–2 hours. Optical density was determined (0.9), and cultures were transferred to a 50mL conical tube and centrifuged for 10 minutes at 4000rpm. Bacterial pellet was washed three times with PBS. After final wash, bacterial pellet was resuspended in PBS containing 25% glycerol to a concentration of 5×108 CFU/ml and frozen at −80 degrees. Prior to infection, glycerol stocks were thawed and diluted in sterile PBS to a concentration of 1×10ˆ8 CFU/ml, and mice were gavaged with 100μl (1×10ˆ7 CFU). Inoculums were confirmed for every experiment by plating dilutions on agar plates and quantifying CFUs. Infections with fresh cultures of each strain were also done to validate that the phenotypes are the same as what we report with the glycerol stock preparations.
Methods Details
Mouse infection models
Mice were orally infected with 10ˆ7 CFU of either wt Salmonella Typhimurium strain SL1344 or a mutant SlrP (ΔslrP) strain. For systemic infections, mice were injected through the intraperitoneal cavity (IP) with 150 CFU wt or ΔslrP Salmonella. For experiments investigating the effects of IL-1β, wt-infected mice were given IP injections of 0.05μg IL-1β (eBioscience) per gram mouse. For food-restriction experiments, at 24 hours post-infection, wt-infected mice were given an amount of food equivalent to that of ΔslrP-infected mice at the start of their anorexic response (see Figure 2E). Every 24 hours after, the amount of food given to the food-restricted group was sequentially reduced. For force feeding experiments, ΔslrP-infected mice were gavaged twice daily with 200ul of liquid diet (TestDiet, LD101). Briefly, 2.5g powder was blended in 7mL dH20 using a TissueRupture (Qiagen). Undissolved powder was pelleted by centrifugation at 900rpm for 5 minutes. Remaining “liquefied” food (approximately 200 calories per 200μl) was used for gavage. For gnotobiotic infections, mice were infected with 10ˆ6 of either strain. Inoculum was plated on LB/agar containing streptomycin after all experiments to confirm infecting concentration.
For horizontal transfer experiments, two mice were housed per cage. One mouse per cage was infected with either wt or ΔslrP Salmonella (primary infection) and the second mouse was the uninfected secondary host. Both mice were monitored for weight loss, and survival. The incidence of fecal shedding of pathogen was also determined for primary and secondary hosts. We measured fecal shedding in the primary hosts every day (beginning with day 1 post-infection) until all the slrP infected primary hosts died. We did not continue to count shedding in the primary hosts of wt Salmonella infected mice after all the primary slrP mice died. The indexes provided indicate the fraction of hosts that shed at least one CFU of Salmonella at any point during that time frame. No host is ever included in the shedding indexes in Figure 6 twice – once assigned a “1” it remains scored as a host that shed at least one CFU at one time point during that time frame, even if we never detect a shedding event in that mouse again.
For the secondary hosts, we measured shedding every day until the termination of the experiment on day 30 post-infection, beginning with day 1 post-infection of the primary host. The indexes provided indicate the fraction of hosts that shed at least one CFU of Salmonella at any point during that time frame. No host is ever included in the shedding indexes in Figure 6 twice – once assigned a “1” it remains scored as a host that shed at least one CFU at one time point during that time frame, even if we never detect a shedding event in that mouse again.
For experiments analyzing the relationship between fecal shedding and dissemination (Figure 6), mice were singly housed during infection with the appropriate strain of bacteria. For calculating the dissemination index, the liver, spleen, PP and MLNs were dissected at 72 hrs postinfection and organs were homogenized and plated for CFU quantification as described below and mice were assigned a binary score of either 0 or 1. A score of 1 indicates that there is at least one CFU in the relevant systemic tissue. A score of 0 indicates no CFU above the limit of detection was detected in the relevant systemic tissues. The average of the scores for each experimental group was determined and plotted as the fraction of hosts with a dissemination event. The total amount of CFUs was also quantified for each tissue. For fecal shedding, a fresh fecal pellet was collected at the indicated time point. Fecal matter for each mouse was weighed, homogenized in PBS/1% triton, diluted and plated. Animals were then assigned a shedding score of 1 or 0. A score of 1 indicates that a particular animal shed at least 1 CFU at the time point indicated. A score of 0 indicates that the animal did not shed at least one CFU above the limit of detection at the time point indicated. CFUs were also quantified and normalized to amount of feces to determine the amount of CFU shed per gram of feces at the indicated time point. We provide scatter plots examining dissemination CFUs (spleen, liver, PP and MLN) vs incidence of fecal shedding at 72 hrs post infection. We also provide fecal shedding CFUs vs incidence of dissemination (spleen, liver, PP and MLN). Because of the binary nature of the phenotypes, data were plotted this way to allow for statistical analyses. We look at 72 hrs because this is the relevant time point at which the dissemination occurs in response to the more severe anorexic response. These data demonstrate that individuals with a shedding event have lower dissemination levels at all extraintestinal sites examined compared to individuals without shedding. Furthermore, these data show that individuals with a dissemination event have lower levels of shedding compared to individuals without a dissemination event. Finally, these data show that slrp-infected B6 fed ad libitum, wt infected B6 mice treated with rIL1B and food restricted wt-infected B6 mice have more dissemination and less shedding than wt-infected B6 and force fed slrp-infected B6 mice.
Survival assay
Time to moribund was used for survival experiments, and morbidity was defined as animals being unable to move when gently touched or unable to right themselves when placed on their side.
Quantification of Salmonella in mouse tissues
Liver, spleen, cecum, colon, and small intestine from infected mice were homogenized in 1mL PBS containing 1% triton X-100 using a Power Lyzer 24 bench top bead-based homogenizer (MoBio). MLNs and PPs were homogenized in 600μl solution. Organ homogenates were serially diluted and plated on LB/agar or brilliant green agar containing streptomycin to quantify bacterial burden.
Metabolic analysis
Infected mice were singly housed in metabolic cages, and O2 consumption, CO2 production and activity data were collected by CLAMS. EchoMRI machine was used to assess lean and fat mass, which were normalized to MRI readings taken prior to infection on day 0 for each animal. For whole body weight, animals were weighed daily using a scale and normalized to their respective weight at day 0 just prior to infection. For feeding assays, animals were given a known amount of chow and every 24 hrs, the amount of chow was measured. Measurements were taken at the same time of day.
