

ABSTRACT
We previously observed that SNAPIN, which is an adaptor protein in the SNARE core complex, was highly expressed in rheumatoid arthritis synovial tissue macrophages, but its role in macrophages and autoimmunity is unknown. To identify SNAPIN's role in these cells, we employed siRNA to silence the expression of SNAPIN in primary human macrophages. Silencing SNAPIN resulted in swollen lysosomes with impaired CTSD (cathepsin D) activation, although total CTSD was not reduced. Neither endosome cargo delivery nor lysosomal fusion with endosomes or autophagosomes was inhibited following the forced silencing of SNAPIN. The acidification of lysosomes and accumulation of autolysosomes in SNAPIN-silenced cells was inhibited, resulting in incomplete lysosomal hydrolysis and impaired macroautophagy/autophagy flux. Mechanistic studies employing ratiometric color fluorescence on living cells demonstrated that the reduction of SNAPIN resulted in a modest reduction of H+ pump activity; however, the more critical mechanism was a lysosomal proton leak. Overall, our results demonstrate that SNAPIN is critical in the maintenance of healthy lysosomes and autophagy through its role in lysosome acidification and autophagosome maturation in macrophages largely through preventing proton leak. These observations suggest an important role for SNAPIN and autophagy in the homeostasis of macrophages, particularly long-lived tissue resident macrophages.
KEYWORDS: acidification, autophagy, CTSD, endosome, LC3, lysosome, macrophage, proton leak, SNAPIN, vacuolar-type H+-ATPase
Introduction
Macrophages are either derived from the yolk sack or by fetal hematopoietic stem cells that differentiate into long-lived tissue-resident macrophages, such as microglia and Kupffer cells, or they differentiate from circulating bone marrow-derived monocytes.1,2 Macrophages are essential in the innate immune response by phagocytizing and digesting microbes. They also recognize microbial pathogens through intracellular and membrane-bound pattern recognition receptors, which results in the release of inflammatory chemokines and cytokines.3 Conversely, macrophages can suppress inflammatory responses by phagocytosis of apoptotic cells.4 Macrophages also play an important role in adaptive immunity by antigen processing and presentation to T cells, and by secreting a variety of cytokines and chemokines capable of recruiting and differentiating other immune cells.3 Although macrophages have a central role in protecting the host, they also contribute to the pathogenesis of inflammatory and degenerative diseases, such as rheumatoid arthritis (RA), in which they are major contributors to joint inflammation and destruction.
Earlier studies, from our laboratory and others, identified endogenous toll-like receptor (TLR) ligands in RA and demonstrated that they are capable of activating RA joint macrophages through TLR2 and TLR4.5,6 In an attempt to identify additional endogenous TLR ligands, we employed a yeast 2-hybrid system using the extracellular domain of TLR2 as the bait, and proteins expressed by RA synovial tissues as the prey.7 We discovered that SNAPIN (SNAP-associated protein) was a TLR2 interacting and signaling protein, which was highly expressed in CD68-positive macrophages in RA synovial tissue.7 SNAPIN was originally identified in neurons as an adaptor protein in the SNARE core complex, reported to bind to SNAP25 (synaptosome associated protein 25) and to facilitate synaptic vesicle fusion and neurotransmitter release.8-11 SNAPIN-deficient mice exhibit perinatal lethality,10 suggesting this protein has a critical and nonredundant function in vivo. Recent data showed that SNAPIN is critical for neuronal homeostasis through its role in coordination of late endosome-to-lysosome trafficking, lysosome maturation and autophagy.12-14
Autophagy is an essential intracellular process that degrades proteins and organelles such as mitochondria, promoting cellular homeostasis. Dysregulation of autophagy may contribute to the pathogenesis of cancer, diabetes, neurodegenerative, and inflammatory diseases.15-17 Autophagy is initiated through the formation of a phagophore, in some cases on the endoplasmic reticulum, in response to starvation or amino acid deprivation.18,19 The phagophore membrane forms around the targeted cargo and expands, with potential contributions from the plasma membrane, mitochondria, Golgi and endosomes, eventually forming a double-membrane autophagosome surrounding the target.18,20,21 The expansion of phagophore involves a number of ATG proteins essential for the formation of the mature autophagosome.22 Subsequent fusion with late endosomes and lysosomes delivers proteolytic enzymes to the autophagosomes, which involves the SNARE proteins STX7 (syntaxin 7), STX8 (syntaxin 8) and VTI1B,23 resulting in an autolysosome, in which the targeted materials are degraded and the amino acids and other nutrients delivered back to the cytoplasm.18,24 Upon autophagy induction, LC3 is lipidated and incorporated into the phagophore membrane. The lipidated form, LC3-II, migrates more rapidly in SDS-PAGE than its cytosolic form LC3-I, and thus can be used as a marker for autophagy activity.25
Given that SNAPIN was highly expressed in RA synovial tissue macrophages, we explored the function of SNAPIN in macrophages. The silencing of SNAPIN in macrophages resulted in the formation and accumulation of LC3-II-positive puncta and SQSTM1/p62, an autophagy cargo protein. The macrophages developed large autophagic vacuoles (AVs) that were incapable of digesting the engulfed target material. The lysosomes became swollen, with reduced acidification and CTSD (cathepsin D) activation. However, when SNAPIN was silenced, we did not observe a defect in endosome-to-lysosome fusion or of lysosome-to-autophagosome fusion in macrophages. Our results suggest that in macrophages SNAPIN is critical for lysosome acidification and CTSD activation, and is necessary for autophagosome maturation and digestion of the targeted materials.
Results
Silencing of SNAPIN in macrophages results in accumulation of late autophagic vacuoles
To determine the role of SNAPIN in macrophages, in vitro monocyte-differentiated human macrophages were transfected with nonspecific (NS) or SNAPIN siRNA. By immunofluorescence microscopy, in NS siRNA-transfected cells, SNAPIN was diffusely distributed throughout the cytosol and concentrated in the perinuclear region, while small LC3B puncta were observed in most cells (Fig. 1A, left panel). Following transfection with SNAPIN siRNA, in those cells in which SNAPIN was silenced, LC3 puncta were increased both in size and number (Fig. 1A, right panel). Similar results were observed with a second SNAPIN-C siRNA (Fig. S1A, B). The red:green fluorescence intensity ratio was increased (p < 0.0001) in the SNAPIN-C siRNA-transfected cells (Fig. S1C). Examined by electron microscopy, NS siRNA-treated macrophages demonstrated typical autophagosomes, 0.5–1 μm in diameter, with double membranes encircling the targeted material (Fig. 1B, left panel). Following the silencing of SNAPIN the accumulation of large AVs, ranging from 2 to 2.5 μm, containing undigested material including organelles, resembling autolysosomes,26 was observed (Fig. 1B, middle and right panels). These AVs had only one limiting membrane, and contained multiple cytoplasmic cargos (red arrows), some at different stages of degradation (green arrows). Similar results were observed in the murine macrophage cell line J774A1 stably expressing a Snapin shRNA, which resulted in a significant (p < 0.01) increase of large LC3-positive puncta (Fig. S2A-C). These data demonstrate that the silencing of SNAPIN in macrophages results in the accumulation of large AVs, resembling autolysosomes containing partially or undigested cargos.
Figure 1.
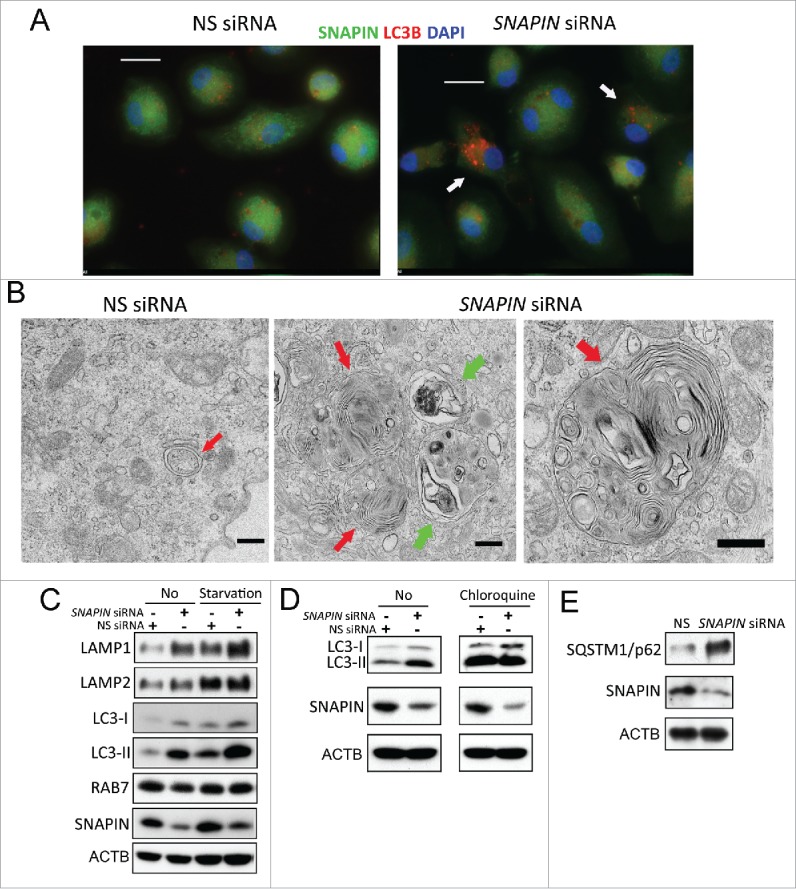
Silencing SNAPIN by siRNA resulted in accumulation of late autophagic vacuoles in human macrophages. (A) Immunofluorescence microscopy of human macrophages following transfection with NS or SNAPIN siRNA, and staining with antibodies to SNAPIN (green) or LC3 (red). Arrows on the right identify cells in which SNAPIN was reduced. Scale bar: 20 μm. (B) Transmission electron microscopy of NS siRNA- (left) and SNAPIN siRNA-treated macrophages (middle and right). A normal autophagosome (red arrow, left panel) is identified in the NS siRNA-transfected cells. In the SNAPIN siRNA-treated macrophages, nondigested organelles (red arrows) or partially digested material were identified in autolysosomes (green arrows). Scale bar: 500 nm. Immunoblot analysis of macrophages following transfection with NS or SNAPIN siRNAs before (No) and after starvation for 2 h, (C) after chloroquine treatment for 2 h (D), or left untreated (E). Panel (A) is representative of 5 and panels (B-E) are representative of 3 independent experiments.