Histological analysis
Ileum from infected mice was isolated, inflated with PBS and fixed in 10% formalin for 24 hours before transfer to 70% ethanol. The ileum was swiss rolled, embedded in paraffin, sectioned at 5μm and stained with hematoxylin and eosin. Inflammation, submucosal edema and epithelial damaged were assessed by a blinded pathologist. Liver and spleens were harvested, fixed in 10% formalin and embedded in paraffin. Multiple, stepwise, four (4) millimeter sections were stained with H&E and analyzed by the pathologist who was blinded for groups. The entire liver surface was scored for inflammation and necrosis on a scale of 0 to 4 (0: absent; 1: focal to multifocal, portal, lobular and perivascular inflammation without necrosis; 2: multifocal to widespread inflammation associated with minimal necrosis; 3: multifocal to widespread inflammation associated with focally extensive necrosis; 4: multifocal to widespread inflammation associated with multifocal and coalescing necrosis). Spleen sections were scored for inflammation, necrosis/abscess formation, and thrombus formation. Each parameter was scored on a score of 0 to 4 (0: absent; 1: mild; 2: moderate; 3: severe; 4: severe). Thrombi were scored as follows: 0: no thrombi; 1: 1–4 thrombi; 2: 5–9 thrombi; 3: 10–15 thrombi; 4: more than 15. The maximum total spleen inflammation score was 12.
Cytokine measurements
Serum and organ homogenates were used for cytokine measurements determined by ELISA. The blood from euthanized mice was obtained by cardiac puncture and centrifuged for 20 minutes at 8000rpm to separate serum. Organ homogenates were centrifuged for 5 minutes at 1200rpm. IL-1β was detected using an anti-mouse purified IL-1β clone (B122) and a biotin-conjugated anti-mouse IL-1β clone. IL-6 was detected using an anti-mouse purified IL-6 clone (MP5-20F3) and a biotin-conjugated anti-mouse IL-6 antibody (MP5-32611). TNFα was measured with an anti-mouse purified TNFα antibody (1F3F304) and a biotin-conjugated anti-mouse TNFα antibody (XT3/XT22). All antibodies were purchased from eBioscience. For immobilization of TNFa, IL-1β and IL-1a, anti-mouse purified IL-1β clone (B122), anti-mouse purified TNFα antibody (1F3F304) and anti-mouse IL-1α clone (ALF-161) were used.
IL-1β bioassay
HEK-Blue-IL1R reporter cell line (Invivogen) was used to measure production of biologically active IL-1β. This reporter cell line stably expressed mouse IL-1 receptor and a secreted alkaline phosphatase (SEAP) reporter gene under the control of a minimal IFN-β promoter fused to NFκB and AP-1 binding sited. Cells were maintained according to manufacturer’s protocol in growth medium containing DMEM, 4.5 g/L glucose (Life Technologies), 2mM L-glutamine (Life Technologies), 10% FBS (Life Technologies), 50 U/ml penicillin (Life Technologies), 50 μg/ml streptomycin (Life Technologies), 100 μg/ml Normocin (Invivogen), 200 μg/ml hygromycin B gold (Invivogen), 1 μg/ml puromycin (Invivogen) and 100 μg/ml Zeocin (Invivogen). Tissue homogenates or supernatants from in vitro infection assays were diluted in PBS (Life Technologies), and 20 μl of dilutant was added to a flat bottom 96 well plate. In separate wells, 20 μl of a positive control rmIL1-β and a negative control human rTNFα were added. As these cells also respond to murine IL-1α and TNFα, as controls, an aliquot of each sample was incubated on plates coated with a mouse anti IL-1α, mouse anti-TNFα or mouse anti-IL-1β to immobilize any IL-1α, TNFα or IL-1β in the samples. The samples were then subjected to the HEK blue assay as follows: 180 μl of HEK cell suspension (~50,000 cells) resuspended in test medium (same as growth medium without hygromycin, puromycin and zeocin) were added to each well. Cells were incubated with samples overnight at 37°C in 5% CO2. The next day, 20 μl of media from each well was added to a new 96 well plate. 180 μl of resuspended QUANTI-Blue reagent was then added to each well. Samples were incubated at 37°C for 30 minutes after which plates were read on a spectrophotometer at 650 nm. As secondary mode of validation, we performed ELISAs on the samples to measure TNFα, IL-1β and IL-1α concentrations.
Intestinal cell preparations and Western blot analysis
Western analysis was performed on lysates obtained from intestinal epithelial cells and intestinal lamina propria cells. Briefly, small intestine was incubated in epithelial strip buffer (PBS containing 5mM EDTA, 1mM DTT, 5% fetal bovine serum, and antibiotic) at 37 degrees with gentle shaking. Epithelial cells were isolated from solution by spinning in a Percoll gradient for 20 minutes at 600g. The remaining intestinal tissue was digested for 30 minutes at 37 degrees with gentle shaking in digest buffer (RPMI 10% fetal bovine serum, antibiotic, 1mM sodium pyruvate, 20mM HEPES, 1mg/ml Collagenase VIII [Sigma] and 20ug/ml DNase I [Roche]). The solution of digested tissue was filtered, subjected to Percoll gradient, and centrifuged for 20 minutes at 600g for isolation of lamina propria leukocytes. Epithelial cells and leukocytes were pelleted and lysed in RIPA lysis buffer (150mM NaCl, 50mM Tris, 0.5% sodium deoxycholate, 1% triton X-100, and protease inhibitor cocktail [Sigma]) for 20 minutes on ice. Lysates were centrifuged at 4 degrees for 10 minutes at 10,000rpm and subjected to Western analysis of CASPASE-1 (Adipogen) and β-actin (Cell Signaling).
Gene expression analysis
RNA was extracted from bead-beaten hypothalamus and from epithelial and lamina propria cells using the Qiagen All Prep RNA/DNA Mini Kit. Briefly, sample was lysed in 600μl RLT buffer containing β-mercaptoethanol and centrifuged for 3 minutes at 6,000rpm in a microcentrifuge. Supernatant was transferred to an AllPrep DNA column and centrifuged for 30 seconds at 10,000rpm. Supernatant was mixed with 70% ethanol and transferred to an RNeasy spin column and centrifuged for 15 seconds. The supernatant flow-through was discarded, and the column containing RNA was washed with 350μl RW1 buffer. Flow-through was discarded, and column was incubated for 15 minutes with DNaseI solution. Column was washed with RW1 buffer, and flow-through was discarded. Column was washed twice with 500μl RPE buffer. After final wash, column was placed in a new Eppendorf tube, and RNA was eluted with 40μl RNase-free water. cDNA was synthesized using the SuperScript III kit (Invitrogen) from 350ng RNA. Real-time quantitative PCR was performed using iTaq SYBR Green Mix (BioRad) on a ViiA7 RT PCR Machine (Applied Biosystems). The comparative Ct method was used to analyze gene expression relative to an uninfected control. β-actin was used as the endogenous control for hypothalamus samples, and rps17 was used for intestinal samples. See Table S2 for primer sequences. For RNA-seq, libraries were prepared using a strand-specific protocol and sequenced on an Illumina HiSeq2500 Instrument. For each sample, at least 22.5 million 51-bp single-end reads were produced. The reads were mapped to mm10 reference genome using STAR 2.3.0e, resulting in at least 20 million uniquely mapped reads per sample. Differential gene expression was calculated using Cufflinks 2.2.1 with UCSC mm9 genome annotation from Illumina’s iGenomes collection. Functional enrichment analysis of differentially expressed genes was carried out using DAVID.
Organ preparations for flow cytometric analysis
Spleen, liver, and MLNs from infected animals were mashed through a 70μM mesh filter using the plunger from a 1-mL syringe. Cells were washed with complete media (DMEM, 10% fetal bovine serum, antibiotic, 1mM sodium pyruvate, 20mM HEPES) and centrifuged for 5 minutes at 1500rpm. Pelleted liver was resuspended in 15mL of 35% Percoll. Solution was underlaid with 75% Percoll and centrifuged for 20 minutes at 1000g. Leukocytes were harvested from the interphase, washed with complete media, and centrifuged for 5 minutes at 1500rpm. Red blood cells were lysed for 2 minutes with ACK lysing buffer. Remaining leukocytes were washed, centrifuged, and counted.
Flow cytometric analysis
Lamina propria, liver, MLN, and spleen leukocytes isolated from infected mice were incubated in a 96-well V-bottom plate with Zombie-UV (diluted 1:800 in PBS) for 20 minutes. Cells were washed with FACS buffer (PBS containing 2% fetal bovine serum) and incubated for 15 minutes with Fc blocking reagent (24G2, diluted 1:200 in FACS buffer). Following Fc block, cells were stained for 20 minutes with the following conjugated antibodies: Ly6G-FITC (1A8), Ly6C-PerCP/Cy5.5 (HK1.4), CX3CR1-BV421 (SA011f11), MHCII-BV510 (M5/114.15.2), CD11b-BV785 (M1/70), CD103-APC (2E7), CD45.2-AF700 (104), CD3-APC/Cy7 (145-2C11), CD19-APC/Cy7 (6D5), CD64-PE (X54-5/7.1), F4/80-APC (BM8), and CD11c-PE/Cy7 (N418). Antibodies to CD11c, F4/80, and CD45.2 were diluted 1:100 in FACS buffer. The remaining antibodies were diluted 1:200. All steps were performed at 4 degrees. Antibodies were purchased from BioLegend. Cells were analyzed on a LSR II (BD) using FACSDiva software, and data were analyzed using Flowjo software (TreeStar).
In vitro infection
Lamina propria leukocytes were isolated and combined from naïve B6, Il1β−/−, Nlrc4−/−, or Caspase1/11−/− mice and stained with antibodies to CD45.2, CD3, and CD19. Live (DAPI negative) myeloid cells were isolated (CD45.2+CD3−CD19−) on a BD Influx™ cell sorter (100μm nozzle, sheath pressure set to 17.5 PSI). After sorting, cells were plated in a 96-well round-bottom plate in RPMI media containing 10% fetal bovine serum, antibiotic, 1mM sodium pyruvate, and 20mM HEPES and allowed to rest overnight. Cells were infected with MOI 1 of wt or ΔslrP Salmonella by centrifuging for 10 minutes at 1400rpm at room temperature and were returned to 37 degrees for 6 hours. Supernatant was harvested for IL-1β analysis by ELISA, and cells were lysed in RIPA buffer for CFU analysis or for Western blot analysis. We also measured TNFα levels by ELISA on infected cells at 6 hrs post-infection. TNFα was induced at comparable levels in cells infected with either ST strain (data not shown due to space constraints).
Complementation of S. Typhimurium ΔslrP mutant
The ThermoFisher GeneArt Gene Synthesis and subcloning technology was used to generate a low copy vector containing the wildtype copy of slrP along with its native promoter for our complementation analyses. A 3067 bp piece of DNA containing the wildtype copy of SL1344 slrP along with its native promoter flanked by SalI and SphI restriction sites was synthesized. The fragment was ligated into the multiple cloning site of the pMA Gene Art cloning vector, which contains ampicillin resistance. Competent E. coli were transformed with the pMA-slrP construct and positive transformants were confirmed by sequencing. The plasmid was extracted and digested using the endonucleases SalI and SphI, which cut solely in the 5′ and 3′ regions flanking the gene and its regulatory regions, ensuring the restriction enzymes did not cut within the gene and disrupt functionality. The digested fragments were ligated into the low copy number plasmid pACYC184, which was also double-digested with SalI and SphI. Ligation reactions were transformed into competent E. coli. The recombinant plasmid was sequenced to verify the gene of interest was present and in the correct orientation. The plasmid was extracted from transformed E. coli and transformed by electroporation according to established protocols into the ΔslrP mutant to generate the strain ΔslrP -pACYC184-slrP. As a control, self-ligated pACYC184 vectors were similarly electroporated into the ΔslrP mutant to generate the strain ΔslrP-pACYC184-Empty. Positive transformants were selected for on brilliant green agar supplemented with streptomycin (100 μg/ml), kanamycin (40 μg/ml) and chloramphenicol (25 μg/ml). Prior to in vivo analysis, plasmids were extracted from positive S. Typhimurium transformants. The presence of the slrP gene within the vector (or absence for empty vector) was confirmed by restriction digest analysis and sequencing of the plasmid from the 5′ and 3′ ends.
We determined whether the ΔslrP -pACYC184-slrP and ΔslrP-pACYC184-Empty were maintained during an in vivo infection. B6 mice were orally infected with 1×107 CFU of ΔslrP-pACYC184-Empty or ΔslrP-pACYC184-slrP. Every 24 hrs the small intestine, cecum, colon, liver, spleen, PPs and MLNs were harvested, homogenized, serially diluted and plated on brilliant green agar supplemented with Streptomycin (100 μg/ml) and Kanamycin (40 μg/ml). The dilutants were also plated on brilliant green plates supplemented with streptomycin (100 μg/ml), kanamycin (40 μg/ml) and chloramphenicol (25 μg/ml). The fraction of pathogen containing plasmid was determined by normalizing the amount of CFUs quantified on strep/kan/chlor plates to the amount of CFUs quantified on strep/kan plates. Comparable levels of pathogen containing either the pACYC184-Empty or pACYC184-slrP vectors were recovered within the first 48 hrs post-infection. By 72 hrs post-infection, there was no detectable vector in either strain, and we therefore focused our complementation analyses on phenotypes within the first 48 hrs of infection – anorexia and levels of IL-1β as follows: B6 mice were orally infected with 1×107 CFU of wt SL1344, ΔslrP-pACYC184-Empty or ΔslrP -pACYC184-slrP. Food consumption was measured every 24hrs post-infection as described above. At 24 and 48 hrs post-infection, tissues were harvested, homogenized and subjected to CFU analysis and bioactive IL-1β analyses as described above. We found that complementation effectively enabled the ΔslrP strain to inhibit IL-1β maturation in the small intestine and the anorexic response.