To determine if silencing SNAPIN expression increased autophagy or blocked autophagosome maturation, macrophages transfected with NS or SNAPIN siRNA were serum starved to promote autophagy. Both the silencing of SNAPIN and starvation resulted in increased LC3-II and both LAMP1 and LAMP2, whereas no change in the late endosomal marker RAB7 was observed by immunoblot analysis (Fig. 1C). Starvation plus the silencing of SNAPIN resulted in more LC3-II, LAMP1 and LAMP2 than either treatment alone (Fig. 1C). Additional experiments employed chloroquine, which is lysosomotropic,27 elevates lysosomal pH, and blocks autophagic efflux, resulting in the accumulation of AVs.28 Chloroquine increased LC3-II in macrophages, however, the combination of chloroquine and SNAPIN siRNA did not show an additive effect (Fig. 1D), suggesting that SNAPIN and chloroquine both target a later stage of autophagy. Further, reduction of SNAPIN increased SQSTM1, a polyubiquitin- and LC3-binding protein, which is a marker for inhibition of autophagy,29-31 suggesting a compromise in hydrolytic digestion (Fig. 1E). Together, these observations suggest that the reduction of SNAPIN in macrophages does not interfere with the initiation of starvation-induced autophagy, but rather leads to defective degradative capacity in autolysosomes.
SNAPIN is essential for CTSD activation in macrophages
Since the accumulated autolysosomes in SNAPIN-silenced macrophages demonstrated incomplete digestion of their cargos, hydrolase activity was examined. CTSD, a major lysosomal aspartic protease that requires an acidic environment for activation,32 was examined employing BODIPY FL-pepstatin A, that preferentially binds to activated CTSD. Bafilomycin A1, a specific inhibitor of the vacuolar-type H+-translocating ATPase (V-ATPase) that prevents lysosome acidification and CTSD activation, served as a positive control. Similar to bafilomycin A1 treatment, siRNA silencing of SNAPIN resulted in reduction of BODIPY FL-pepstatin A staining, suggesting decreased CTSD activation (Fig. 2A). Confirming this observation, by immunoblot analysis, both SNAPIN silencing and bafilomycin A1 each resulted in diminished mature activated CTSD (Fig. 2B, C). In contrast, active CTSB (cathepsin B), which is a marker for macrophage differentiation,33 was reduced by bafilomycin A1 but not by silencing SNAPIN (Fig. 2B, D). Similar results were observed employing either total cell lysates or lysosome-enriched fractions from J774A1 murine macrophages stably transfected with a Snapin shRNA (Fig. S3). Further, following treatment with SCR shRNA, CTSD identified with an antibody that recognizes both proCTSD and the 28-kDa activated form colocalized with BODIPY FL-pepstatin A (Fig. S4), whereas after silencing SNAPIN, large puncta of CTSD were identified that did not colocalize with BODIPY FL-pepstatin A. These observations suggest that the reduction of CTSD is due to decreased activation rather than diminished production.
Figure 2.
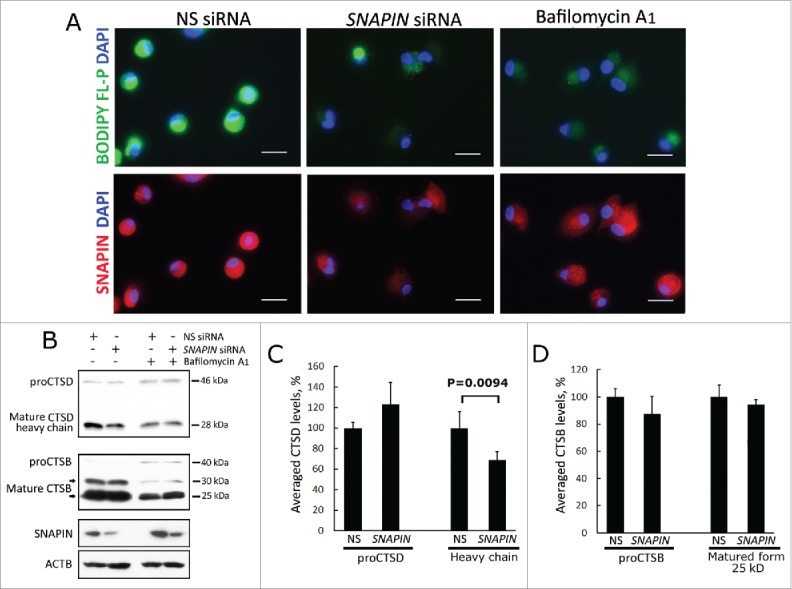
Silencing SNAPIN by siRNA reduced CTSD activation in human macrophages. Macrophages were transfected with NS or SNAPIN siRNAs or treated with bafilomycin A1 for 2 h and examined by immunofluorescence microscopy and immunoblot analysis for active CTSD. (A) Macrophages were stained with antibodies to SNAPIN (red) and BODIPY FL-pepstatin A (green), which binds to the active form of CTSD. Scale bar: 20 μm. Results are representative of 3 independent experiments. (B-D) proCTSD and mature CTSD (heavy chain, active form), and as a control proCTSB and mature CTSB were detected in whole cell lysates by immunoblot analysis (panel B) and quantified by densitometric analysis of immunoblots normalized to ACTB (n = 5) (panel D). Changes were presented as percentage of pro- and active forms of the CTSD with the results of the NS siRNA-treated cells set at 100%. The p value was determined by Student t test, comparing SNAPIN- to NS-siRNA reduced macrophages. Error bar: ± 1 SEM.
SNAPIN is enriched in isolated lysosomal fractions and the reduction of SNAPIN results in swollen lysosomes in macrophages
Because lysosomes are specialized to generate an acidic environment and the silencing of SNAPIN resulted in reduced active CTSD, the localization of SNAPIN was examined by immunofluorescence microscopy in macrophages. Although SNAPIN was broadly distributed, it colocalized with the lysosomal protein LAMP1, in a perinuclear pattern in NS siRNA-treated macrophages (Fig. 3A, left panel, and Fig. S5A, left panel). Lysosome enriched fractions were isolated by ultracentrifugation and gradient separation from J774A1 macrophages. Fraction 1 (F1) was enriched in lysosomes represented by the high content of LAMP1 and activated CTSB, with little contamination by markers of the endoplasmic reticulum (HSPA5/BIP, F2–4) or mitochondria (NDUFS3, F3–4) (Fig. S6A). Notably, F1 also contained markers for late endosomes (RAB7) and autophagosomes (LC3 and SQSTM1) (Fig. S6A). Next, structures present in F1 isolated with an anti-LC3 antibody conjugated to Dynabeads contained both LAMP1 and SNAPIN suggesting that SNAPIN interacted with lysosomes and/or AVs, such as autophagosomes or autolysosomes (Fig. S6B). Furthermore, immunoprecipitation of SNAPIN from total cell lysates co-purified LAMP1, but not LC3 or RAB7, from cells transfected with NS siRNA, whereas reduced LAMP1 was co-purified in SNAPIN siRNA-transfected macrophages, suggesting that SNAPIN may directly interact with LAMP1 in human macrophages (Fig. S6C). However, we were unable to pull down SNAPIN with an antibody to LAMP1, most likely due to the relatively low level of SNAPIN. Together these observations suggest that SNAPIN interacts with LAMP1-positive lysosomes; however, we cannot exclude an indirect interaction with LAMP1.
Figure 3.

Silencing of SNAPIN in macrophages induced the formation of swollen lysosomes. (A) Macrophages transfected with NS (left) or SNAPIN (middle and right) siRNAs were examined with antibodies to SNAPIN (green) and LAMP1 (red) by immunofluorescence microscopy. Large LAMP1-positive organelles are marked by arrowheads. The cell marked by a square in the middle panel was enlarged in the right panel. Macrophages transfected with (B) NS or (C) SNAPIN siRNAs were examined by immunofluorescence microscopy with antibodies to LAMP1 (magenta staining) and active CTSD (BODIPY FL-pepstatin A, green), with colocalization indicated in white. Arrows identify active CTSD within the LAMP1-positive lysosomes. Scale bar: 13.3 μm. The images were processed by deconvolution with NIS-element imaging software. The results are representative of 3 independent experiments.
The effect of SNAPIN reduction on lysosomal morphology was next examined. The forced reduction of SNAPIN in macrophages resulted in swollen LAMP1-positive organelles (Fig. 3A, middle and right panels, and similar results were seen with SNAPIN-C siRNA, Fig. S5A, right panel). In macrophages transfected with NS siRNA, active CTSD, as detected by BODIPY FL-pepstatin A, colocalized with LAMP1 (Fig. 3B). When SNAPIN was silenced, although diminished, residual active CTSD was identified within the swollen LAMP1-positive lysosomes (Fig. 3C, and with SNAPIN-C siRNA, Fig. S5B). Overall these observations suggest that SNAPIN interacts with lysosomes and that silencing SNAPIN results in swollen lysosomes that fail to activate CTSD.
Silencing SNAPIN does not inhibit late endosome-lysosome fusion
In order to identify the mechanism responsible for the lysosomal dysfunction, endosome trafficking to lysosomes was examined. Under homeostatic conditions, following transfection with NS siRNA, limited colocalization of LAMP1 and RAB7 was identified, although both were enriched in the perinuclear region (Fig. 4A). Following the silencing of SNAPIN, large LAMP1-positive lysosomes where identified which partially colocalized with RAB7 (Fig. 4B, C), suggesting that endosome-lysosome fusion occurred. These observations were confirmed with SNAPIN-C siRNA (Fig. S7A). Further, endocytic trafficking to lysosomes was examined by incubation of macrophages with Alexa Fluor 488 dextran for 2 h, followed by a 16-h chase. In the presence of NS siRNA, dextran colocalized with LAMP1, indicating efficient trafficking of the dextran to lysosomes (Fig. 4D). Likewise, following transfection with SNAPIN siRNA, dextran was readily observed within the swollen LAMP1-positive lysosomes (Fig. 4D, E, and with SNAPIN-C siRNA, Fig. S7B). These observations, together with the lack of reduction of pro-CTSD and CTSB in the lysosomal fractions following the silencing of SNAPIN (Fig. 3D, E), suggest that diminished endosomal trafficking to the swollen lysosomes was not the cause of the impaired lysosomal function following the silencing of SNAPIN in macrophages.
Figure 4.
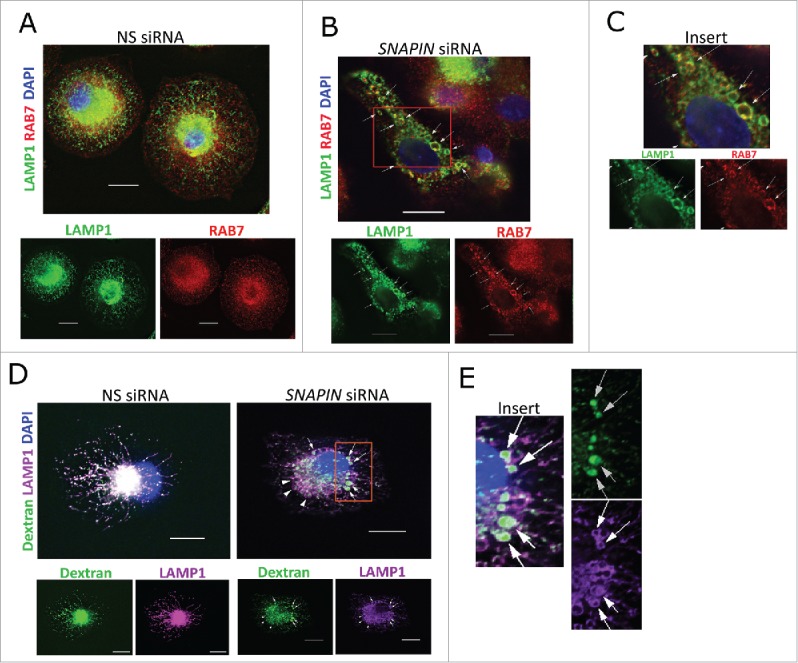
Silencing of SNAPIN in human macrophages did not affect fusion of late endosomes with lysosomes. Macrophages were transfected with NS (A) or SNAPIN siRNAs (B) and then examined by immunofluorescence microscopy employing antibodies to LAMP1 (green) and RAB7 (red). Both molecules exhibited perinuclear localization, however, colocalization was not apparent (left panel). The area marked in panel (B), was expanded in panel (C). Following SNAPIN siRNA transfection, a portion of the LAMP1-positive lysosomes incorporated RAB7 (arrow). (D) Human primary macrophages transfected with siRNAs were incubated with Alexa Fluor 488 dextran (green) for 2 h, chased for 16 h and stained with LAMP1. Swollen LAMP1-positive lysosomes containing dextran are marked by arrows. (E) Enlarged area indicated in panel (D). Scale bar: 13.3 μm. The images were processed by deconvolution with NIS-element imaging software. Results in this figure are representative of 3 independent experiments.