Quantification and Statistical Analysis
Statistical tests were done using Prism version 6.0. The statistical test used, sample sizes and p values are indicated for each figure. All experiments were repeated at least twice. Data shown are either a single representative experiment or data are combined from independent experiments and are indicated in the figure legends.
Supplementary Material
(A–B) Rate of oxygen consumption (VO2) (A) and rate of CO2 production (VCO2) (B) were determined by CLAMS 72hr after oral infection of B6 mice with stated strain of S. Typhimurium. Black bar indicates dark/night cycle, and white bar indicates light/day cycle. n=6/group.
(C) Food consumption of B6 mice orally infected with wt ST, ΔslrP ST with an empty control vector, or ΔslrP complemented with a vector expressing wt slrP under the native promoter. Grams of food per animal was normalized to consumption of uninfected animals. Data are two independent experiments combined, n=15–30 animals per group.
(D) CFU in the SI of B6 mice infected with wt ST, ΔslrP ST with an control empty vector, or ΔslrP ST complemented with wt slrP under the native promoter. n=6–8 per group and representative of two independent experiments. (E–F) Uninfected B6 mice were food restricted as described in methods, and weight (E) and survival (F) were monitored. n=5 mice.
* indicates p<0.05 and ** indicates p<0.005 by unpaired student’s t test; error bars indicate +/− SEM.
(A) Histological scoring of spleen, liver, and ileum 72hr after oral infection of B6 mice with stated strain of S. Typhimurium. n=5/group. “ND” indicates none detected – no pathology above limit of detection.
(B) Gating strategy for flow cytometric analysis of leukocyte populations in the small intestine LP: T/B lymphocytes (P1), neutrophils (P2), phagocytic macrophages (P3), inflammatory monocytes (P4).
(C) Gating strategy for flow cytometric analysis of migratory dendritic cells (P5) in the MLNs.
(D) Flow cytometric analysis of percentages of LP and MLN leukocytes 48hr after oral infection of B6 mice with stated strain of S. Typhimurium. Animal numbers shown.
(E) Total cellularity (left) and flow cytometric analysis of numbers (top) and percentages (bottom) of LP and MLN leukocytes 24hr after oral infection of B6 mice with stated strain of S. Typhimurium. n=8–9 mice per group.
(F) Gating strategy for flow cytometric analysis of leukocyte populations in the spleen and liver: T/B lymphocytes (P1), neutrophils (P2), macrophages (P3), inflammatory monocytes (P4), and dendritic cells (P5).
(G) Total cellularity (left) and flow cytometric analysis of numbers (top) and percentages (bottom) of spleen and liver leukocytes 24hr after oral infection of B6 mice with stated strain of S. Typhimurium. n=5 mice per group.
(H) Total cellularity (left) and flow cytometric analysis of numbers (top) and percentages (bottom) of spleen and liver leukocytes 48hr after oral infection of B6 mice with stated strain of S. Typhimurium. n=5 mice per group.
(I) ELISA measurements of IL-6 in the indicated tissues 24hr after oral infection of B6 mice with wt or ΔslrP S. Typhimurium. n=4–7 mice per group. ND denotes Not Detected. NS denotes Not Significant.
Error bars indicate +/− SEM. All comparisons not significant by unpaired student t-test.
(A) Levels of active/mature IL-1β determined by IL-1β bioassay of whole organ homogenates in liver, spleen and serum of B6 mice 24hr post-infection (n=9–13/group). Data represent 2 independent experiments combined. A. U. indicates arbitrary units of reporter activation.
(B) Levels of active/mature IL-1β determined by IL-1β bioassay of the stomach and pancreas 24hr and 48hr after oral infection of B6 mice with wt and ΔslrP S. Typhimurium. n=5 infected mice per group and n=3 vehicle (PBS)-gavaged mice. A.U. indicates arbitrary units of reporter activity.
(C) Levels of active/mature IL-1β determined by IL-1β bioassay of the SI 24hr and 48hr after oral infection of B6 mice with wt ST, ΔslrP ST with an empty control vector, or ΔslrP ST complemented with a vector expressing wt slrP under the native promoter. Data are two independent experiments combined, n=15–30 animals per group. * indicates p<0.05 and *** indicates p<0.0005 by unpaired t-test. A.U. indicates arbitrary units of reporter activity.
(D–F) Food consumption (D), weight loss (E), and survival (F) of ΔslrP infected B6 (n=9) and Il1β−/− (n=10) mice. (C) Grams of food per animal was normalized to consumption of uninfected animals. Bedding from B6 and Il1β−/− cages was mixed to control for differences in microbiota as described in methods.
(G–H) B6 (n=14) and Il1β−/− (n=15) mice were orally infected with wt S. Typhimurium, and weight (G) and survival
(H) were monitored. Data represent two experiments combined.
(I) Original Western blot images from Figure 4.
(J) Cleaved Caspase-1 p20 from Western blots was quantified as percent of full-length Caspase-1 using ImageJ densitometry analysis. n=2/group for 24hr and n=3/group for 48hr.
(K) Myeloid cells (CD45+CD3−CD19−) were sorted from LP preparations from naïve B6, Casp1/11−/− or Il1β−/− mice and infected in vitro with MOI 1 of wt or ΔslrP S. Typhimurium. Mature IL-1β levels in supernatant were determined by IL-1β bioassay 6hr post-infection. Graph depicts 2 independent experiments for cells from B6 mice and represents 2 independent experiments for cells from genetic knockout mice.
(L) Intracellular CFU in sorted myeloid cells from the indicated genotypes infected for 6hr in vitro with wt or ΔslrP S. Typhimurium. n=3–4 per group.
(M) Western blot analysis of CASPASE-1 cleavage (p20) in in vitro infected myeloid cells from (J). Blot representative of 2 independent experiments. Due to space constraints, blot was cropped to remove membrane not relevant for the manuscript.
(N) Quantification of CASPASE-1 p20 from (l) normalized to β-actin using ImageJ densitometry analysis. Graph depicts quantification from 2 independent experiments for each genotype.
Error bars indicate +/− SEM. *p<0.05, unpaired student t-test; ***p<0.0005 by log rank analysis.
(A) Il1β−/− mice infected with ΔslrP were given food ad libitum (n=6) or given restricted amounts of food (n=6) as described in methods and Figure 2E. ΔslrP-infected B6 (n=5) were fed ad libitum. Grams of food per animal was normalized to consumption of uninfected animals of appropriate genotype.
(B–C) Uninfected Il1β−/− mice were food restricted as described in methods and Figure 2E. Weight (B) and survival
(C) were monitored. n = 6.