SNAPIN is required for lysosome acidification
Since activation of CTSD was reduced and endosomal trafficking to the lysosome was not impaired in macrophages following the silencing of SNAPIN, lysosomal acidification was examined employing dextran labeled with the pH sensitive pHrodo green or pH insensitive Alexa Fluor 546 red fluorescence dyes. In macrophages transfected with the NS siRNA the dextran that accumulated in the lysosomes emitted green and red fluorescence, which colocalized (Fig. 5A), indicating that the lysosomes were acidified. In contrast, the dextran that accumulated in macrophages transfected with SNAPIN siRNA, emitted little green fluorescence (Fig. 5B), consistent with impaired lysosomal acidification. Comparable results were observed following incubation with bafilomycin A1 (Fig. 5C). Quantification of the ratio of green to red fluorescence demonstrated significant (p < 0.001) reduction of lysosomal acidification following the silencing of SNAPIN or treatment with bafilomycin A1 (Fig. 5D, E). These observations were confirmed with SNAPIN-C siRNA (Fig. S8). Together, these observations demonstrate an essential role of SNAPIN in lysosomal acidification.
Figure 5.
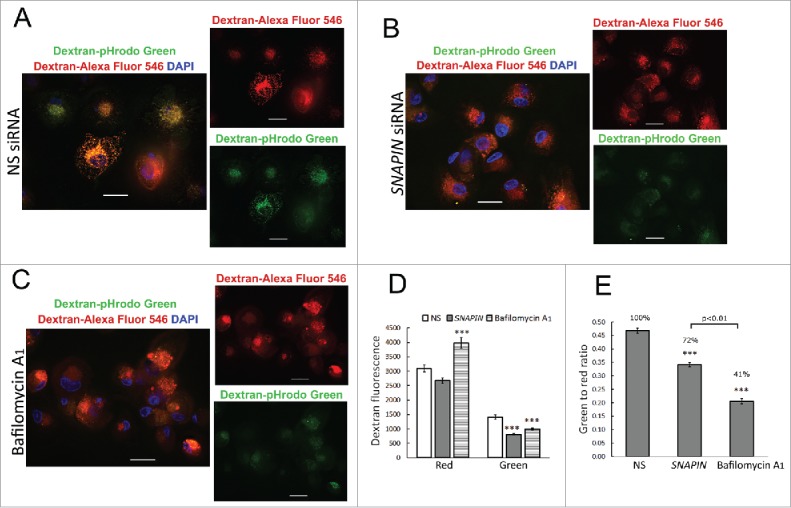
SNAPIN silencing in macrophages inhibited lysosome acidification. Human macrophages were transfected with NS (A) or SNAPIN siRNAs (B), then incubated for 2 h with dextran labeled with pH-sensitive pHrodo green dye and with dextran labeled with pH-insensitive Alexa Fluor 546 (red) and then chased for 6 h. (C) Bafilomycin A1 (100 nM) was added to cells that were not transfected with siRNA, for 2 h prior to harvesting the cells. Scale bar: 20 μm. (D) Green and red fluorescence mean intensity in each macrophage was measured with Nikon NIS element imaging software. The green:red ratios were calculated and the results summarized in the bar graph. The number of macrophages employed to measure fluorescence intensity was: NS = 160; SNAPIN = 166; bafilomycin A1 = 142. The ratio of green:red fluorescence was calculated from the data in panel (D). *** represents p<0.001, compare with NS siRNA-transfected macrophages following statistical analysis by AVONA. The results in this figure were obtained from 2 independent experiments.
SNAPIN is required for autophagosome acidification and maturation
Because the silencing of SNAPIN in macrophages resulted in reduced lysosomal acidification, the effect on autophagosome acidification was examined using a tandem mRFP-EGFP-LC3-expressing plasmid. During autophagosome formation, the tandem fluorescence-fused LC3 is recruited to and incorporated into the membrane of phagophores. The GFP signal is sensitive to the acidic and/or proteolytic condition of the lysosome, whereas mRFP is more stable,34 thus, the ratio of GFP to mRFP fluorescence reflects acidification resulting from autophagosome-lysosome fusion.35 Since we were not able to adequately transfect plasmids into macrophages, to perform these experiments, HEK293 cells stably transformed by SCR or Snapin shRNA lentiviral constructs were used to transiently express the tandem mRFP-GFP-LC3 protein. In the control SCR shRNA-transfected cells red puncta dominated, while the GFP signal was quenched, suggesting normal autophagosome-lysosome fusion and lysosomal acidification (Fig. 6A, C). The silencing of SNAPIN not only increased the LC3-II level (Fig. 6E) and the number of LC3 puncta, but also significantly elevated the green fluorescence intensity of LC3 puncta (Fig. 6B, C) and the ratio of green:red puncta (p < 0.01) (Fig. 6D), suggesting that silencing SNAPIN prevented fusion and/or increased the pH in autolysosomes. Overall, these results demonstrate that SNAPIN plays a key role in autophagosomal acidification and maturation.
Figure 6.
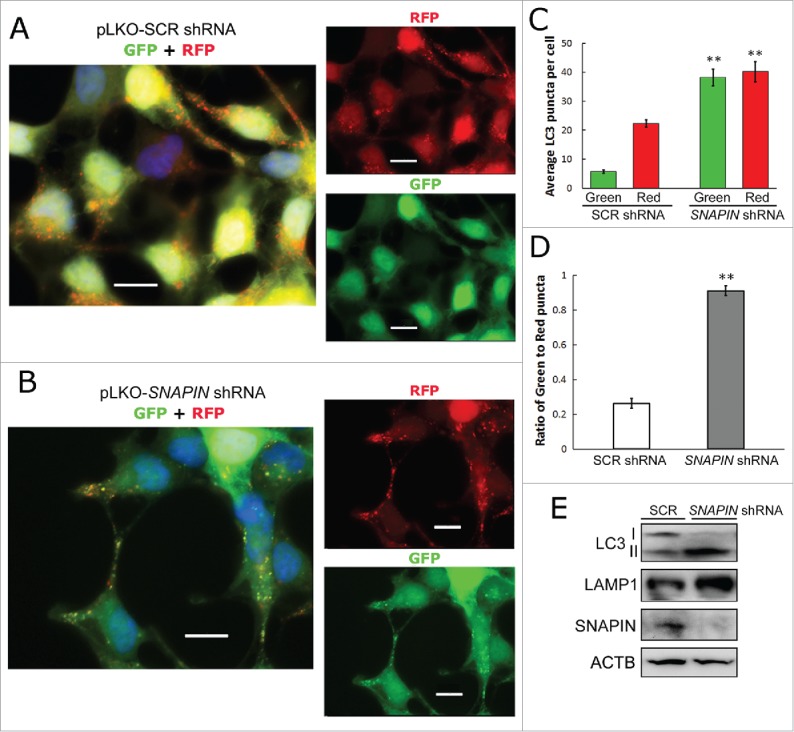
SNAPIN is important for autophagosome acidification. HEK293 cell lines stably transduced with scrambled (SCR)- (panel A) or SNAPIN shRNA (panel B)-expressing lenti-viruses, were transfected with the ptf-LC3 plasmid, which expresses LC3 tagged with tandem fluorescent RFP and GFP. The red fluorescence is expressed regardless of the pH, while the green fluorescence is quenched at low pH. (C) The average numbers of GFP- and RFP-positive puncta from SCR or SNAPIN shRNA-expressing cells are shown as a bar graph (n = 20; **p < 0.01, compared with the same colored puncta in SCR shRNA HEK293 cells). (D) The ratio of green:red puncta in cells was calculated and presented (n = 20 cells; **p < 0.01), comparing SCR and SNAPIN shRNA-expressing HEK293 cells. (E) Immunoblot analysis of the 2 cell lines. Scale bar: 13.3 μm. Results (panels A-D) were obtained from 4 independent experiments.
SNAPIN silencing in macrophages did not affect lysosome-autophagosome fusion
Since, recent observations demonstrated that autophagosome-lysosome fusion is independent of lysosomal acidification,36 the role of SNAPIN in this fusion process was examined by quantifying LC3 and LAMP1 double-positive puncta. Human macrophages were transfected with NS or SNAPIN siRNA, or incubated with rapamycin (an MTOR inhibitor and autophagy inducer) or bafilomycin A1. Total LC3 puncta, which includes those that are both LC3-only and LC3 and LAMP1-double positive, were significantly (p < 0.001) increased in macrophages after SNAPIN silencing, similar to treatment with rapamycin or bafilomycin A1 (Fig. 7A, B). The percentage of LC3+ LAMP1+ to total LC3 puncta is a measure of lysosome-autophagosome fusion. Rapamycin treatment did not affect lysosome-autophagosome fusion, even though total AVs were increased. However, SNAPIN silencing or bafilomycin A1 slightly increased the percentage of puncta that were LC3 and LAMP1 double positive, when compared with NS siRNA-transfected cells (p<0.001, Fig. 7C). However, colocalization of LC3 and LAMP1 in macrophages, measured by Pearson's correlation coefficient, showed no difference among cells treated with NS siRNA, rapamycin or SNAPIN siRNA (Fig. 7D), although bafilomycin A1-treated macrophages showed a slight increase in colocalization. Together, these observations demonstrate no defect in autophagosome-lysosome fusion following the silencing of SNAPIN.
Figure 7.