(D) Quantitative PCR validation of expression of genes from Fig 5C on RNA isolated from hypothalamus of B6 or Il1β−/− mice infected with wt or ΔslrP at 48hr. n=5–10 mice/group.
Error bars indicate +/− SEM. *p<0.05 for B6 wt vs ΔslrP unpaired student t-test.
(A) Total amount of grams of food consumed per mouse in 24 hours under uninfected conditions of vagotomized (n=5) and sham-vagotomized (n=4) mice.
(B) Quantitative PCR validation of expression of genes from Fig 5C on RNA isolated from hypothalamus of sham or vagotomized B6 mice infected with ΔslrP for 48hr. n=4–5 mice/group.
Error bars indicate +/− SEM. *p<0.05 unpaired student t-test.
(A) Splenic and liver burden of B6 mice infected IP with wt and ΔslrP S. Typhimurium for 48hr (left) and 96hr (right). n=7–8 mice per group.
(B) Orally-infected animals from the indicated groups were assigned a dissemination score of “1” (at least 1 CFU at the level of detection in spleen) or “0” (no CFU detected in spleen) at 72 hrs post infection. Dissemination index refers to splenic burden 72hr post-infection. n=10/group
(C) CFU in spleen of mice from (B) was plotted against fecal shedding index for each individual mouse. “−“ denotes mice with no fecal shedding, and “+” denotes mice with fecal burden at 72 hrs postinfection. Dotted line indicates limit of detection, and mice with no CFU detected in the spleen are indicated by symbols below this line.
(D) CFU detected in feces of mice from (C) was plotted against spleen dissemination index for each individual mouse. “−“ denotes mice with no spleen dissemination, and “+” denotes mice with spleen burden at 72 hrs postinfection. Dotted line indicates limit of detection, and mice with no CFU detected in the feces are indicated by symbols below this line. This graph looks the same as shown in Figure 6D, because the animals used for analysis of fecal burden and spleen/liver dissemination are the same and the same animals with liver dissemination event had a spleen dissemination event yielding similar plots.
(E) Dissemination index for the PP and MLN 72hr post-infection of the indicated groups. n=10/group
(F) CFU in PP (left) and MLN (right) of mice from (E) was plotted against fecal shedding index for each individual mouse. “−“ denotes mice with no fecal shedding, and “+” denotes mice with fecal burden. Dotted line indicates limit of detection, and mice with no CFU detected in the indicated tissue are indicated by symbols below this line.
(G) CFU detected in feces of mice from (F) was plotted against PP dissemination index (left) and MLN dissemination index (right) for each individual mouse. “−“ denotes mice with no organ dissemination, and “+” denotes mice with organ burden. Dotted line indicates limit of detection, and mice with no CFU detected in the feces are indicated by symbols below this line.
(H) Dissemination index of indicated organs of ΔslrP-infected Il1β−/− or B6mice that were fed ad libitum or ΔslrP-infected Il1β−/− food-restricted. n=13–20 per group. Graph represents 2–3 experiments combined.
(I) CFU in indicated tissue of the mice from (H).
(J) Quantitative PCR analysis of Cldn2 and Cldn4 expression in intestinal epithelial cells 48hr after infection of B6 mice with indicated strain of S. Typhimurium. n=5/group. Data are representative of 2 individual experiments.
Error bars indicate +/− SEM. ****p<0.0001; **p<0.01; *p<0.05 by unpaired student t test.
Table S1. RNAseq analysis of hypothalamus of wt and ΔslrP infected B6 mice at 48hr post-infection. Related to Figure 5.
Table S2. Primers used in this study. Related to STAR Methods.
Highlights.
Feeding status of host regulates pathogen virulence and transmission
Pathogen inhibition of IL-1β prevents anorexia via the gut-brain axis
Pathogen inhibition of anorexia promotes host survival and pathogen transmission
Sickness-induced anorexia creates trade-offs between virulence and transmission
Pathogens can interfere with the gut-brain circuits that control host anorexia during infection to promote survival while facilitating disease transmission
Acknowledgments
We thank R. Evans and B. Collins for use of the CLAMS equipment; M. Montminy for use of the EchoMRI; C. Fitzpatrick for help with FACS; G. Sulli for reagents; M. Abt for technical advice. This work was supported by NIH grant R01AI114929 (J.S.A), The Nomis Foundation, the Searle Scholar Foundation (J.S.A), the Ray Thomas Edward Foundation (J.S.A.), CA014195 and a Nomis Foundation Postdoctoral Fellowship (S.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
J.S.A conceived and designed the study. S.R. and J.S.A. designed and oversaw all experiments. S.R., A.M.P.S. and J.S.A. performed experiments. C.P.O. provided consultation, technical and analytical assistance for experiments involving flow cytometry. M.L. performed the pathology analysis. D.M. designed and conducted breeding and helped with germ free mouse experiments. S.R. and J.S.A analyzed the data. S.R. and J.S.A. wrote the paper.
The authors declare no conflicts of interest.