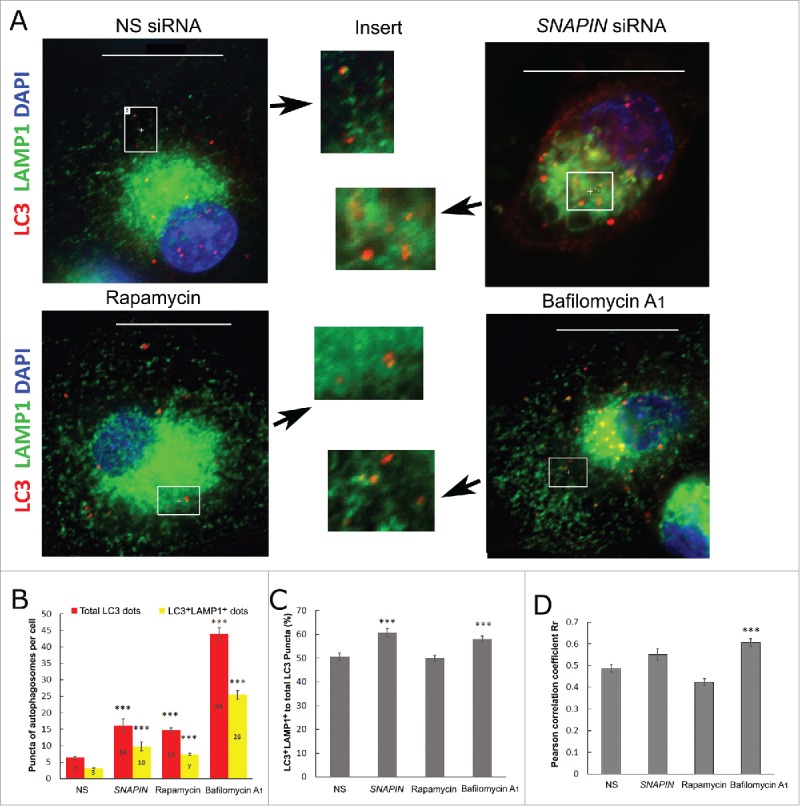
SNAPIN silencing in macrophages did not affect lysosome-autophagosome fusion. (A) Human macrophages were transfected with NS or SNAPIN siRNAs, incubated with 2 μM of rapamycin or 100 nM bafilomycin A1 for 2 h. Cells were fixed with methanol for 20 min and stained with antibodies of LC3 (red) and LAMP1 (green). The highlighted areas in each panel were enlarged and shown in the inserted panels as indicated by the arrows. (B) Total LC3-positive puncta (red bars) and LC3 and LAMP1 colocalized puncta (yellow bars) were counted (numbers indicated in the bars) in 2 independent experiments. (C) The percentage of LC3+ LAMP1+ puncta to total LC3 puncta was calculated and presented. (D) Colocalization between LAMP1 and LC3 was measured by Pearson's correlation coefficient Rr employing NIS-element imaging software. *** represents p<0.001, following analysis by AVONA compare with NS-transfected cells. The presented images were processed by deconvolution.
SNAPIN maintains lysosomal integrity preventing proton dissipation
In order to identify a mechanism by which SNAPIN maintains lysosomal homeostasis, J774A1 macrophage cell lines stably transformed with SCR or Snapin shRNAs were employed. The cells were incubated overnight with the pH-sensitive pHrodo Red and the pH-insensitive Alexa Fluor 488 green dextrans. An increased red:green ratio is indicative of a lower pH. After washing and incubation for an additional 8 h to allow the dextrans to localize to the lysosomes, the cells were incubated with the V-ATPase inhibitor concanamycin A (CcA) and the change of the red:green ratio was documented over time.37 As expected the red:green ratio was significantly lower in the Snapin shRNA-expressing cells prior to the addition of CcA (Fig. 8). After treatment with CcA the ratio was maintained in the control cells, while the ratio rapidly declined in the Snapin shRNA-expressing cells. These observations indicate that SNAPIN is necessary to maintain lysosomal homeostasis by preventing proton leakage from lysosomes.
Figure 8.
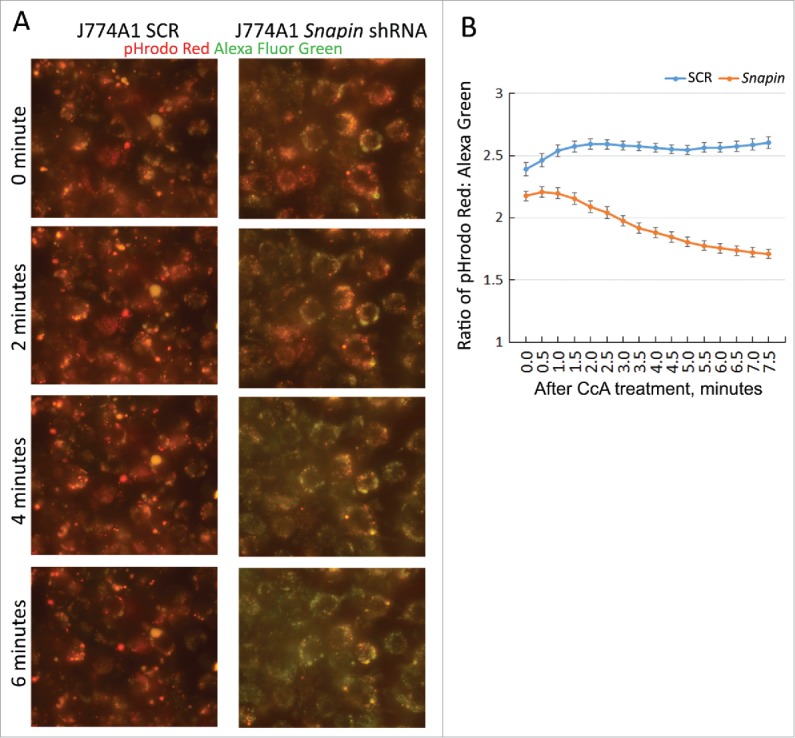
SNAPIN is important in maintaining lysosomal acidic pH. J774A1 macrophage cell line stably expressing a SCR shRNA or a Snapin shRNA was incubated overnight with a pH-sensitive pHrodo Red dextran and a pH-insensitive Alexa Fluor 488 green dextran. Then, dyes were replaced with fresh medium and the dextrans were chased for 8 h, allowing delivery to lysosomes. The H+-pump was inhibited by incubating cells in a Na+ buffer containing 2 μM concanamycin A (CcA). The leak of lysosomal H+ was assessed by the ratio change of red:green fluorescence in J774A1 living cells by photos taken automatically in half-minute intervals. (A) Representative photos of red:green color change in the J774A1 cell lines after CcA was added. (B) The ratio of red:green fluorescence intensity in J774A1 cells was calculated and presented. Data were obtained from 36 cells for the J774A1 SCR cell line and 42 cells for the J774A Snapin shRNA cell line. Results presented above are representative of 2 independent experiments. Every paired time point between SCR and Snapin shRNA cells was statistically different following ANOVA and Tukey's test (p < 0.05 at time 0 and p < 0.01–0.001 at all other time points).
Further studies were performed to determine the capacity of the V-ATPase proton pump. Stably transformed J774A1 cells were incubated with carbonyl cyanide m-chlorophenyl hydrazone (CCCP) in order to diminish the pH gradient of the lysosomal membrane.37 Once the pH was increased, the CCCP was removed and the lysosomes allowed to reacidify, using the change in the red:green ratio as a measure of pH change. Following the addition of CCCP, the red:green ratio decreased in both the SCR and Snapin shRNA-transfected cells (Fig. 9A). There was no difference in the rate of change of the ratios between the cell lines (Fig. 9B). The difference in the expression of SNAPIN between the lines is presented in Fig. 9D. Following the removal of CCCP the red:green ratio increased in both lines (Fig. 9A). The rate of change was significantly (p < 0.001) reduced during the first minute after removal of the CCCP in the Snapin-shRNA transformed cells (Fig. 9B, C). However, after the first minute there was no difference in the rate of increase of the red:green ratio between the lines, and red:green ratio of the Snapin shRNA-transformed line remained reduced compared with the control line (Fig. 9B, C). Together these observations suggest that while SNAPIN may have a modest effect on proton pump activity, its role in preventing proton leakage from the lysosome is more important in maintaining lysosomal homeostasis.
Figure 9.
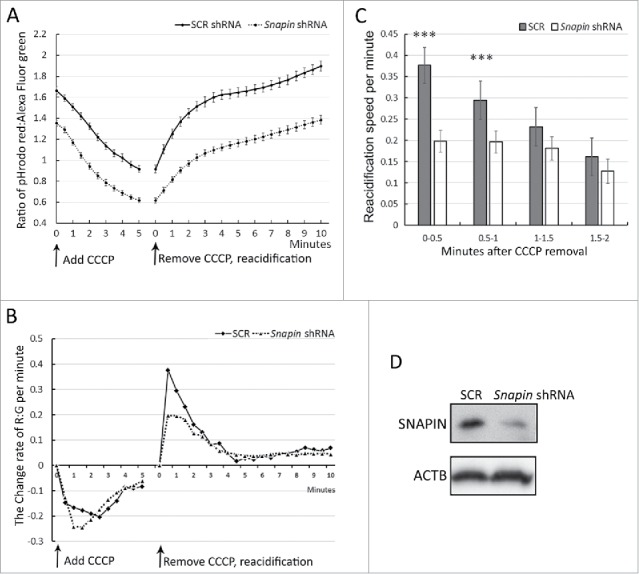
SNAPIN facilitates lysosomal acidification. J774A1 lines transformed with SCR or Snapin shRNAs were incubated overnight with a pH-sensitive pHrodo Red dextran and a pH-insensitive Alexa Fluor 488 green dextran. Afterwards, dextrans in J774A1 cells were chased for 8 h. J774A1 live-cell fluorescence imaging was taken automatically in half-minute intervals under a fluorescence microscope. (A) J774A1 cells were incubated with K+ buffer containing 450 μM carbonyl cyanide m-chlorophenyl hydrazone (CCCP), a protonophore capable of dissipating the lysosomal transmembrane ΔpH. The ratio of red:green fluorescence, which is inversely related to pH in lysosomes, was measured. Re-acidification of lysosomes started promptly after removing CCCP, replacing with Na+ buffer alone. The ratio of red:green was calculated and presented. (B) The rate of change of the red:green fluorescence per minute was calculated (ratiot2-ratiot1)/0.5 min) and the results plotted over time. (C) The rate of re-acidification in the first 2 min after CCCP removal was calculated and was summarized. *** represents p < 0.001 following ANOVA and Tukey's test. (D) The image demonstrates SNAPIN expression in the J774A1 cell lines by immunoblot analysis.
Discussion
We previously demonstrated that SNAPIN was significantly increased in the synovial tissue macrophages of patients with RA.7 This study documents for the first time an important role for SNAPIN in the homeostasis of human macrophages. Silencing of SNAPIN resulted in the accumulation of autolysosomes incapable of appropriately degrading their cargos. Increased autolysosomes may result either from autophagy induction, such as nutrient deprivation,38,39 or blockage of autophagy efflux, as occurs following inhibition or deficiency of lysosome hydrolase activity.40-42 AVs accumulated in SNAPIN-silenced macrophages, which demonstrated the typical signs of late stage blockage:43 one membrane (i.e., an autolysosome), insufficient cargo degradation, increased SQSTM144 and the identification of multilamellar and multivesicular structures.45 The effects of starvation and the silencing of SNAPIN were additive, whereas the addition of chloroquine, which blocks lysosomal acidification, was not. These findings suggest that SNAPIN is critical for efficient autophagic efflux and that the AVs induced following SNAPIN silencing were accumulated autolysosomes defective in lytic enzyme digestive capacity, which resulted in blockage at a late stage of autophagy flux.