References
- Alexander M. Why microbial predators and parasites do not eliminate their prey and hosts. Annu Rev Microbiol. 1981;35:113–133. doi: 10.1146/annurev.mi.35.100181.000553. [DOI] [PubMed] [Google Scholar]
- Ayres JS. Inflammasome-microbiota interplay in host physiologies. Cell host & microbe. 2013;14:491–497. doi: 10.1016/j.chom.2013.10.013. [DOI] [PubMed] [Google Scholar]
- Ayres JS. Cooperative Microbial Tolerance Behaviors in Host-Microbiota Mutualism. Cell. 2016;165:1323–1331. doi: 10.1016/j.cell.2016.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres JS, Schneider DS. The role of anorexia in resistance and tolerance to infections in Drosophila. PLoS biology. 2009;7:e1000150. doi: 10.1371/journal.pbio.1000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedoyan JK, Patil CS, Kyriakides TR, Spence KD. Effect of Excess Dietary Glucose on Growth and Immune-Response of Manduca-Sexta. J Insect Physiol. 1992;38:525–532. [Google Scholar]
- Boyer L, Lemichez E. Targeting of host-cell ubiquitin and ubiquitin-like pathways by bacterial factors. Nat Rev Microbiol. 2004;2:779–788. doi: 10.1038/nrmicro1005. [DOI] [PubMed] [Google Scholar]
- Brandt SM, Dionne MS, Khush RS, Pham LN, Vigdal TJ, Schneider DS. Secreted Bacterial Effectors and Host-Produced Eiger/TNF Drive Death in aSalmonella-Infected Fruit Fly. PLoS biology. 2004;2:e418. doi: 10.1371/journal.pbio.0020418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. Journal of Experimental Medicine. 2010;207:1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger JM, Hwangbo DS, Corby-Harris V, Promislow DE. The functional costs and benefits of dietary restriction in Drosophila. Aging Cell. 2007;6:63–71. doi: 10.1111/j.1474-9726.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- Dantzer R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin North Am. 2009;29:247–264. doi: 10.1016/j.iac.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, Lur G, Dimaline R, Varro A, Raybould H, Dockray GJ. EGR1 Is a target for cooperative interactions between cholecystokinin and leptin, and inhibition by ghrelin, in vagal afferent neurons. Endocrinology. 2010;151:3589–3599. doi: 10.1210/en.2010-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn PE, Bohnert TJ, Russell V. Regulation of antibacterial protein synthesis following infection and during metamorphosis of Manduca sexta. Ann N Y Acad Sci. 1994;712:117–130. doi: 10.1111/j.1749-6632.1994.tb33567.x. [DOI] [PubMed] [Google Scholar]
- Ewald PW. Evolution of infectious disease. Response. J Hist Behav Sci. 1996;32:234–235. [Google Scholar]
- Exton MS. Infection-induced anorexia: active host defence strategy. Appetite. 1997;29:369–383. doi: 10.1006/appe.1997.0116. [DOI] [PubMed] [Google Scholar]
- Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12:489–495. doi: 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- Galan JE. Molecular genetic bases of Salmonella entry into host cells. Mol Microbiol. 1996;20:263–271. doi: 10.1111/j.1365-2958.1996.tb02615.x. [DOI] [PubMed] [Google Scholar]
- Goehler LE, Gaykema RPA, Nguyen KT, Lee JE, Tilders FJH, Maier SF, Watkins LR. Interleukin-1 beta in immune cells of the abdominal vagus nerve: a link between the immune and nervous systems? Journal of Neuroscience. 1999;19:2799–2806. doi: 10.1523/JNEUROSCI.19-07-02799.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JI, Dewey KG, Mills DA, Medzhitov RM. The Human Gut Microbiota and Undernutrition. Sci Transl Med. 2012;4 doi: 10.1126/scitranslmed.3004347. [DOI] [PubMed] [Google Scholar]
- Hansen MK, O’Connor KA, Goehler LE, Watkins LR, Maier SF. The contribution of the vagus nerve in interleukin-1 beta-induced fever is dependent on dose. Am J Physiol-Reg I. 2001;280:R929–R934. doi: 10.1152/ajpregu.2001.280.4.R929. [DOI] [PubMed] [Google Scholar]
- Hart BL. Biological basis of the behavior of sick animals. Neuroscience and biobehavioral reviews. 1988;12:123–137. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- Hensel M, Shea JE, Waterman SR, Mundy R, Nikolaus T, Banks G, Vazquez-Torres A, Gleeson C, Fang FC, Holden DW. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol Microbiol. 1998;30:163–174. doi: 10.1046/j.1365-2958.1998.01047.x. [DOI] [PubMed] [Google Scholar]
- Johnstone LE, Fong TM, Leng G. Neuronal activation in the hypothalamus and brainstem during feeding in rats. Cell Metab. 2006;4:313–321. doi: 10.1016/j.cmet.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Kristan DM. Chronic calorie restriction increases susceptibility of laboratory mice (Mus musculus) to a primary intestinal parasite infection. Aging Cell. 2007;6:817–825. doi: 10.1111/j.1474-9726.2007.00345.x. [DOI] [PubMed] [Google Scholar]
- Kyriazakis II, Tolkamp BJ, Hutchings MR. Towards a functional explanation for the occurrence of anorexia during parasitic infections. Anim Behav. 1998;56:265–274. doi: 10.1006/anbe.1998.0761. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, Flavell RA, Galan JE. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. Journal of Experimental Medicine. 2006;203:1407–1412. doi: 10.1084/jem.20060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRock CN, Cookson BT. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell host & microbe. 2012;12:799–805. doi: 10.1016/j.chom.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libert S, Chao Y, Zwiener J, Pletcher SD. Realized immune response is enhanced in long-lived puc and chico mutants but is unaffected by dietary restriction. Mol Immunol. 2008;45:810–817. doi: 10.1016/j.molimm.2007.06.353. [DOI] [PubMed] [Google Scholar]
- Mackinnon MJ, Gandon S, Read AF. Virulence evolution in response to vaccination: the case of malaria. Vaccine. 2008;26(Suppl 3):C42–52. doi: 10.1016/j.vaccine.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maculins T, Fiskin E, Bhogaraju S, Dikic I. Bacteria-host relationship: ubiquitin ligases as weapons of invasion. Cell Res. 2016;26:499–510. doi: 10.1038/cr.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH, Quast MJ, Gorenstein D, Chen KH, Chan L. Absence of perilipin results in leanness and reverses obesity in Lepr(db/db) mice. Nat Genet. 2000;26:474–479. doi: 10.1038/82630. [DOI] [PubMed] [Google Scholar]
- McLaughlin LM, Xu H, Carden SE, Fisher S, Reyes M, Heilshorn SC, Monack DM. A microfluidic-based genetic screen to identify microbial virulence factors that inhibit dendritic cell migration. Integr Biol (Camb) 2014;6:438–449. doi: 10.1039/c3ib40177d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335:936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Scherer CA, Tsolis RM, Kingsley RA, Adams LG, Baumler AJ, Miller SI. Salmonella typhimurium leucine-rich repeat proteins are targeted to the SPI1 and SPI2 type III secretion systems. Mol Microbiol. 1999;34:850–864. doi: 10.1046/j.1365-2958.1999.01651.x. [DOI] [PubMed] [Google Scholar]
- Monack DM, Mueller A, Falkow S. Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat Rev Microbiol. 2004;2:747–765. doi: 10.1038/nrmicro955. [DOI] [PubMed] [Google Scholar]
- Muller AJ, Hoffmann C, Galle M, Van Den Broeke A, Heikenwalder M, Falter L, Misselwitz B, Kremer M, Beyaert R, Hardt WD. The S. Typhimurium Effector SopE Induces Caspase-1 Activation in Stromal Cells to Initiate Gut Inflammation. Cell Host & Microbe. 2009;6:125–136. doi: 10.1016/j.chom.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Murray MJ, Murray AB. Anorexia of Infection as a Mechanism of Host Defense. Am J Clin Nutr. 1979;32:593–596. doi: 10.1093/ajcn/32.3.593. [DOI] [PubMed] [Google Scholar]