Following the silencing of SNAPIN in macrophages, swollen LAMP1-positive structures consistent with lysosomes were observed because they were LAMP1 positive, contain CTSD and take up dextran after an overnight chase. The lysosomes were enlarged and a marker of late endosomes (RAB7) was also present on lysosomal membranes of SNAPIN-silenced macrophages. The swollen lysosomes exhibited impaired acidification and reduced activation of CTSD46 Swollen lysosomes and endosomes have also been noted following modulation of RAB family proteins including RAB5 and RAB7, and interference with the PtdIns3K pathway46-48 or an increase of lysosomal pH by chloroquine or other lysosomotropic weakly basic agents.49,50 These observations demonstrate that SNAPIN is necessary for lysosome homeostasis in macrophages.
Recent studies examining the mechanism of AV turnover have identified the importance of lysosome biogenesis in the autophagic process.24 Most cathepsins, including CTSD and CTSB, are synthesized as nonactive zymogens and are activated in the acidic environment of the lysosome. Inhibition of lysosome acidification reduces cathepsin activation, as well as their proteolytic abilities, since these enzymes operate primarily at low pH.51 The inhibition of lysosome protease activity by leupeptin and pepstatin A resulted in AV accumulation.52 CTSE (cathepsin E)−/− murine macrophages exhibit increased accumulation of AVs, as well as LC3 and SQSTM1.53 CTSD−/− and mice that are deficient in both CTSB and CTSL show abnormal accumulation of AVs in neurons,54 emphasizing the importance of these proteases in autophagosome biogenesis. Because incomplete digestion of cytoplasmic materials in accumulated AVs was observed in SNAPIN silenced macrophages, we examined the hydrolase activity in these cells. In SNAPIN-silenced cells, lysosome and autolysosome acidification was impaired, and CTSD activation was reduced, consistent with the results of SNAPIN depletion in neurons.12
Our data demonstrated impaired activation of CTSD, but no reduction of active CTSB following the silencing of SNAPIN, while both were impaired following treatment with bafilomycin A1. CTSD requires a more acidic pH to become proteolytically activated compare with the cysteine lysosomal enzymes such as CTSB and CTSL.55 The auto-activation of CTSD in vitro requires an acidic pH between 4 and 5, while the fully active 28-kDa version is observed at a pH of 3.56 CTSB is activated in a less acidic environment, and even at neutral pH.57 In order to more directly assess lysosomal pH, dextrans labeled with pH-sensitive and -insensitive fluorophores were employed,5859 and their ratio served as an indicator of the relative pH of the lysosomes.60 A 2-h incubation with bafilomycin A1, a V-ATPase inhibitor, reduced the ratio in macrophages by about 60%, while this ratio was reduced by 28% following the silencing of SNAPIN. These observations, together with those documenting impaired CTSD activation, indicate that SNAPIN deficiency results in an elevated lysosomal pH, although the increase of pH was not as great as observed with bafilomycin A1 treatment, and this may be due to variable reduction of SNAPIN in the macrophages.
Prior studies in neurons identified SNAPIN as an important mediator of late endosome transport.12 SNAPIN was also reported to interact with late endosomal SNAREs,61 and we previously identified partial colocalization of SNAPIN with RAB7 in macrophages.7 Late endosomes are involved in lysosome maturation by delivery of newly synthesized hydrolases to lysosomes. Several models have been proposed to explain how endosomes and lysosomes interact, such as direct fusion, kiss-and-run, or forming hybrid organelles.62,63 Since SNARE proteins STX7, STX8 and VAMP8 are required for late endosome-lysosome docking and fusion,64,65 the role of SNAPIN, a SNARE complex adaptor protein, in endosome-lysosomal fusion was examined. Under homeostatic conditions RAB7 did not substantially colocalize with LAMP1, nor with the active CTSD within lysosomes (data not shown), in macrophages. However, following the silencing of SNAPIN, RAB7 was readily identified incorporated on swollen LAMP1-positive lysosomes, suggesting the presence of active endosome-lysosome fusion. Further, no defect in the delivery of CTSD or dextran to swollen LAMP1-positive lysosomes was identified in SNAPIN-silenced macrophages. In lipid storage disorders, endosome-lysosome fusion is impaired, identified by the reduced incorporation of dextran into lysosomes.66 Our observations appear consistent with the results observed in snapin−/− mouse embryonic fibroblasts which exhibit increased colocalization of the endosome marker IGF2R/CI-MPR and LAMP1, suggesting efficient endosome-lysosome fusion in these cells.12 In contrast, in neurons, the speed of retrograde delivery of endosomes from peripheral axons to the central soma is reduced, potentially impairing endosome-lysosome fusion.12,13 RAB7 is necessary for endosome-lysosome fusion,67 and in macrophages, RAB7 levels are not changed in SNAPIN-silenced macrophages, suggesting no increase of late endosome accumulation. Additionally, depletion of RAB7 in HeLa cells impairs cargo transfer from endosomes to the lysosomes, suggesting RAB7 is required for endosome-to-lysosome delivery.68 These observations, together with the lack of reduction of CTSB and proCTSD in lysosomes, in SNAPIN-deficient macrophages, suggest that endosome-lysosome fusion is not impaired and was not the cause of the impaired lysosomal maturation, suggesting cell type-specific differences in the mechanisms by which SNAPIN controls lysosomal homeostasis.
Bafilomycin A1 has also been reported to prevent maturation of AVs by inhibition of fusion between AVs and lysosomes.69 Recent data,36 however, demonstrates that the inhibitory effect of bafilomycin A1 on V-ATPase activity is independent from AV-lysosome fusion. Our results confirm that treatment with bafilomycin A1, as well as silencing SNAPIN, dramatically increased LC3 puncta but did not affect colocalization of LAMP1 and LC3, suggesting no defect in AV-lysosome fusion. Similar results were reported employing Chinese hamster ovary cells treated with 100 nM bafilomycin A1 for 2 h.70 In contrast, longer treatment for 6–12 h with bafilomycin A1 was required to identify its effect on blocking autophagosome-lysosome fusion.71 These observations suggest that the effects of bafilomycin A1 observed in this study were due to increased pH and not reduced AV-lysosome fusion.
Studies were performed to identify potential mechanism(s) by which SNAPIN contributes to the maintenance of lysosomal acidity. Although there was a modest effect on proton pump activity, the more striking observation was the need for SNAPIN to prevent the loss of protons from lysosomes. The mechanisms regulating the leak of lysosomal protons are incompletely understood. Mutations of the ion channel MCOLN1/TRPML1 (mucolipin 1) are responsible for mucolipidosis type IV, which results in the accumulation of membranes and lipids in cytosolic organelles.72 MCOLIN1-deficient cells obtained from patients with mucolipidosis type IV demonstrate increased acidification, suggesting that MCOLIN1 may act as a proton channel releasing protons from lysosomes.72 It is possible that SNAPIN may attenuate the activity of the MCOLIN1 channel, in which case the reduction of SNAPIN would increase proton loss. Additionally, lysosomal cholesterol may be important in preventing proton leak since treatment of lysosomes with methyl-β-cyclodextrin results in the loss of cholesterol and increased pH through increased exchange of H+ and K+.73 However, there has been no description of how SNAPIN might affect cholesterol content of any organelle. Recently it has been demonstrated the position of lysosomes within cells relative to the nucleus was a determinant of the lysosomal pH. 37 Lysosomes in the periphery demonstrate increased pH, compared with those in the perinuclear region, due to increased permeability of protons and to decreased V-ATPase activity.37 In macrophages, the change of pH in SNAPIN-deficient cells is not strictly due to mobilization to the periphery as noted with Arl8b-transfected cells.37 The precise molecular interaction by which SNAPIN regulates lysosomal permeability remains to be determined.
Our data demonstrate that SNAPIN in macrophages is important in maintaining healthy lysosomes, which is necessary for efficient autophagy. The role for the increased SNAPIN in macrophages observed in inflammatory conditions such as RA7 may relate to the increased need for autophagy to promote survival or to accommodate critical functions of macrophages. Oxidative stress, which is present in RA synovial tissue, increases autophagy in macrophages.74 Autophagy-deficient macrophages demonstrate a decreased ability to protect against active tuberculosis7576 and atherosclerosis.77,78 Further, mice deficient in autophagy in myeloid cells, particularly macrophages, demonstrate increased spontaneous inflammation of the lungs79,80 by enhancing NFKB activation.81 Additionally, in TNF transgenic mice, autophagy-deficient macrophages caused less joint destruction due to decreased osteoclast differentiation.82 Also, autophagy is important in presenting pathogenic citrullinated antigens by macrophages and dendritic cells to CD4+ T cells,83 which may be an important mechanism in the pathogenesis of RA. Whether the increased expression of SNAPIN as observed in RA macrophages protects or promotes disease pathogenesis remains to be determined. However, the role of SNAPIN and autophagy may be particularly important in long-lived tissue-resident macrophages,2 which are important in suppressing inflammation, and may depend on autophagy for survival, in contrast to monocyte-derived macrophages, which are readily replaced.84
Materials and methods
Cell isolation and culture
Human monocytes were isolated from the buffy coats (Lifesource, CP2D BC) by countercurrent centrifugal elutriation (JE-6B, Beckman Coulter, Brea, CA, USA), as previously described.85 Monocytes were allowed to adhere to plastic plates for 1 h in RPMI medium (Life Technologies, 11875–093) without serum and were differentiated to macrophages in vitro for 7 d in RPMI containing 20% fetal bovine serum, 1 μg/ml polymyxin B sulfate (Sigma, 1405–20–5), 120 units/ml penicillin and streptomycin.86 The mouse J774A1 macrophage-like cell line and the HEK 293 cell line were purchased from ATCC (TIB-67 and CRL-1573) and cultured in vitro in RPMI1640 medium (J774A1) or DMEM (HEK293) containing 10% fetal bovine serum.