- Page AL, de Rekeneire N, Sayadi S, Aberrane S, Janssens AC, Rieux C, Djibo A, Manuguerra JC, Ducou-le-Pointe H, Grais RF, et al. Infections in Children Admitted with Complicated Severe Acute Malnutrition in Niger. Plos One. 2013;8 doi: 10.1371/journal.pone.0068699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul REL, Lafond T, Muller-Graf CDM, Nithiuthai S, Brey PT, Koella JC. Experimental evaluation of the relationship between lethal or non-lethal virulence and transmission success in malaria parasite infections. Bmc Evol Biol. 2004;4 doi: 10.1186/1471-2148-4-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov VA, Tracey KJ. Neural regulators of innate immune responses and inflammation. Cell Mol Life Sci. 2004;61:2322–2331. doi: 10.1007/s00018-004-4102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex-linking immunity and metabolism. Nat Rev Endocrinol. 2012;8:743–754. doi: 10.1038/nrendo.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, Bogatyrev SR, Ismagilov RF, Pamer EG, Turnbaugh PJ, et al. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature. 2014;514:638–641. doi: 10.1038/nature13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raupach B, Peuschel SK, Monack DM, Zychlinsky A. Caspase-1-mediated activation of interleukin-1 beta (IL-1 beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar typhimurium infection. Infection and Immunity. 2006;74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz BW, Aktan I, Nogusa S, Gardner EM. Energy restriction impairs natural killer cell function and increases the severity of influenza infection in young adult male C57BL/6 mice. J Nutr. 2008;138:2269–2275. doi: 10.3945/jn.108.093633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby T, McLaughlin L, Gopinath S, Monack D. Salmonella’s long-term relationship with its host. FEMS Microbiol Rev. 2012;36:600–615. doi: 10.1111/j.1574-6976.2012.00332.x. [DOI] [PubMed] [Google Scholar]
- Schieber AM, Lee YM, Chang MW, Leblanc M, Collins B, Downes M, Evans RM, Ayres JS. Disease tolerance mediated by microbiome E. coli involves inflammasome and IGF-1 signaling. Science. 2015;350:558–563. doi: 10.1126/science.aac6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Nielsen K. Animal physiology : adaptation and environment. 5th. Cambridge England; New York, NY, USA: Cambridge University Press; 1997. [Google Scholar]
- Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol. 2008;8:889–895. doi: 10.1038/nri2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin ME, Muller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, Maslowski KM, Hardt WD. Epithelium-Intrinsic NAIP/NLRC4 Inflammasome Drives Infected Enterocyte Expulsion to Restrict Salmonella Replication in the Intestinal Mucosa. Cell Host & Microbe. 2014;16:237–248. doi: 10.1016/j.chom.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Steeb B, Claudi B, Burton NA, Tienz P, Schmidt A, Farhan H, Maze A, Bumann D. Parallel Exploitation of Diverse Host Nutrients Enhances Salmonella Virulence. Plos Pathogens. 2013;9 doi: 10.1371/journal.ppat.1003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Muthukumar AR, Lawrence RA, Fernandes G. Effects of calorie restriction on polymicrobial peritonitis induced by cecum ligation and puncture in young C57BL/6 mice. Clin Diagn Lab Immunol. 2001;8:1003–1011. doi: 10.1128/CDLI.8.5.1003-1011.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsolis RM, Townsend SM, Miao EA, Miller SI, Ficht TA, Adams LG, Baumler AJ. Identification of a putative Salmonella enterica serotype typhimurium host range factor with homology to IpaH and YopM by signature-tagged mutagenesis. Infection and immunity. 1999;67:6385–6393. doi: 10.1128/iai.67.12.6385-6393.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annual review of immunology. 2013;31:73–106. doi: 10.1146/annurev-immunol-032712-095944. [DOI] [PubMed] [Google Scholar]
- Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot JD, Booth CJ, Medzhitov R. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell. 2016;166:1512–1525 e1512. doi: 10.1016/j.cell.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham ME, Brown NF, Boyle EC, Coombes BK, Finlay BB. Virulence is positively selected by transmission success between mammalian hosts. Current biology : CB. 2007;17:783–788. doi: 10.1016/j.cub.2007.03.067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A–B) Rate of oxygen consumption (VO2) (A) and rate of CO2 production (VCO2) (B) were determined by CLAMS 72hr after oral infection of B6 mice with stated strain of S. Typhimurium. Black bar indicates dark/night cycle, and white bar indicates light/day cycle. n=6/group.
(C) Food consumption of B6 mice orally infected with wt ST, ΔslrP ST with an empty control vector, or ΔslrP complemented with a vector expressing wt slrP under the native promoter. Grams of food per animal was normalized to consumption of uninfected animals. Data are two independent experiments combined, n=15–30 animals per group.
(D) CFU in the SI of B6 mice infected with wt ST, ΔslrP ST with an control empty vector, or ΔslrP ST complemented with wt slrP under the native promoter. n=6–8 per group and representative of two independent experiments. (E–F) Uninfected B6 mice were food restricted as described in methods, and weight (E) and survival (F) were monitored. n=5 mice.
* indicates p<0.05 and ** indicates p<0.005 by unpaired student’s t test; error bars indicate +/− SEM.
(A) Histological scoring of spleen, liver, and ileum 72hr after oral infection of B6 mice with stated strain of S. Typhimurium. n=5/group. “ND” indicates none detected – no pathology above limit of detection.
(B) Gating strategy for flow cytometric analysis of leukocyte populations in the small intestine LP: T/B lymphocytes (P1), neutrophils (P2), phagocytic macrophages (P3), inflammatory monocytes (P4).
(C) Gating strategy for flow cytometric analysis of migratory dendritic cells (P5) in the MLNs.
(D) Flow cytometric analysis of percentages of LP and MLN leukocytes 48hr after oral infection of B6 mice with stated strain of S. Typhimurium. Animal numbers shown.
(E) Total cellularity (left) and flow cytometric analysis of numbers (top) and percentages (bottom) of LP and MLN leukocytes 24hr after oral infection of B6 mice with stated strain of S. Typhimurium. n=8–9 mice per group.
(F) Gating strategy for flow cytometric analysis of leukocyte populations in the spleen and liver: T/B lymphocytes (P1), neutrophils (P2), macrophages (P3), inflammatory monocytes (P4), and dendritic cells (P5).
(G) Total cellularity (left) and flow cytometric analysis of numbers (top) and percentages (bottom) of spleen and liver leukocytes 24hr after oral infection of B6 mice with stated strain of S. Typhimurium. n=5 mice per group.
(H) Total cellularity (left) and flow cytometric analysis of numbers (top) and percentages (bottom) of spleen and liver leukocytes 48hr after oral infection of B6 mice with stated strain of S. Typhimurium. n=5 mice per group.
(I) ELISA measurements of IL-6 in the indicated tissues 24hr after oral infection of B6 mice with wt or ΔslrP S. Typhimurium. n=4–7 mice per group. ND denotes Not Detected. NS denotes Not Significant.
Error bars indicate +/− SEM. All comparisons not significant by unpaired student t-test.
(A) Levels of active/mature IL-1β determined by IL-1β bioassay of whole organ homogenates in liver, spleen and serum of B6 mice 24hr post-infection (n=9–13/group). Data represent 2 independent experiments combined. A. U. indicates arbitrary units of reporter activation.