The silencing of SNAPIN
The forced reduction of SNAPIN and CTSD in primary macrophages was achieved by siRNA transfection. In brief, macrophages were transfected with 200 nM of a nonspecific (NS) siRNA or SNAPIN siRNA (GACUGAGACGGCUAAACCA; Sigma, SASI-Hs01–00205757), SNAPIN-C siRNA (CAGUAGCAGUGUUGAUAGATT; Santa Cruz Biotechnology, sc-45545C), CTSD siRNA (Qiagen, SI00029813) with Lipofectamine 2000 (Life Technologies, 12566014), following the vender's protocol. Forty-eight h after siRNA transfection, macrophages were collected for experiments. The HEK293 SNAPIN and J774A1 Snapin shRNA cell lines were established by infection of lentivirus pLKO.1 puro vector (Addgene, 8453, deposited by Bob Weinberg) expressing SNAPIN shRNA (ACGACUGAGACGGCUAAACCA) for HEK293 and Snapin shRNA (ACGACUAAGGCGGUUAAACCA) for J774A1 cells. A pLKO.1 puro vector expressing a scrambled shRNA (SCR) served as control.87
Immunoblot analysis and co-immunoprecipitation (co-IP)
Immunoblot analysis employed whole-cell protein extracts (60 μg) prepared from macrophages or cell lines, which were electrophoresed on SDS-PAGE 12% polyacrylamide gels and transferred to Immobilon-P membranes (Millipore, IPSN07852) employing a semidry electro blotter (Bio-Rad, Hercules, CA, USA). The membranes were then blocked in 5% nonfat milk in PBST (phosphate-buffered saline [PBS; Corning cellgro, 21–031-CV] containing 0.2% Tween-20 [Sigma, P1379]) and subsequently incubated overnight at 4°C with primary antibody. Membranes were washed in PBST and incubated with either donkey anti-rabbit (NA935V) or anti-mouse secondary antibodies (NA931V) conjugated with horseradish peroxidase (1:3300 dilution; Amersham Pharmacia Biotech). The specific proteins were detected employing the Enhanced Chemiluminescent Detection Reagent (Amersham Pharmacia Biotech, RPN2232).7 The following primary antibodies were used: a mouse monoclonal anti-human SNAPIN (UC Davis, NeuroMab Facility, 75–045); polyclonal antibodies recognizing mouse and human SNAPIN (Proteintech, 10055–1-AP); monoclonal anti-human LAMP1 (H4A3), LAMP2 (H4B4) (both from the Developmental Studies Hybridoma Bank, University of Iowa), anti-mouse CTSD (Abcam, ab6313), anti-ACTB (Sigma, A2066), and mouse anti-HSPA5/BIP (BD Transduction Lab, 610978). Polyclonal antibodies to human LAMP1 (9091), anti-LC3B (3868), RAB7 (9367), SQSTM1 (5114), and CTSD (2284) were purchased from Cell Signaling Technology. The anti-CTSB (Abcam, ab33538) and anti-NDUFS3 antibodies (Abcam, ab110246) recognize both human and mouse species.
Co-IP was performed employing NS- or SNAPIN-siRNA-transfected macrophage whole cell lysates (1000 μg) that were immunoprecipitated employing a mouse monoclonal anti-SNAPIN antibody (10 μg) which was conjugated to protein A agarose (Roche, 11719408001). Following electrophoresis the blots were probed with antibodies to SNAPIN, LAMP1, LC3 and RAB7. Reciprocal co-IP was performed using anti-LAMP1 antibodies.
Electron microscopy
Monocytes isolated from buffy coat were seeded onto Thermanox plastic coverslips (NuncLabware product, GG-12-pre) and allowed to differentiate to macrophages, as described above. Macrophages were then transfected with NS or SNAPIN siRNAs, and after 48 h cells were fixed overnight at 4°C in a 0.2 M sodium cacodylate buffer (pH 7.4) containing 2% glutaraldehyde. Cells were then post-fixed in cacodylate-buffered 1% osmium tetroxide, dehydrated, and embedded in Epon 812 (Nacalai Tesque, Inc.) for ultrathin sectioning. The cells were stained with uranyl acetate and lead citrate and examined employing a FEI Tecnai Spirit G2 transmission electron microscope (FEI, Hillsboro, OR, USA).
Lysosome enrichment and autophagic vacuole precipitation
The lysosomes from the J774A1 macrophage cell line were enriched in fraction 1 (F1) by gradient ultracentrifuation employing a Lysosome Enrichment Kit (Thermo scientific, 89839)88 following cell homogenization according to the vender's recommendations. Since LC3-II was enriched in F1, AVs were enriched employing Dynabeads protein A (Life Technology, 10006D) conjugated with anti-LC3 antibody according to the company's instructions, which were subjected to immunoblot analysis.
Immunofluorescence microscopy
Cells were allowed to attach to pre-cleaned 12-mm glass coverslips, fixed with 4% paraformaldehyde and permeabilized with acetone for 10 min on ice, and then blocked for 30 min with blocking buffer in 1×PBS containing 0.3% Triton X-100 (Sigma, 9002–93–1), 3% BSA (Sigma, A3059), 5% normal goat serum (Sigma, G9023) and immunostained with the following antibodies alone or in combination: mouse monoclonal human anti-SNAPIN (1:200), rabbit monoclonal anti-LAMP1 (9091), anti-RAB7 (9367) and polyclonal anti-CTSD (2284). Secondary antibodies were Alexa Fluor 488-, 546-, or 647-conjugated goat antibodies to mouse IgG or rabbit IgG (Thermo Fisher, A-11017, A-11071, A-20990). For LC3 staining, cells were fixed with ice-cold methanol for 20 min, followed by LC3B (D11) staining (1:200). Nuclei were counterstained with DAPI (4,6-diamidino-2-phenylindole; Invitrogen, D1306). BODIPY FL-pepstatin A (1 μM; Thermo Fisher, P12271) was employed to stain the active form of CTSD. Cells were washed with PBS containing 0.1% Triton X-100, and coverslips were mounted using Fluoromount-G (Southern Biotech, 0100–01). Images were acquired by fluorescence microscopy on a Nikon TE2000E2-PFS (Nikon) with a 60× oil immersion objective and some images were analyzed following deconvolution, as identified in the figures. Nikon NIS-Elements imaging software was employed.7
Evaluation of lysosomal pH
Dextran is delivered to lysosomes via endocytosis. In order to assess lysosomal pH, dextran labeled with pH-sensitive or -insensitive fluorescence dyes was employed.89 Human macrophages transfected with NS or SNAPIN siRNAs, or incubated 2 h with 100 nM of bafilomycin A1 (Calbiochem, 196000), were then incubated for 2 h with the pH-sensitive dextran pHrodo green (12 μg/ml; Thermo Fisher, P35368) and the dextran labeled with pH-insensitive Alexa Fluor 546 (12 μg/ml; Thermo Fisher, D22911) and chased for 6 h. Cells were fixed with 4% paraformaldehyde overnight and nuclei stained with DAPI for 5 min. Red and green fluorescence in single macrophages was measured with the Nikon NIS element imaging software and the green to red fluorescence ratio was calculated.
Acidification of autophagic vacuoles
HEK293 SNAPIN shRNA and SCR shRNA cell lines were transfected with ptf-LC3 plasmid (Addgene, 21074; deposited by Tamotsu Yoshimori). Ptf-LC3 expresses LC3 fused with both GFP and RFP fluorescent tags. AVs incorporate LC3 and appear as fluorescent puncta.34 In acidified AVs, such as autolysosomes, the fluorescence of RFP-GFP-LC3 is red dominant, because green fluorescence is quenched while RFP remains stable in a low pH environment. The ratio of green to red fluorescence reflects the acidification in AVs.35
Lysosome-autophagosome fusion
After formation, autophagosomes fuse with lysosomes forming an autolysosome, in which LAMP1 will remain on the outer membrane and LC3B will localize either on the outer membrane or inside the autolysosomes.90 To evaluate lysosome-autophagosome fusion, macrophages were fixed with ice-cold methanol for 20 min, and stained overnight with a LC3B and LAMP1 antibodies, respectively incubated with a secondary antibody labeled with red or green fluorescent tags. Single-positive LC3 (red) or double-positive LC3 and LAMP1 (yellow) puncta were counted. The total number of LC3 punta in each cell included those that were LC3 only, as well as those that were LC3 and LAMP1 double positive. The percentage of LC3 and LAMP1 double-positive:LC3 single-positive puncta assesses lysosome-autopagosome fusion. The Pearson's correlation coefficient,91 which is a measure of colocalization between LAMP1 and LC3 was measured by NIS-element imaging software.
Endosome to lysosome trafficking
Endocytic trafficking to lysosomes was examined by Alexa Fluor 488 green-labeled dextran.92 Primary human macrophages were incubated with 0.1 mg/ml of Alexa Fluor 488-Dextran (10,000MW; Life Technology, 1441205) for 2 h at 37°C, washed with PBS, and incubated in fresh medium for 16 h, then fixed with 4% paraformaldehyde and examined by immunofluorescence microscopy.
Lysosomal H+ leak employing J774A1 macrophages
J774A1 macrophage cell lines stably expressing a SCR or Snapin shRNA were employed. The cells were seeded on an 8-chambered coverglass plate (Lab-Tek, 155411) and incubated overnight with 8 μg/ml of a pH-sensitive pHrodo Red dextran (Life Technologies, P10361) and 30 μg/ml of a pH-insensitive Alexa Fluor 488 green dextran (Life Technolegies, D22910). The medium was refreshed with normal culture medium and cells were chased for 8 h allowing dextran delivery to lysosomes. The H+-pump was inhibited by incubating cells in isotonic Na+ buffer (140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM glucose, 20 mM HEPES [ThermoFisher 15630080], pH 7.4) containing 2 μM V-ATPase inhibitor concanamycin A (CcA; Santa Cruz Biotechnology, sc-202111A). The leak of lysosomal H+ in J7774A1 cells was assessed by the ratio change of red to green fluorescence after the V-ATPase was inhibited. Live cell ratiometric fluorescence microscopy was performed employing a Nikon TE2000E2-PFS miscroscope. Immediately after adding CcA in Na+ buffer, the cells were photographed automatically in half-minute intervals for 5 min. The ratio of red to green fluorescence intensity in each J774A1 cell, which is inversely related to lysosome pH, was calculated in the imaging from each time point.
Lysosome re-acidification after dissipation of lysosomal transmembrane ΔpH
J774A1 SCR shRNA and Snapin shRNA-expressing cell lines were incubated and chased with pHrodo red dextran and Alexa Fluor 488 dextran and underwent live cell imaging, as described above. Lysosomal transmembrane ΔpH of J774A1 cells was dissipated by incubating for 5 min in a K+ buffer (143 mM KCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM glucose, 20 mM HEPES, pH 7.4) containing 450 μM carbonyl cyanide m-chlorophenyl hydrazone (CCCP; Calbiochem, 215911), a protonophore that moves protons across lipid bilayers. Re-acidification of lysosomes was initiated by replacing CCCP in K+ buffer with a Na+ buffer (defined above). The live cell images were taken in half-minute intervals for 10 min. The red and green fluorescence intensity in each cell at each time point was recorded and the ratio of red:green was calculated and presented to represent the pH change in the lysosomes.
Statistical analysis
Experimental data are presented as the mean ± 1 SEM. The statistical differences between groups were determined by Student t test and for multiple parameters by ANOVA followed by Tukey analysis. P values less than 0.05 were considered statistically significant.