(B) Levels of active/mature IL-1β determined by IL-1β bioassay of the stomach and pancreas 24hr and 48hr after oral infection of B6 mice with wt and ΔslrP S. Typhimurium. n=5 infected mice per group and n=3 vehicle (PBS)-gavaged mice. A.U. indicates arbitrary units of reporter activity.
(C) Levels of active/mature IL-1β determined by IL-1β bioassay of the SI 24hr and 48hr after oral infection of B6 mice with wt ST, ΔslrP ST with an empty control vector, or ΔslrP ST complemented with a vector expressing wt slrP under the native promoter. Data are two independent experiments combined, n=15–30 animals per group. * indicates p<0.05 and *** indicates p<0.0005 by unpaired t-test. A.U. indicates arbitrary units of reporter activity.
(D–F) Food consumption (D), weight loss (E), and survival (F) of ΔslrP infected B6 (n=9) and Il1β−/− (n=10) mice. (C) Grams of food per animal was normalized to consumption of uninfected animals. Bedding from B6 and Il1β−/− cages was mixed to control for differences in microbiota as described in methods.
(G–H) B6 (n=14) and Il1β−/− (n=15) mice were orally infected with wt S. Typhimurium, and weight (G) and survival
(H) were monitored. Data represent two experiments combined.
(I) Original Western blot images from Figure 4.
(J) Cleaved Caspase-1 p20 from Western blots was quantified as percent of full-length Caspase-1 using ImageJ densitometry analysis. n=2/group for 24hr and n=3/group for 48hr.
(K) Myeloid cells (CD45+CD3−CD19−) were sorted from LP preparations from naïve B6, Casp1/11−/− or Il1β−/− mice and infected in vitro with MOI 1 of wt or ΔslrP S. Typhimurium. Mature IL-1β levels in supernatant were determined by IL-1β bioassay 6hr post-infection. Graph depicts 2 independent experiments for cells from B6 mice and represents 2 independent experiments for cells from genetic knockout mice.
(L) Intracellular CFU in sorted myeloid cells from the indicated genotypes infected for 6hr in vitro with wt or ΔslrP S. Typhimurium. n=3–4 per group.
(M) Western blot analysis of CASPASE-1 cleavage (p20) in in vitro infected myeloid cells from (J). Blot representative of 2 independent experiments. Due to space constraints, blot was cropped to remove membrane not relevant for the manuscript.
(N) Quantification of CASPASE-1 p20 from (l) normalized to β-actin using ImageJ densitometry analysis. Graph depicts quantification from 2 independent experiments for each genotype.
Error bars indicate +/− SEM. *p<0.05, unpaired student t-test; ***p<0.0005 by log rank analysis.
(A) Il1β−/− mice infected with ΔslrP were given food ad libitum (n=6) or given restricted amounts of food (n=6) as described in methods and Figure 2E. ΔslrP-infected B6 (n=5) were fed ad libitum. Grams of food per animal was normalized to consumption of uninfected animals of appropriate genotype.
(B–C) Uninfected Il1β−/− mice were food restricted as described in methods and Figure 2E. Weight (B) and survival
(C) were monitored. n = 6.
(D) Quantitative PCR validation of expression of genes from Fig 5C on RNA isolated from hypothalamus of B6 or Il1β−/− mice infected with wt or ΔslrP at 48hr. n=5–10 mice/group.
Error bars indicate +/− SEM. *p<0.05 for B6 wt vs ΔslrP unpaired student t-test.
(A) Total amount of grams of food consumed per mouse in 24 hours under uninfected conditions of vagotomized (n=5) and sham-vagotomized (n=4) mice.
(B) Quantitative PCR validation of expression of genes from Fig 5C on RNA isolated from hypothalamus of sham or vagotomized B6 mice infected with ΔslrP for 48hr. n=4–5 mice/group.
Error bars indicate +/− SEM. *p<0.05 unpaired student t-test.
(A) Splenic and liver burden of B6 mice infected IP with wt and ΔslrP S. Typhimurium for 48hr (left) and 96hr (right). n=7–8 mice per group.
(B) Orally-infected animals from the indicated groups were assigned a dissemination score of “1” (at least 1 CFU at the level of detection in spleen) or “0” (no CFU detected in spleen) at 72 hrs post infection. Dissemination index refers to splenic burden 72hr post-infection. n=10/group
(C) CFU in spleen of mice from (B) was plotted against fecal shedding index for each individual mouse. “−“ denotes mice with no fecal shedding, and “+” denotes mice with fecal burden at 72 hrs postinfection. Dotted line indicates limit of detection, and mice with no CFU detected in the spleen are indicated by symbols below this line.
(D) CFU detected in feces of mice from (C) was plotted against spleen dissemination index for each individual mouse. “−“ denotes mice with no spleen dissemination, and “+” denotes mice with spleen burden at 72 hrs postinfection. Dotted line indicates limit of detection, and mice with no CFU detected in the feces are indicated by symbols below this line. This graph looks the same as shown in Figure 6D, because the animals used for analysis of fecal burden and spleen/liver dissemination are the same and the same animals with liver dissemination event had a spleen dissemination event yielding similar plots.
(E) Dissemination index for the PP and MLN 72hr post-infection of the indicated groups. n=10/group
(F) CFU in PP (left) and MLN (right) of mice from (E) was plotted against fecal shedding index for each individual mouse. “−“ denotes mice with no fecal shedding, and “+” denotes mice with fecal burden. Dotted line indicates limit of detection, and mice with no CFU detected in the indicated tissue are indicated by symbols below this line.
(G) CFU detected in feces of mice from (F) was plotted against PP dissemination index (left) and MLN dissemination index (right) for each individual mouse. “−“ denotes mice with no organ dissemination, and “+” denotes mice with organ burden. Dotted line indicates limit of detection, and mice with no CFU detected in the feces are indicated by symbols below this line.
(H) Dissemination index of indicated organs of ΔslrP-infected Il1β−/− or B6mice that were fed ad libitum or ΔslrP-infected Il1β−/− food-restricted. n=13–20 per group. Graph represents 2–3 experiments combined.
(I) CFU in indicated tissue of the mice from (H).
(J) Quantitative PCR analysis of Cldn2 and Cldn4 expression in intestinal epithelial cells 48hr after infection of B6 mice with indicated strain of S. Typhimurium. n=5/group. Data are representative of 2 individual experiments.
Error bars indicate +/− SEM. ****p<0.0001; **p<0.01; *p<0.05 by unpaired student t test.
Table S1. RNAseq analysis of hypothalamus of wt and ΔslrP infected B6 mice at 48hr post-infection. Related to Figure 5.
Table S2. Primers used in this study. Related to STAR Methods.