Supplementary Material
Abbreviations
- ATG
autophagy related
- AV
autophagic vacuole
- CcA
Concanamycin A
- CCCP
carbonyl cyanide m-chlorophenyl hydrazone
- EGFP
enhanced green fluorescent protein
- F1
fraction 1
- LAMP1
lysosomal-associated membrane protein 1
- MAP1LC3/LC3
microtubule-associated proteins 1 light chain 3
- mRFP
monomeric red fluorescent protein
- MTOR
mechanistic target of rapamycin
- NDUFS3
NADH:ubiquinone oxidoreductase core subunit S3
- NFKB
nuclear factor kappa B subunit
- NS
nonspecific
- PtdIns3K
class III phosphatidylinositol 3-kinase
- RA
rheumatoid arthritis
- SCR shRNA
scrambled shRNA
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- shRNA
small hairpin RNA
- siRNA
small interfering RNA
- SNAP25
synaptosome associated protein 25
- SNAPIN
SNAP associated protein
- SNARE
soluble NSF attachment protein receptor
- SQSTM1/p62
sequestosome 1
- TLR2
toll-like receptor 2
- TLR4
toll like receptor 4
- V-ATPase
vacuolar H+-translocating ATPase
- VTI1B
vesicle transport through interaction with t-SNAREs 1B
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the grants from National Institute of Health AR065076 (RMP), AR064349 (CS), DK094980 (CH), AR066739 (AD) and the grant from the Rheumatology Research Foundation and support from the Solovy Arthritis Research Society of Northwestern University (RMP).
References
- [1].Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 2014; 14:392-404; PMID:24854589; http://dx.doi.org/ 10.1038/nri3671 [DOI] [PubMed] [Google Scholar]
- [2].Sheng J, Ruedl C, Karjalainen K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015; 43:382-93; PMID:26287683; http://dx.doi.org/ 10.1016/j.immuni.2015.07.016 [DOI] [PubMed] [Google Scholar]
- [3].Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011; 11:723-37; PMID:21997792; http://dx.doi.org/ 10.1038/nri3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol 2013; 5:a008748; PMID:23284042; http://dx.doi.org/ 10.1101/cshperspect.a008748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, et al.. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 2009; 15:774-80; PMID:19561617; http://dx.doi.org/ 10.1038/nm.1987 [DOI] [PubMed] [Google Scholar]
- [6].Huang QQ, Koessler RE, Birkett R, Dorfleutner A, Perlman H, Haines GK 3rd, Stehlik C, Nicchitta CV, Pope RM. Glycoprotein 96 perpetuates the persistent inflammation of rheumatoid arthritis. Arthritis Rheum 2012; 64:3638-48; PMID:22777994; http://dx.doi.org/ 10.1002/art.34610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shi B, Huang Q, Tak PP, Vervoordeldonk MJ, Huang CC, Dorfleutner A, Stehlik C, Pope RM. SNAPIN: an endogenous Toll-like receptor ligand in rheumatoid arthritis. Ann Rheum Dis 2012; 71:1411-7; PMID:22523426; http://dx.doi.org/ 10.1136/annrheumdis-2011-200899 [DOI] [PubMed] [Google Scholar]
- [8].Buxton P, Zhang XM, Walsh B, Sriratana A, Schenberg I, Manickam E, Rowe T. Identification and characterization of Snapin as a ubiquitously expressed SNARE-binding protein that interacts with SNAP23 in non-neuronal cells. Biochem J 2003; 375:433-40; PMID:12877659; http://dx.doi.org/ 10.1042/bj20030427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chheda MG, Ashery U, Thakur P, Rettig J, Sheng ZH. Phosphorylation of Snapin by PKA modulates its interaction with the SNARE complex. Nat Cell Biol 2001; 3:331-8; PMID:11283605; http://dx.doi.org/ 10.1038/35070000 [DOI] [PubMed] [Google Scholar]
- [10].Ruder C, Reimer T, Delgado-Martinez I, Hermosilla R, Engelsberg A, Nehring R, Dörken B, Rehm A. EBAG9 adds a new layer of control on large dense-core vesicle exocytosis via interaction with Snapin. Mol Biol Cell 2005; 16:1245-57; PMID:15635093; http://dx.doi.org/ 10.1091/mbc.E04-09-0817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cadwell K, Stappenbeck TS, Virgin HW. Role of autophagy and autophagy genes in inflammatory bowel disease. Curr Top Microbiol Immunol 2009; 335:141-67; PMID:19802564 [DOI] [PubMed] [Google Scholar]
- [12].Cai Q, Lu L, Tian JH, Zhu YB, Qiao H, Sheng ZH. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron 2010; 68:73-86; PMID:20920792; http://dx.doi.org/ 10.1016/j.neuron.2010.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Di Giovanni J, Sheng ZH. Regulation of synaptic activity by snapin-mediated endolysosomal transport and sorting. EMBO J 2015; 34:2059-77; PMID:26108535; http://dx.doi.org/ 10.15252/embj.201591125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cheng XT, Zhou B, Lin MY, Cai Q, Sheng ZH. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol 2015; 209:377-86; PMID:25940348; http://dx.doi.org/ 10.1083/jcb.201412046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15:713-20; PMID:23817233; http://dx.doi.org/ 10.1038/ncb2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jiang P, Mizushima N. Autophagy and human diseases. Cell Res 2014; 24:69-79; PMID:24323045; http://dx.doi.org/ 10.1038/cr.2013.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Navone F, Genevini P, Borgese N. Autophagy and neurodegeneration: insights from a cultured cell model of ALS. Cells 2015; 4:354-86; PMID:26287246; http://dx.doi.org/ 10.3390/cells4030354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 2013; 14:759-74; PMID:24201109; http://dx.doi.org/ 10.1038/nrm3696 [DOI] [PubMed] [Google Scholar]
- [19].Shibutani ST, Yoshimori T. A current perspective of autophagosome biogenesis. Cell Res 2014; 24:58-68; PMID:24296784; http://dx.doi.org/ 10.1038/cr.2013.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656-67; PMID:20478256; http://dx.doi.org/ 10.1016/j.cell.2010.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].van der Vaart A, Reggiori F. The Golgi complex as a source for yeast autophagosomal membranes. Autophagy 2010; 6:800-1; PMID:20714226; http://dx.doi.org/ 10.4161/auto.6.6.12575 [DOI] [PubMed] [Google Scholar]
- [22].Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 2012; 23:1860-73; PMID:22456507; http://dx.doi.org/ 10.1091/mbc.E11-09-0746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moreau K, Renna M, Rubinsztein DC. Connections between SNAREs and autophagy. Trends Biochem Sci 2013; 38:57-63; PMID:23306003; http://dx.doi.org/ 10.1016/j.tibs.2012.11.004 [DOI] [PubMed] [Google Scholar]
- [24].Shen HM, Mizushima N. At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci 2014; 39:61-71; PMID:24369758; http://dx.doi.org/ 10.1016/j.tibs.2013.12.001 [DOI] [PubMed] [Google Scholar]
- [25].Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805-12; PMID:15169837; http://dx.doi.org/ 10.1242/jcs.01131 [DOI] [PubMed] [Google Scholar]
- [26].Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al.. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Myers BM, Tietz PS, Tarara JE, LaRusso NF. Dynamic measurements of the acute and chronic effects of lysosomotropic agents on hepatocyte lysosomal pH using flow cytometry. Hepatology 1995; 22:1519-26; PMID:7590671 [PubMed] [Google Scholar]
- [28].Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 2011; 7:188-204; PMID:21107021; http://dx.doi.org/ 10.4161/auto.7.2.14181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181-97; PMID:19200883; http://dx.doi.org/ 10.1016/S0076-6879(08)03612-4 [DOI] [PubMed] [Google Scholar]
- [30].Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/ 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al.. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/ 10.1016/j.cell.2007.10.035 [DOI] [PubMed] [Google Scholar]
- [32].Mindell JA. Lysosomal acidification mechanisms. Ann Rev Physiol 2012; 74:69-86; PMID:22335796; http://dx.doi.org/ 10.1146/annurev-physiol-012110-142317 [DOI] [PubMed] [Google Scholar]
- [33].Jacquel A, Obba S, Boyer L, Dufies M, Robert G, Gounon P, Lemichez E, Luciano F, Solary E, Auberger P. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 2012; 119:4527-31; PMID:22452982; http://dx.doi.org/ 10.1182/blood-2011-11-392167 [DOI] [PubMed] [Google Scholar]
- [34].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/ 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- [35].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mauvezin C, Nagy P, Juhasz G, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun 2015; 6:7007; PMID:25959678; http://dx.doi.org/ 10.1038/ncomms8007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Johnson DE, Ostrowski P, Jaumouille V, Grinstein S. The position of lysosomes within the cell determines their luminal pH. J Cell Biol 2016; 212:677-92; PMID:26975849; http://dx.doi.org/ 10.1083/jcb.201507112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/ 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- [39].McLeland CB, Rodriguez J, Stern ST. Autophagy monitoring assay: qualitative analysis of MAP LC3-I to II conversion by immunoblot. Methods Mol Biol 2011; 697:199-206; PMID:21116969; http://dx.doi.org/ 10.1007/978-1-60327-198-1_21 [DOI] [PubMed] [Google Scholar]
- [40].Kovacs AL, Seglen PO. Inhibition of hepatocytic protein degradation by inducers of autophagosome accumulation. Acta biologica et medica Germanica 1982; 41:125-30; PMID:7113543 [PubMed] [Google Scholar]
- [41].Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al.. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141:1146-58; PMID:20541250; http://dx.doi.org/ 10.1016/j.cell.2010.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C, Rubinsztein DC, Ballabio A. A block of autophagy in lysosomal storage disorders. Hum Mol Genet 2008; 17:119-29; PMID:17913701; http://dx.doi.org/ 10.1093/hmg/ddm289 [DOI] [PubMed] [Google Scholar]
- [43].Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy 2005; 1:1-10; PMID:16874026; http://dx.doi.org/ 10.4161/auto.1.1.1270 [DOI] [PubMed] [Google Scholar]
- [44].Bartlett BJ, Isakson P, Lewerenz J, Sanchez H, Kotzebue RW, Cumming RC, Harris GL, Nezis IP, Schubert DR, Simonsen A, et al.. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 2011; 7:572-83; PMID:21325881; http://dx.doi.org/ 10.4161/auto.7.6.14943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hariri M, Millane G, Guimond MP, Guay G, Dennis JW, Nabi IR. Biogenesis of multilamellar bodies via autophagy. Mol Biol Cell 2000; 11:255-68; PMID:10637306; http://dx.doi.org/ 10.1091/mbc.11.1.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Johnson EE, Overmeyer JH, Gunning WT, Maltese WA. Gene silencing reveals a specific function of hVps34 phosphatidylinositol 3-kinase in late versus early endosomes. J Cell Sci 2006; 119:1219-32; PMID:16522686; http://dx.doi.org/ 10.1242/jcs.02833 [DOI] [PubMed] [Google Scholar]
- [47].Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell 2000; 11:467-80; PMID:10679007; http://dx.doi.org/ 10.1091/mbc.11.2.467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hirota Y, Kuronita T, Fujita H, Tanaka Y. A role for Rab5 activity in the biogenesis of endosomal and lysosomal compartments. Biochem Biophys Res Commun 2007; 364:40-7; PMID:17927960; http://dx.doi.org/ 10.1016/j.bbrc.2007.09.089 [DOI] [PubMed] [Google Scholar]
- [49].Ohkuma S, Poole B. Cytoplasmic vacuolation of mouse peritoneal macrophages and the uptake into lysosomes of weakly basic substances. J Cell Biol 1981; 90:656-64; PMID:7287819; http://dx.doi.org/ 10.1083/jcb.90.3.656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yoon YH, Cho KS, Hwang JJ, Lee SJ, Choi JA, Koh JY. Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest Ophthalmol Vis Sci 2010; 51:6030-7; PMID:20574031; http://dx.doi.org/ 10.1167/iovs.10-5278 [DOI] [PubMed] [Google Scholar]
- [51].Guha S, Padh H. Cathepsins: fundamental effectors of endolysosomal proteolysis. Indian J Biochem Biophys 2008; 45:75-90; PMID:21086720 [PubMed] [Google Scholar]
- [52].Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci 2008; 28:6926-37; PMID:18596167; http://dx.doi.org/ 10.1523/JNEUROSCI.0800-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tsukuba T, Yanagawa M, Kadowaki T, Takii R, Okamoto Y, Sakai E, Okamoto K, Yamamoto K. Cathepsin E deficiency impairs autophagic proteolysis in macrophages. PloS One 2013; 8:e82415; PMID:24340026; http://dx.doi.org/ 10.1371/journal.pone.0082415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Koike M, Shibata M, Waguri S, Yoshimura K, Tanida I, Kominami E, Gotow T, Peters C, von Figura K, Mizushima N, et al.. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am J Pathol 2005; 167:1713-28; PMID:16314482; http://dx.doi.org/ 10.1016/S0002-9440(10)61253-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Liaudet-Coopman E, Beaujouin M, Derocq D, Garcia M, Glondu-Lassis M, Laurent-Matha V, Prébois C, Rochefort H, Vignon F. Cathepsin D: newly discovered functions of a long-standing aspartic protease in cancer and apoptosis. Cancer Lett 2006; 237:167-79; PMID:16046058; http://dx.doi.org/ 10.1016/j.canlet.2005.06.007 [DOI] [PubMed] [Google Scholar]
- [56].Briozzo P, Morisset M, Capony F, Rougeot C, Rochefort H. In vitro degradation of extracellular matrix with Mr 52,000 cathepsin D secreted by breast cancer cells. Cancer Res 1988; 48:3688-92; PMID:3378211 [PubMed] [Google Scholar]
- [57].Linebaugh BE, Sameni M, Day NA, Sloane BF, Keppler D. Exocytosis of active cathepsin B enzyme activity at pH 7.0, inhibition and molecular mass. Eur J Biochem 1999; 264:100-9; http://dx.doi.org/ 10.1046/j.1432-1327.1999.00582.x [DOI] [PubMed] [Google Scholar]
- [58].Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer's disease and the role of defective lysosomal acidification. Eur J Neurosci 2013; 37:1949-61; PMID:23773064; http://dx.doi.org/ 10.1111/ejn.12169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Thiery J, Keefe D, Boulant S, Boucrot E, Walch M, Martinvalet D, Goping IS, Bleackley RC, Kirchhausen T, Lieberman J. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat Immunol 2011; 12:770-7; PMID:21685908; http://dx.doi.org/ 10.1038/ni.2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].DiCiccio JE, Steinberg BE. Lysosomal pH and analysis of the counter ion pathways that support acidification. J Gen Physiol 2011; 137:385-90; PMID:21402887; http://dx.doi.org/ 10.1085/jgp.201110596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lu L, Cai Q, Tian JH, Sheng ZH. Snapin associates with late endocytic compartments and interacts with late endosomal SNAREs. Biosci Rep 2009; 29:261-9; PMID:19335339; http://dx.doi.org/ 10.1042/BSR20090043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Luzio JP, Pryor PR, Bright NA. Lysosomes: fusion and function. Nat Rev Mol Cell Biol 2007; 8:622-32; PMID:17637737; http://dx.doi.org/ 10.1038/nrm2217 [DOI] [PubMed] [Google Scholar]
- [63].Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 2009; 10:623-35; PMID:19672277; http://dx.doi.org/ 10.1038/nrm2745 [DOI] [PubMed] [Google Scholar]
- [64].Antonin W, Holroyd C, Fasshauer D, Pabst S, Von Mollard GF, Jahn R. A SNARE complex mediating fusion of late endosomes defines conserved properties of SNARE structure and function. EMBO J 2000; 19:6453-64; PMID:11101518; http://dx.doi.org/ 10.1093/emboj/19.23.6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Pryor PR, Mullock BM, Bright NA, Lindsay MR, Gray SR, Richardson SC, Stewart A, James DE, Piper RC, Luzio JP. Combinatorial SNARE complexes with VAMP7 or VAMP8 define different late endocytic fusion events. EMBO Rep 2004; 5:590-5; PMID:15133481; http://dx.doi.org/ 10.1038/sj.embor.7400150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Fraldi A, Annunziata F, Lombardi A, Kaiser HJ, Medina DL, Spampanato C, Fedele AO, Polishchuk R, Sorrentino NC, Simons K, et al.. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J 2010; 29:3607-20; PMID:20871593; http://dx.doi.org/ 10.1038/emboj.2010.237 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [67].Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell 2011; 42:731-43; PMID:21700220; http://dx.doi.org/ 10.1016/j.molcel.2011.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Vanlandingham PA, Ceresa BP. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J Biol Chem 2009; 284:12110-24; PMID:19265192; http://dx.doi.org/ 10.1074/jbc.M809277200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23:33-42; PMID:9639028; http://dx.doi.org/ 10.1247/csf.23.33 [DOI] [PubMed] [Google Scholar]
- [70].Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem 2006; 281:36303-16; PMID:16963441; http://dx.doi.org/ 10.1074/jbc.M607031200 [DOI] [PubMed] [Google Scholar]
- [71].Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 2008; 4:849-50; PMID:18758232; http://dx.doi.org/ 10.4161/auto.6845 [DOI] [PubMed] [Google Scholar]
- [72].Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, Kiselyov K. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem 2006; 281:7294-301; PMID:16361256; http://dx.doi.org/ 10.1074/jbc.M508211200 [DOI] [PubMed] [Google Scholar]
- [73].Deng D, Jiang N, Hao SJ, Sun H, Zhang GJ. Loss of membrane cholesterol influences lysosomal permeability to potassium ions and protons. Biochim Biophys Acta 2009; 1788:470-6; PMID:19109925; http://dx.doi.org/ 10.1016/j.bbamem.2008.11.018 [DOI] [PubMed] [Google Scholar]
- [74].Perrotta I, Carito V, Russo E, Tripepi S, Aquila S, Donato G. Macrophage autophagy and oxidative stress: an ultrastructural and immunoelectron microscopical study. Oxid Med Cell Longev 2011; 2011:282739; PMID:21922037; http://dx.doi.org/ 10.1155/2011/282739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bonilla DL, Bhattacharya A, Sha Y, Xu Y, Xiang Q, Kan A, Jagannath C, Komatsu M, Eissa NT. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 2013; 39:537-47; PMID:24035364; http://dx.doi.org/ 10.1016/j.immuni.2013.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, et al.. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A 2012; 109:E3168-76; PMID:23093667; http://dx.doi.org/ 10.1073/pnas.1210500109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 2012; 15:534-44; PMID:22440612; http://dx.doi.org/ 10.1016/j.cmet.2012.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Sergin I, Bhattacharya S, Emanuel R, Esen E, Stokes CJ, Evans TD, Arif B, Curci JA, Razani B. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci Signal 2016; 9:ra2; PMID:26732762; http://dx.doi.org/ 10.1126/scisignal.aad5614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kanayama M, He YW, Shinohara ML. The lung is protected from spontaneous inflammation by autophagy in myeloid cells. J Immunol 2015; 194:5465-71; PMID:25911758; http://dx.doi.org/ 10.4049/jimmunol.1403249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Abdel Fattah E, Bhattacharya A, Herron A, Safdar Z, Eissa NT. Critical role for IL-18 in spontaneous lung inflammation caused by autophagy deficiency. J Immunol 2015; 194:5407-16; PMID:25888640; http://dx.doi.org/ 10.4049/jimmunol.1402277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Kanayama M, Inoue M, Danzaki K, Hammer G, He YW, Shinohara ML. Autophagy enhances NFkappaB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun 2015; 6:5779; PMID:25609235; http://dx.doi.org/ 10.1038/ncomms6779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lin NY, Beyer C, Giessl A, Kireva T, Scholtysek C, Uderhardt S, Munoz LE, Dees C, Distler A, Wirtz S, et al.. Autophagy regulates TNFalpha-mediated joint destruction in experimental arthritis. Ann Rheum Dis 2013; 72:761-8; PMID:22975756; http://dx.doi.org/ 10.1136/annrheumdis-2012-201671 [DOI] [PubMed] [Google Scholar]
- [83].Ireland JM, Unanue ER. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med 2011; 208:2625-32; PMID:22162830; http://dx.doi.org/ 10.1084/jem.20110640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, Berdnikovs S, Filer A, Clark AR, Buckley CD, et al.. Nonclassical Ly6C(−) monocytes drive the development of inflammatory arthritis in mice. Cell Rep 2014; 9:591-604; PMID:25373902; http://dx.doi.org/ 10.1016/j.celrep.2014.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Liu H, Ma Y, Pagliari LJ, Perlman H, Yu C, Lin A, Pope RM. TNF-alpha-induced apoptosis of macrophages following inhibition of NF-kappa B: a central role for disruption of mitochondria. J Immunol 2004; 172:1907-15; PMID:14734776; http://dx.doi.org/ 10.4049/jimmunol.172.3.1907 [DOI] [PubMed] [Google Scholar]
- [86].Ma Y, Temkin V, Liu H, Pope RM. NF-kappaB protects macrophages from lipopolysaccharide-induced cell death: the role of caspase 8 and receptor-interacting protein. J Biol Chem 2005; 280:41827-34; PMID:16246838; http://dx.doi.org/ 10.1074/jbc.M510849200 [DOI] [PubMed] [Google Scholar]
- [87].de Almeida L, Khare S, Misharin AV, Patel R, Ratsimandresy RA, Wallin MC, Perlman H, Greaves DR, Hoffman HM, Dorfleutner A, et al.. The PYRIN Domain-only Protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity 2015; 43:264-76; PMID:26275995; http://dx.doi.org/ 10.1016/j.immuni.2015.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Yogalingam G, Hwang S, Ferreira JC, Mochly-Rosen D. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cdelta (PKCdelta) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J Biol Chem 2013; 288:18947-60; PMID:23653351; http://dx.doi.org/ 10.1074/jbc.M113.466870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ogawa M, Kosaka N, Regino CA, Mitsunaga M, Choyke PL, Kobayashi H. High sensitivity detection of cancer in vivo using a dual-controlled activation fluorescent imaging probe based on H-dimer formation and pH activation. Mol Biosyst 2010; 6:888-93; PMID:20567775; http://dx.doi.org/ 10.1039/b917876g [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry A 2010; 77:733-42; PMID:20653013; http://dx.doi.org/ 10.1002/cyto.a.20896 [DOI] [PubMed] [Google Scholar]
- [92].Baravalle G, Schober D, Huber M, Bayer N, Murphy RF, Fuchs R. Transferrin recycling and dextran transport to lysosomes is differentially affected by bafilomycin, nocodazole, and low temperature. Cell Tissue Res 2005; 320:99-113; PMID:15714281; http://dx.doi.org/ 10.1007/s00441-004-1060-x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.